

Abstract
Purpose
To report changes in the tapetal-like reflex in a female carrier of RPGR ORF15 c.3395delA X-linked retinitis pigmentosa (XLRP) between examinations at 16 and 22 years of age, and to report the observation that the tapetal-like reflex faded due to exposure to daylight and reappeared with prolonged dark adaptation at 22 years of age.
Methods
Clinical examination, kinetic Goldmann perimetry, dark adaptometry, fundus autofluorescence photography, spectral domain optical coherence tomography (SD-OCT), full-field electroretinography (ffERG), and multifocal electroretinography (mfERG) were performed.
Results
A female carrier of RPGR XLRP presented with a tapetal-like reflex at age 16. At age 22, the tapetal-like reflex was absent upon examination in daylight; however, the reflex reappeared after 12 h of dark adaptation. Fundus autofluorescence was unremarkable and did not change after prolonged dark adaptation. Full-field electroretinography and dark adaptometry at age 22 demonstrated reduced rod and cone function compared to at age 16.
Conclusions
Dark adaptation before fundus photography may enable the detection of a tapetal-like reflex where it is otherwise invisible. The light-dependent fluctuation of a disease-related substance in the photoreceptors should prompt further study of the potential role of light as a modulator of the progression of RPGR XLRP.
Introduction
Altered fundus reflexes are observed in some retinal degenerative diseases; however, their pathophysiological cause and their potential role in disease progression remain largely unknown. Examples of such altered fundus reflexes include the golden fundus reflex observed in X-linked retinoschisis and Oguchi disease [1-3], a form of congenital stationary night blindness. In the latter, the characteristic fundus reflex disappears with dark adaptation for 2 to 3 h [4-6]. For both disease entities, studies suggest that changes in the potassium concentration in the retina are involved in the underlying pathophysiology [1-3,7]. Sheen retinal dystrophy and the early retinal changes observed in men affected with X-linked retinitis pigmentosa (XLRP) may also present as increased abnormal fundus reflexes [8,9]. The fundus characteristics of women carrying an XLRP mutation may range from normal to widespread degeneration of the retinal periphery [10]. A subtle intermediate fundus abnormality for female carriers of XLRP is the so-called tapetal-like reflex, a pattern of multiple brightly scintillating, partially confluent patches spread over the posterior pole, with relative sparing of the fovea [11]. The tapetal-like reflex is generally considered specific for the carrier condition, although we recently have reported a case of tapetal-like reflex in a healthy man [12]. Fundus imaging studies indicate that the tapetal-like reflex is attributable to increased reflectivity of a component of the photoreceptors [11,13]. The heritability of the condition and the approximately even distribution of bright and dark patches of the fundus fit the assumption that the pattern is caused by the random inactivation of the X chromosome in women [14,15].
Little information is available about the time of onset, natural development, and prognostic value of the tapetal-like reflex in female XLRP carriers except that a retrospective longitudinal study by Grover et al. suggested the tapetal-like reflex was associated with a better outcome [10]. In this report, we describe a female carrier of XLRP where the tapetal-like reflex was clearly present after light exposure at 16 years of age, but much less so at 22 years of age when it did, however, reappear after prolonged dark adaptation and was attenuated by renewed light exposure.
Methods
This was a retrospective study of a female carrier of XLRP, a condition that can be associated with a variable degree of retinal degeneration that in itself warrants ophthalmological attention. Institutional approval is not required and cannot be obtained for retrospective studies under Danish law. Informed consent was obtained. The study adhered to the tenets of the Declaration of Helsinki and the ARVO statement on human subjects.
Standard clinical investigations performed when the proband was 16 and 22 years of age included automated refractioning and keratometry (Righton, Retinomax 3), determination of best-corrected visual acuity (BCVA), Ishihara color vision testing (performed only at 22 years of age), kinetic Goldmann perimetry (object size IV/4e and I/4e), Goldmann-Weeker dark adaptometry, slit-lamp biomicroscopy, and dilated funduscopy. Spectral domain optical coherence tomography (SD-OCT) and fundus autofluorescence photography were performed at 22 years of age in the light-adapted state and after 2 h of dark adaptation using a combined SD-OCT and scanning laser fundus camera (Spectralis HRA-OCT, Heidelberg Engineering, Heidelberg, Germany). For examinations after 2 h of prolonged dark adaptation, the patched proband was positioned in the darkened examination room, where only dimmed red light was used for the examiner’s spatial orientation, and the infrared fundus photo function was used before the light-emitting examinations to secure the best possible focus and alignment. Fundus autofluorescence imaging was made with averaging over 15 frames. First, images were obtained directly after the period of dark adaptation and thereafter successively recorded during a period of up to 30 min of light adaptation.
