

Abstract
Multiple growth pathways lead to enhanced proliferation in malignant cells. However, how the core machinery of DNA replication is regulated by growth signaling remains largely unclear. The sliding clamp PCNA (Proliferating Cell Nuclear Antigen) is an indispensable component of the DNA machinery responsible for replicating the genome and maintaining genomic integrity. We previously reported that epidermal growth factor receptor (EGFR) triggered tyrosine 211 (Y211) phosphorylation of PCNA, which in turn stabilized PCNA on chromatin to promote cell proliferation. Here we show that the phosphorylation can also be catalyzed by the non-receptor tyrosine kinase c-Abl. We further demonstrate that, in the absence of EGFR, signaling to PCNA can be attained through the activation of the Ron receptor tyrosine kinase and the downstream non-receptor tyrosine kinase c-Abl. We show that Ron and c-Abl form a complex, and that activation of Ron by its ligand, HGFL, stimulates c-Abl kinase activity, which in turn directly phosphorylates PCNA at Y211 and leads to an increased level of chromatin-associated PCNA. Correspondingly, HGFL-induced Ron activation resulted in Y211 phosphorylation of PCNA while silencing of c-Abl blocked this effect. We show that cAbl and Y211 phosphorylation of PCNA is an important axis downstream of Ron, which is required for cell proliferation. Treatment with a specific peptide which inhibits Y211 phosphorylation of PCNA or with the c-Abl pharmacological inhibitor imatinib suppressed HGFL-induced cell proliferation. Our findings identify the pathway of Ron-cAbl-PCNA as a mechanism of oncogene-induced cell proliferation, with potentially important implications for development of combination therapy of breast cancer.
Keywords: PCNA, Ron, receptor tyrosine kinase, tyrosine phosphorylation, Y211, tyrosine kinase, c-Abl
INTRODUCTION
Proliferating cell nuclear antigen (PCNA) is an indispensable protein for cell proliferation. PCNA functions as a sliding clamp encircling the DNA double helix (Ayyagari et al., 1995; Essers et al., 2005; Lehmann, 2006; Maga & Hubscher, 2003; Marti et al., 2002; Matunis, 2002; Moldovan et al., 2007; Paunesku et al., 2001; Ulrich, 2004). Three molecules of PCNA form a homotrimeric ring on chromatin and act as the molecular platform to recruit other proteins involved in DNA synthesis, cell-cycle control, and DNA damage response and repair (Kelman, 1997; Komatsu et al., 2000; Maga & Hubscher, 2003; Moldovan et al., 2007; Paunesku et al., 2001). While it is generally agreed that these tasks of PCNA are coordinated by many different binding partners and post-translational modifications of the sliding clamp, the molecular mechanisms coordinating PCNA functions remain largely unaddressed. We previously reported that PCNA is phosphorylated at the tyrosine residue 211 (Y211) which resulted in stabilization of PCNA on chromatin in breast cancer cells overexpressing epidermal growth factor receptor (EGFR) (Wang et al., 2006). Inhibition of EGFR by its kinase inhibitor blocked Y211 phosphorylation and resulted in degradation of PCNA on chromatin. It remained to be determined whether Y211 phosphorylation takes place in EGFR-negative cells and, if so, through what signaling mechanisms.
To address this issue, we screened a microarray containing functional protein domains with a fluorescence-labeled Y211 sequence peptide probe, and identified that the phospho-Y211 motif is specifically associated with the Src homologue 2 (SH2) domain derived from c-Abl (Zhao et al., 2012). The c-Abl proto-oncogene is a ubiquitous non-receptor tyrosine kinase that shuttles between the cytoplasm and the nucleus (Taagepera et al., 1998). It functions in multiple cellular activities, including normal development (Baker et al., 2009), gene expression (Barila et al., 2000; Baskaran et al., 1999), cell morphology and migration (Stuart et al., 2006; Woodring et al., 2003), response to DNA damage stress (Baskaran et al., 1997; Gong et al., 1999; Kharbanda et al., 1998; Kim et al., 2007; Leong et al., 2007; Levav-Cohen et al., 2005; Mantovani et al., 2004; Nehme et al., 1997; Shafman et al., 1997; Shaul & Ben-Yehoyada, 2005; Truong et al., 2003; Tsai & Yuan, 2003; Yi et al., 2006; Yoshida et al., 2005; Yuan et al., 1997; Yuan et al., 1996; Yuan et al., 1999; Zhao et al., 2012), survival (Agami et al., 1999; Wang & Ki, 2001; Yogalingam & Pendergast, 2008), and proliferation (Yogalingam & Pendergast, 2008). In breast cancer, activated c-Abl kinase promotes cell survival and invasion (Sirvent et al., 2007; Srinivasan & Plattner, 2006; Srinivasan et al., 2008; Zhao et al., 2012) and resistance to anti-estrogen treatment (Zhao et al., 2011b; Zhao et al., 2010). Our recent studies have also shown that c-Abl overexpression is a frequent event in breast cancer (~40%) and plays a role in advanced breast cancer (Zhao et al., 2010). c-Abl may function as a signaling switchboard in cancer cells and may be an important target for cancer therapy (Lin & Arlinghaus, 2008; Srinivasan et al., 2008). c-Abl is subject to a tight regulation in normal cells. However, in cancer cells, this regulation is abolished and the deregulated c-Abl contributes to tumor development (Lin & Arlinghaus, 2008; Lin et al., 2007; Srinivasan & Plattner, 2006; Srinivasan et al., 2008). EGFR is known to interact with c-Abl and the cross talk between these two kinases results in c-Abl activation (Srinivasan et al., 2009; Tanos & Pendergast, 2006). However, how c-Abl is activated in the absence of EGFR is not known.