For all SD-OCT recordings, an excitation wavelength of 870 nm, acquisition speed of 40,000 A-scans per second, scan depth 1.8 mm, and a depth resolution of approximately 3.5 µm/pixel were applied. Furthermore, 19 transversal B-scans were obtained in the 1, 3, and 6 mm Early Treatment of Diabetic Retinopathy Study (ETDRS) modes with the HRA2 Spectralis Family Acquisition 5.3.3.0 Module. For all fundus autofluorescence measurements, the excitation wavelength was 488 nm, and the retina field applied was 55 degrees [16,17]. Color fundus photography was performed in the light-adapted state at the age of 16 and 22 years of age, and at 22 years of age after prolonged dark adaptation for 2 and 12 h on separate occasions. For the dark-adapted fundus photography, the proband was positioned in a darkened examination room, and a dim red light was used for the examiner’s spatial orientation. Preflash fundus illumination before fundus photography was minimized. In all fundus photography sessions, the subject fixated on the camera-mounted adjustable red light diode with the contralateral eye. When the proband was 16 and 22 years of age, full-field electroretinography (ffERG) was recorded on a Viking IV workstation (Nicolet Biomedical Instruments, Madison, WI) according to the standards of the International Society for Clinical Electrophysiology of Vision (ISCEV) [18] after pupil dilation, 30 min of dark adaptation, topical anesthesia, and a bipolar Burian-Allen cornea electrode and a reference electrode on the ipsilateral earlobe. Full-field electroretinography was repeated at 22 years of age, including examination after 6 h of prolonged dark adaptation using this method. For each response, at least two similar waveforms were required, to demonstrate reproducibility. Multifocal electroretinography (mfERG) was performed in the light-adapted state at 22 years of age after pupil dilation and topical anesthesia using a bipolar Burian-Allen cornea electrode and a reference electrode on the forehead, with stimuli (103 hexagons, full contrast, 20 degrees eccentricity) generated by the VERIS Science instrument using ver. 5.0 software (VERIS Science Electro Diagnostics Imaging, San Mateo, CA). Correct fixation during mfERG examination was ensured by continuous fundus monitoring with an infrared fundus camera integrated in the device and feed-back being given to the patient during the recording period.
The technique applied for dark adaptation before examination in the dark-adapted state was conventional dense light-proof patching. When prolonged dark adaptation for 2, 6, or 12 h was applied, mydriatics were administered in a completely darkened room and thorough patching reapplied 30 min before examination.
Results
The proband was a female carrier of X-linked retinitis pigmentosa from a known Danish family who was seen in connection with genetic counseling. Affected men from five known generations of the family suffer from severe retinitis pigmentosa and are typically blind in their fourth decade. One affected man has been subject of molecular genetic analysis, and the disease-causing mutation in the family has been identified as a c.3395delA single base-pair deletion at the 3′ end of the ORF15 exon in RPGR (Gene ID 6103; OMIM 312610). The mutation results in removal of adenosine 62 bp upstream of the ORF15 stop codon, thus inducing a frameshift and subsequently a premature stop codon affecting the last 21 amino acids of the RPGR gene product (Figure 1). A large 6,419-bp deletion including a segment of the downstream non-coding part of the ORF15 exon and exons 15a and 15b in the RPGR gene is also found in the family, but since this deletion does not affect the coding region of ORF15, the deletion is not believed to be pathogenic. These results were previously published by Neidhardt et al. [19]. Subsequent molecular genetic analysis of the proband confirmed her carrier status. Another female carrier who was examined at the age of 44 years at that time had retinitis pigmentosa-like fundus abnormalities, tunnel vision, night blindness, and subnormal visual acuity. The proband was a nullipara who at the age of 6 years had been treated for hypermetropia and strabismic and astigmatic amblyopia in her left eye (Figure 1, Table 1). She was first seen at our clinic at the age of 16 years when slit-lamp biomicroscopy was unremarkable and ophthalmoscopy revealed normal optic discs, normal retinal vessels, and normal fundi including the periphery, except that a marked tapetal-like reflex was found, most conspicuously temporal of the fovea. Relative sparing of the fovea was observed. No fundus hyperpigmentation or atrophy was seen (Figure 2A–D). Goldmann visual fields (object sizes IV/4e and I/4e) were normal, Goldmann-Weekers dark adaptometry showed a normal biphasic response (Table 1, Figure 3), and standard full-field ERG [9] showed normal scotopic and photopic response configurations, implicit times, and amplitudes (Table 2, Figure 4). We saw the proband again when she was 22 years old, and BCVA was 1.0 (decimal acuity scale) in her right eye and 0.66 (decimal acuity scale) in her left eye (Table 1). She had no visual complaints, slit-lamp biomicroscopy was unremarkable, Goldmann fields (object sizes IV/4e and I/4e) were normal, and the patient had a biphasic Goldmann-Weekers dark adaptometry response, albeit delayed in reaching the final threshold (Table 1, Figure 3). Standard full-field ERG [18] after 30 min of dark adaptation showed normal scotopic and photopic response configurations and implicit times, except the patient had slightly longer than normal implicit times of the scotopic rod-cone b-wave and the oscillatory potentials (reference values from 85 eyes in 85 healthy adults). The amplitudes were within normal range except the rod a-wave (which represents activation of the photoreceptors), which was borderline subnormal. The amplitudes were, however, generally lower when examined at 22 years compared to 16 years of age (Table 2, Figure 4). When the proband was 22, ffERG was performed twice, after 30-min and 6-h dark adaptation, respectively. Compared to the ffERG at age 16, rod response was reduced by 28% at the age of 22. The combined rod-cone response a wave (resulting from activity of the photoreceptors) and b wave (resulting from activity of the bipolar and Muller cells) were reduced by 39% and 17%, respectively. Under photopic conditions, single flash cone response was reduced by about 41% and 30-Hz flicker response by 36%. No significant changes were recorded after the 6-h dark adaptation. Multifocal electroretinography on the right eye after light exposure showed normal first-order latencies and amplitude values throughout the 103 hexagons, within 20 degrees eccentricity (Figure 5).
Figure 1.
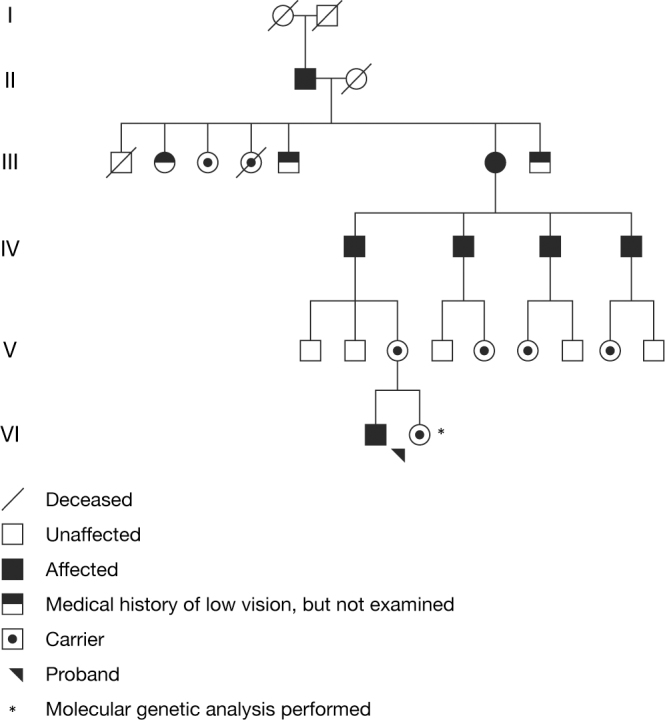
Family pedigree of RPGR ORF15 X-linked retinitis pigmentosa. Affected men from five known generations of the family suffer from severe retinitis pigmentosa and are typically blind in their fourth decade. The disease-causing family mutation in RPGR ORF15 was identified with molecular genetic analysis of one affected male subject (fourth generation, third individual from the left). A woman with the mutation examined at the age of 44 years had retinitis pigmentosa-like fundus abnormalities, tunnel vision, night blindness, and subnormal visual acuity (third generation, sixth individual from the left). The proband is a non-obligate carrier who at 16 years of age presented with a tapetal-like reflex in the light adapted state, but at reexamination at 22 years of age showed marked attenuation of the tapetal-like reflex in the light adapted state and reappearance of the tapetal-like reflex with prolonged dark adaptation.
Table 1. Clinical characteristics of a female carrier of RPGR X-linked retinitis pigmentosa.
| Age | Right/Left | BCVA* | Refraction** | Slit lamp biomicroscopy | IOP (mmHg) | Goldmann perimetry (IV/4e and I/4e) | Goldmann dark adaptometry |
Color vision (Ishihara) | |
|---|---|---|---|---|---|---|---|---|---|
| Pattern | Final threshold | ||||||||
| 16 years |
RE |
1 |
+6.0/–4.0/10° |
Normal |
N/A |
No scotomata, and normal outer boundaries |
Biphasic |
Normal |
N/A |
| LE |
0.5 |
+7.0/-5.0/170° |
Normal |
N/A |
No scotomata, and normal outer boundaries |
N/A |
|||
| 22 years |
RE |
1 |
+6.5/-5.0/5° |
Normal |
12 |
No scotomata, and normal outer boundaries |
Biphasic |
Normal |
Normal |
| LE | 0.66 | +7.25/-4.5/174° | Normal | 12 | No scotomata, and normal outer boundaries | Normal | |||
RE: Right eye, LE: Left eye, BCVA: Best corrected visual acuity, IOP: Intra ocular pressure, mmHg: millimeter Mercury, N/A: Data not available, * Decimal acuity scale, ** Refraction values spherical/cylindrical/axis
Figure 2.

Color fundus photographs of the right eye of female carrier of RPGR ORF15 X-linked retinitis pigmentosa. A: After light exposure at the age of 16 years, a tapetal-like reflex is present, most conspicuously temporal of the fovea. B: After light exposure at the age of 22 years, no tapetal-like reflex can be seen. C: After 2 h of dark adaptation at the age of 22 years, a discreet tapetal-like reflex is present. D: After 12 h of dark adaptation at the age of 22 years, a clearly visible tapetal-like reflex is present, most conspicuously temporal of the fovea, in a pattern similar to the initial presentation at the age of 16 years.
Figure 3.

Goldmann-Weeker dark adaptation curves at the age 16 and 22 in a female carrier of RPGR ORF15 X-linked retinitis pigmentosa. The rod sensitivity after 30-min dark adaptation is similar at age 16 and age 22. The threshold value is about 2.6 for both curves. However, there is a delay of 3–5 min in the blue curve performed at age 22. This delay occurs about 10 min after the dark adaptation and lasts for about 7 min. Furthermore, at age 16, the dark adaptation final threshold is reached after 14 min, compared to 17 min at age 22. This may indicate a progressive delay in the rod activation kinetics.
Table 2. Full-field electroretinography examination results (right eye) in a female carrier of RPGR ORF15 X-linked retinitis pigmentosa at age 16 and 22 years.
| Parameters |
Scotopic |
Photopic |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Stimulus | Rod | Rod-cone a-wave | Rod-cone b-wave | Oscillatory potentials | Single flash cone response | 30 Hz Flicker | Red | Green | Blue |
| Normal values (n=85) | |||||||||
| Median amplitude (µV) |
210 |
205 |
427 |
77 |
136 |
101 |
105.2 |
114.6 |
4.4 |
| (5–95th percentile) |
(124–302) |
(111–312) |
(285–674) |
(29–125) |
(61–154) |
(61–154) |
(64.5–172.9) |
(62–181) |
(0.9–8.9) |
| Median implicit times (ms) |
103 |
23 |
48 |
25.2 |
29 |
26 |
29.2 |
28 |
43.6 |
| (5–95th percentile) |
(81–123) |
(21.0–25.0) |
(41.0–55.0) |
(24.0–27.2) |
(27.0–32.0) |
(25.0–30.0) |
(27.0–32.0) |
(26.0–32.2) |
(39.4–51.0) |
| 16 years* | |||||||||
| Amplitudes (µV) |
335 |
237 |
538 |
94.9 |
201 |
139 |
159 |
184 |
2.91 |
| Implicit times (ms) |
88.5 |
21.5 |
57.5 |
24.6 |
31.5 |
30.5 |
30.8 |
30.6 |
44 |
| 22 years* |
|||||||||
| Amplitudes (µV) |
240 |
145 |
448 |
31.1 |
118 |
89.2 |
109 |
114 |
2.41 |
| Implicit times (ms) |
100 |
21.5 |
62 |
31.6 |
32.5 |
27.5 |
31.2 |
31.6 |
46 |
| 22 years | |||||||||
| (6 h dark adaptation) |
|||||||||
| Amplitudes (µV) |
230 |
155 |
415 |
41.8 |
107 |
92 |
96.8 |
119 |
N/A |
| Implicit times (ms) | 94.5 | 23 | 55 | 26 | 34 | 32.5 | 32.6 | 33 | N/A |
µV=microvolts ms=milliseconds * Full-field electroretinography was performed after standard dark adaptation for 30 min. N/A: Data not available.
Figure 4.

Full-field electroretinography (ffERG) performed at the age of 16 and six years later in a female carrier of RPGR ORF15 X-linked retinitis pigmentosa. At the age of 22, ffERG was performed twice: after 30 min and 6 h of dark adaptation, respectively. Compared to the ffERG at age 16, rod response was reduced by 28% at the age of 22. The combined rod-cone response a wave (resulting from activity of the photoreceptors) and b wave (resulting from activity of the bipolar and Muller cells) were reduced by 39% and 17%, respectively. Under photopic conditions, single flash cone response was reduced by about 41% and 30-Hz flicker response by 36%. No significant changes were recorded after the 6-h dark adaptation. The figure corresponds to Table 2. cd.s/m2=candela seconds per square meter. µV=microvolts. ms=milliseconds.
Figure 5.

Multifocal electroretinography of the right eye female carrier of RPGR ORF15 X-linked retinitis pigmentosa demonstrates normal first-order latencies and amplitude values for all 103 hexagons, within 20 degrees eccentricity. ms=milliseconds. nV/deg2=nanovolts per degree squared.
When fundus examination was made at 22 years after at least 30-min light adaptation, the optic discs, retinal vessels, and fundus including the periphery in both eyes were normal, and the previously observed tapetal-like reflex was missing bilaterally. As in the previous examination, no fundus hyperpigmentation or atrophy was seen. This finding was confirmed with color fundus photography (Figure 2B). Transfoveal horizontal spectral-domain optical coherence tomography in the light-adapted state showed an ordered configuration of the photoreceptor/retinal pigment epithelium complex. Renewed ophthalmoscopy after 2 h of dark adaptation showed that the tapetal-like reflex had reappeared in a discrete pattern resembling that seen 6 years earlier (Figure 2C). Spectral-domain optical coherence tomography after 2 h of dark adaptation appeared to be unchanged (Figure 6A). Autofluorescence fundus photography in the light-adapted state at 22 years showed no abnormal pattern and thus no reminiscence of tapetal-like reflex (Figure 6B), and the same was true after 2 h of dark adaptation (Figure 6C). At the age of 22 years, after 12 h of dark adaptation, a pronounced tapetal-like reflex with a pattern identical to that seen at age 16 years was found in both eyes (Figure 2D).
Figure 6.

Spectral-domain optical coherence tomogram and fundus autofluorescence in a female carrier of RPGR ORF15 X-linked retinitis pigmentosa. Horizontal transfoveal spectral-domain optical coherence tomogram (A) from the left eye of the female carrier of XLRP after 2 h of dark adaptation at the age of 22 years. An ordered configuration of the photoreceptor/retinal pigment epithelium complex is seen. Except a slightly undulating ellipsoid line, there seems to be no obvious difference compared to the normal optical coherence tomogram of a healthy subject. Retina layers from bottom upward: RPE=retinal pigment epithelium/Bruch’s membrane complex, ISe=inner segment ellipsoid, ONL=outer nuclear layer, OPL=outer plexiform layer, INL=inner nuclear layer, IPL=inner plexiform layer, NFL=nerve fiber layer. Autofluorescence fundus photography was performed at the age of 22 years. No abnormal pattern and thus no reminiscence of tapetal-like reflex was noted on imaging obtained in the light adapted state (B, last image in a series of five images), or after 2 h of dark adaptation (C, first image in a series of five images). The patient’s autofluorescence fundus images displayed a sensitivity to dark adaptation that is also known from subjects with normal eyes. Thus, a progression was seen from the first to the last image in that the intensity of autofluorescence gradually increased, except in the fovea. This phenomenon is believed to reflect the bleaching of the photoreceptor photopigment as the result of continued exposure to blue light and the shielding of the foveal fluorophores by the xanthophyll pigment. The autofluorescence images showed no trace of the tapetal reflex pattern, neither before nor after bleaching.
Discussion
Our female carrier of RPGR XLRP presented with a tapetal-like reflex after light exposure when first examined at 16 years of age, but when she was examined at 22 years of age, the reflex was repeatedly absent in the light-adapted state during daylight but reappeared after prolonged dark adaptation. Standard examination procedures were the same at the two visits. The only addition at the second visit was the repeated examination after prolonged dark adaptation at age 22 years. Bleaching of the tapetal reflex in a carrier of XLRP and its reappearance after dark adaptation has not previously, to the best of our knowledge, been described in carriers of XLRP. This observation suggests that the tapetal-like reflex may be more common than routine clinical observation suggests and that detection of the carrier state with ophthalmoscopy may be improved by performing fundus photography in the dark-adapted state when relevant.
Although only two examinations were made and at the relatively short interval of 6 years, the finding that the tapetal-like reflex had weakened, combined with a reduction in ffERG rod and cone responses over time, suggests that it is related to the disease process in carriers of RPGR XLRP that will eventually induce photoreceptor loss. Fishman et al. examined the variability in the ffERG and concluded that the threshold for significant increase or decrease in inter-visit ERG amplitudes at a 95% confidence level for patients with RP and visually normal subjects was often at or above 25% [20]. According to this, rod and cone function may have declined significantly in our carrier of RPGR XLRP. Thus, the tapetal like reflex may also hold the key to understanding the pathogenesis of RPGR XLRP, provided that retinal degeneration in carriers is indeed just a weak version of the highly aggressive course of disease in men with RPGR XLRP.
The mechanism that produces the tapetal-like reflex is unknown. A microperimetry study of three XLRP carriers with a tapetal-like reflex by Genead et al. found reduced retinal sensitivity and increased fundus autofluorescence corresponding to the bright patches of the tapetal-like reflex. Optical coherence tomography did not show any structural abnormalities, although increased reflectivity from the RPE-photoreceptor layer complex was observed in two of the three carriers [21]. In our patient, although the tapetal like reflex reappeared after dark adaptation, optical coherence tomography did not change in response to dark adaptation, and thus did not seem to present any further clues as to the anatomic substrate of the this reflex.
Wegscheider et al. [22] reported abnormal fundus autofluorescence in nine of 11 carriers of XLRP, but only two of the nine carriers with abnormal fundus autofluorescence also had a tapetal-like reflex. In one, the tapetal-like reflex was discrete, whereas the abnormal fundus autofluorescence had a radial pattern, and in the carrier with a marked tapetal-like reflex, the fundus autofluorescence was less pronounced and located around the fovea [22]. Thus, no characteristic correlation between the presence of a tapetal-like reflex, fundus autofluorescence abnormalities, and structural abnormalities in the retina has been established. The unremarkable autofluorescence pattern, and normal retinal microstructure and thickness detected in the light-adapted state and after 2 h of dark adaptation in the female carrier reported here, is yet another example of the diverse XLRP carrier phenotype. Furthermore, Sheng et al. reported that female carriers in a large Chinese pedigree of XLRP caused by mutation in the RPGR gene predominantly presented with variable degrees of myopia, but no clinical signs of retinitis pigmentosa or a tapetal-like reflex on examination [23]. This is in contrast to the Danish pedigree presented here, where an obligate carrier presented with retinitis pigmentosa-like fundus abnormalities and clinical symptoms, and the proband presented with a tapetal-like reflex and hypermetropia. Thus, the RPGR XLRP carrier phenotype is variable, and the clinical presentation may range from asymptomatic to severely affected.
It has been suggested that the increase in fundus autofluorescence in XLRP carriers could be caused by accumulation of retinoid derivatives in the photoreceptor inner segment [21]. Our observation of short-term dark adaptation-dependent changes in the tapetal-like reflex support the argument, that the slowly and steadily accumulating fluorophore lipofuscin is not involved in the tapetal-like reflex. The suggested location of fluorophore changes in the photoreceptor is supported by the RPGR gene encoding a guanosine triphosphase (GTPase) regulator that is expressed in the outer segments of human rods and cones and in the cilia connecting the inner and outer segments of the photoreceptors [24-26]. At present, there is no clear link between the type of RPGR mutation and the presence of a tapetal-like reflex. However, the observation that the tapetal-like reflex can reappear after prolonged dark adaptation in an RPGR XLRP carrier suggests that a quantitative deficit in the RPGR gene product leads to some abnormality during dark adaptation, perhaps the accumulation in the photoreceptor inner or outer segments of a component of the visual cycle that is reflective, colored, and, perhaps, fluorescent. The opposite effect, attenuation of the tapetal-like reflex by light adaptation suggests clearance of the accumulated substance during light exposure. If the accumulated substance is pathogenic, then it is obvious that light must be suspected of modulating the progression of XLRP and that dark adaptation may promote disease progression. This hypothesis should clearly be examined in more detail before any therapeutic conclusions can be drawn.
Acknowledgments
The study was supported by the Øjenfonden under the Danish Society for the Visually Disabled, the Dag Lenards Fond, Stiftelsen Kronprinsessan Margaretas Arbetsnämnd för Synskadade and Stiftelsen för synskadade i fd Malmöhus län, Sweden and Cronqvists Stiftelse of the Swedish Society of Medicine. We thank Mr. Robert Nilsson at Landskrona Hospital, Landskrona, Sweden and Mr. Johnny Ring at Lund University, Department of Clinical Sciences, Ophthalmology, Malmö, Sweden, for technical support.
References
- 1.Vincent A, Shetty R, Yadav NK, Shetty BK. Foveal schisis with Mizuo phenomenon: etiopathogenesis of tapetal reflex in X-linked retinoschisis. Eye (Lond) 2009;23:1240–1. doi: 10.1038/eye.2008.170. [DOI] [PubMed] [Google Scholar]
- 2.Robson AG, Mengher LS, Tan MH, Moore AT. An unusual fundus phenotype of inner retinal sheen in X-linked retinoschisis. Eye (Lond) 2009;23:1876–8. doi: 10.1038/eye.2008.358. [DOI] [PubMed] [Google Scholar]
- 3.Miyake Y, Terasaki H. Golden tapetal-like fundus reflex and posterior hyaloid in a patient with x–linked juvenile retinoschisis. Retina. 1999;19:84–6. doi: 10.1097/00006982-199901000-00018. [DOI] [PubMed] [Google Scholar]
- 4.Usui T, Ichibe M, Ueki S, Takagi M, Hasegawa S, Abe H, Sekiya K, Nakazawa M. Mizuo phenomenon observed by scanning laser ophthalmoscopy in a patient with Oguchi disease. Am J Ophthalmol. 2000;130:359–61. doi: 10.1016/s0002-9394(00)00532-8. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet. 1995;10:360–2. doi: 10.1038/ng0795-360. [DOI] [PubMed] [Google Scholar]
- 6.Noble KG, Margolis S, Carr RE. The golden tapetal sheen reflex in retinal disease. Am J Ophthalmol. 1989;107:211–7. doi: 10.1016/0002-9394(89)90302-4. [DOI] [PubMed] [Google Scholar]
- 7.de Jong PT, Zrenner E, van Meel GJ, Keunen JE, van Norren D. Mizuo phenomenon in X-linked retinoschisis. Pathogenesis of the Mizuo phenomenon. Arch Ophthalmol. 1991;109:1104–8. doi: 10.1001/archopht.1991.01080080064029. [DOI] [PubMed] [Google Scholar]
- 8.Goodman G, Ripps H, Siegel IM. Sex-linked ocular disorders: Trait expressivity in males and carrier females. Arch Ophthalmol. 1965;73:387–98. doi: 10.1001/archopht.1965.00970030389018. [DOI] [PubMed] [Google Scholar]
- 9.Polk TD, Gass JD, Green WR, Novak MA, Johnson MW. Familial internal limiting membrane dystrophy. A new sheen retinal dystrophy. Arch Ophthalmol. 1997;115:878–85. doi: 10.1001/archopht.1997.01100160048007. [DOI] [PubMed] [Google Scholar]
- 10.Grover S, Fishman GA, Anderson RJ, Lindeman M. A longitudinal study of visual function in carriers of X-linked recessive retinitis pigmentosa. Ophthalmology. 2000;107:386–96. doi: 10.1016/s0161-6420(99)00045-7. [DOI] [PubMed] [Google Scholar]
- 11.Cideciyan AV, Jacobson SG. Image analysis of the tapetal-like reflex in carriers of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1994;35:3812–24. [PubMed] [Google Scholar]
- 12.Schatz P, Bregnhøj J, Arvidsson H, Sharon D, Mizrahi-Meissonnier L, Sander B, Grønskov K, Larsen M. A tapetal-like fundus reflex in a healthy male: evidence against a role in the pathophysiology of retinal degeneration? Mol Vis. 2012;18:1147–55. [PMC free article] [PubMed] [Google Scholar]
- 13.Berendschot TT, DeLint PJ, van Norren D. Origin of tapetal-like reflexes in carriers of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1996;37:2716–23. rn>. [PubMed] [Google Scholar]
- 14.Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961;190:372–3. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- 15.Krill AE. Observations of carriers of X-chromosomal-linked chorioretinal degenerations. Do these support the “inactivation hypothesis”? Am J Ophthalmol. 1967;64:1029–40. doi: 10.1016/0002-9394(67)93055-3. [DOI] [PubMed] [Google Scholar]
- 16.Helb HM, Charbel Issa P, Fleckenstein M, Schmitz-Valckenberg S, Scholl HP, Meyer CH, Eter N, Holz FG. Clinical Investigation of simultaneous confocal scanning laser ophthalmoscopy imaging combined with high-resolution, spectral-domain optical coherence tomography. Acta Ophthalmol (Copenh) 2010;88:842–9. doi: 10.1111/j.1755-3768.2009.01602.x. [DOI] [PubMed] [Google Scholar]
- 17.Fleckenstein M, Charbel Issa P, Helb HM, Schmitz-Valckenberg S, Finger RP, Scholl HP, Loeffler KU, Holz FG. High-resolution spectral-domain OCT imaging in geographic atrophy associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:4137–44. doi: 10.1167/iovs.08-1967. [DOI] [PubMed] [Google Scholar]
- 18.Marmor MF, Fulton AB, Holder GE, Miyaki Y, Brigell M, Bach M, International Society for Clinical Electrophysiology of Vision ISCEV Standard for clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 19.Neidhardt J, Glaus E, Lorenz B, Netzer C, Li Y, Schambeck M, Wittmer M, Feil S, Kirschner-Schwabe R, Rosenberg T, Cremers FP, Bergen AA, Barthelmes D, Baraki H, Schmid F, Tanner G, Fleischhauer J, Orth U, Becker C, Wegscheider E, Nürnberg G, Nürnberg P, Bolz HJ, Gal A, Berger W. Identification of novel mutations in X-linked retinitis pigmentosa families and implications for diagnostic testing. Mol Vis. 2008;14:1081–93. [PMC free article] [PubMed] [Google Scholar]
- 20.Fishman GA, Chappelow AV, Anderson RJ, Rotenstreich Y, Derlacki DJ. Short-term inter-visit variability of erg amplitudes in normal subjects and patients with retinitis pigmentosa. Retina. 2005;25:1014–21. doi: 10.1097/00006982-200512000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Genead MA, Fishman GA, Lindeman M. Structural and functional characteristics in carriers of X-linked retinitis pigmentosa with a tapetal-like reflex. Retina. 2010;30:1726–33. doi: 10.1097/IAE.0b013e3181dde629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wegscheider E, Preising MN, Lorenz B. Fundus autofluorescence in carriers of X-linked recessive retinitis pigmentosa associated with mutations in RPGR, and correlation with electrophysiological and psychophysical data. Graefes Arch Clin Exp Ophthalmol. 2004;242:501–11. doi: 10.1007/s00417-004-0891-1. [DOI] [PubMed] [Google Scholar]
- 23.Sheng X, Li Z, Zhang X, Wang J, Ren H, Sun Y, Meng R, Rong W, Zhuang W. A novel mutation in retinitis pigmentosa GTPase regulator gene with a distinctive retinitis pigmentosa phenotype in a Chinese family. Mol Vis. 2010;16:1620–8. [PMC free article] [PubMed] [Google Scholar]
- 24.Vervoort R, Lennon A, Bird AC, Tulloch B, Axton R, Miano MG, Meindl A, Meitinger T, Ciccodicola A, Wright AF. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25:462–6. doi: 10.1038/78182. [DOI] [PubMed] [Google Scholar]
- 25.Hong DH, Pawlyk B, Sokolov M, Strissel KJ, Yang J, Tulloch B, Wright AF, Arshavsky VY, Li T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Vis Sci. 2003;44:2413–21. doi: 10.1167/iovs.02-1206. [DOI] [PubMed] [Google Scholar]
- 26.Iannaccone A, Breuer DK, Wang XF, Kuo SF, Normando EM, Filippova E, Baldi A, Hiriyanna S, MacDonald CB, Baldi F, Cosgrove D, Morton CC, Swaroop A, Jablonski MM. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J Med Genet. 2003;40:e118. doi: 10.1136/jmg.40.11.e118. [DOI] [PMC free article] [PubMed] [Google Scholar]