The receptor tyrosine kinase Ron, also known as Macrophage Stimulating Protein 1 Receptor (MST1R), is a cell surface receptor tyrosine kinase of the c-Met family and has been identified as a major player in the development of multiple types of epithelial cancers (Wagh et al., 2008). The Ron receptor contains an extracellular ligand-binding domain, a transmembrane domain, and an intracellular domain harboring tyrosine kinase activity. Activation of the Ron receptor by its only known ligand, hepatocyte growth factor-like protein (HGFL, also known as macrophage-stimulating protein [MSP]), leads to activation of multiple downstream signaling pathways including PI-3K, mitogen-activated protein kinase (MAPK), and Src (Follenzi et al., 2000; Iwama et al., 1996). Forced overexpression of the Ron kinase transformed normal cells and led to increased cell proliferation (Peace et al., 2001). Overexpression of Ron selectively in mouse mammary epithelium caused breast tumor formation with an associated proliferative phenotype while loss of Ron lead to decreased breast tumor cell growth (Peace et al., 2005; Zinser et al., 2006). Although expressed at low level in most normal tissues, up-regulation of Ron expression is a frequent pathological event in breast cancer and has been associated with disease progression and poor outcome (Lee et al., 2005; Leonis et al., 2007; Maggiora et al., 1998; O’Toole et al., 2006; Welm et al., 2007). It remains to be determined how Ron promotes cell proliferation and what signaling pathways downstream from the Ron receptor are important in this process.
In this study, we identify the novel pathway of Ron-Abl-PCNA as a mechanism of oncogene-induced cell proliferation. We demonstrate that PCNA is a new substrate of cAbl and that Ron stimulates Y211 phosphorylation of PCNA through c-Abl in an EGFR-independent manner. Phosphorylation of PCNA at Y211 is a key event downstream of Ron signaling for cell proliferation in vitro and in tumor tissues.
RESULTS
We previously identified that c-Abl interacts with PCNA and enhances cell proliferation in breast cancer cells (Zhao et al., 2012). To determine whether c-Abl can directly phosphorylate PCNA at Y211, a glutathione S-transferase (GST) fusion protein of wild-type PCNA and a mutant PCNA in which the Y211 residue was replaced with phenylalanine (Y211F) were incubated with recombinant c-Abl kinase in the presence of 32P-γ-ATP. A mutant GST fusion protein with all seven tyrosine residues of PCNA substituted with phenylalanine (7F) was used as a negative control (Figure 1A). Wild-type GST-PCNA was significantly phosphorylated by c-Abl. The phosphorylation was abolished in Y211F PCNA to a level as low as that of the 7F mutant, suggesting that Y211 is the sole phosphorylation site in PCNA catalyzed by c-Abl. Consistent with the in vitro kinase assay, the levels of Y211 phosphorylation is lower in MCF-7, which has low level of c-Abl kinase activity, compared with BT474 and T47D, which have relatively higher c-Abl kinase activities (Figure 1B).
Figure 1. c-Abl phosphorylates PCNA at Y211.

(A) Right panel, c-Abl kinase was incubated with purified GST or GST fusion protein with wild-type (WT), Y211F, or tyrosine-less (7F) mutant PCNA in the presence of [γ-32P]ATP. After boiling, the reaction mixes were separated by SDS-PAGE, followed by analysis with a PhosphorImager. Left panel, protein input was visualized by Coomassie Blue staining. (B) Upper panel, the indicated breast cancer cells were lysed and phospho-Y211 PCNA was measured by immunoprecipitation (IP) with the anti-phospho-Y211 antibody, followed by Western analysis using a total PCNA antibody. Lower panel, endogenous c-Abl activity was measured by immunoprecipitation of cellular c-Abl from serum-starved cells and the kinase activities were determined by in vitro kinase assay (ICK). Total c-Abl and GAPDH proteins were shown by Western blotting (WB).
Our previous report showed that EGFR phosphorylates PCNA Y211 in MDA-MB-468 cells, which express high levels of EGFR (Wang et al., 2006). Certain other breast cancer cell lines, such as T47D and BT474, express very low levels of EGFR, but they express similar levels of Y211-phosphorylated PCNA as MDA-MB-468 cells (Figure 2). However, unlike MDA-MB-468 cells, treatment with lapatinib, a potent inhibitor of EGFR family members EGFR and ERBB2/HER2, did not result in downregulation of phospho-Y211 PCNA in T47D and BT474 cells (Figure 2A). The treatment effectively inhibited EGFR and ERBB2 activities as demonstrated by the overall and receptor-specific tyrosine phosphorylation (Figure 2B). These results indicate that Y211 phosphorylation of PCNA can be induced via an EGFR- and ERBB2-independent mechanism in T47D and BT474 cells. We noticed that c-Abl is expressed at higher levels in the T47D and BT474 cell lines than in MDA-MB-468 cells (Figure 2C). Phosphorylation of the endogenous adaptor protein CrkL at tyrosine 207, a well-known signaling event directly catalyzed by c-Abl (Andoniou et al., 1996), was also increased in these two cell lines in comparison with MDA-MB-468 (Fig. 2C). These results suggested that Y211 phosphorylation of PCNA in BT474 and T47D cells with low EGFR expression is mediated by a pathway in which the c-Abl kinase is activated. To further establish the causal relationship between c-Abl and the signaling event, mouse embryonic fibroblasts (MEFs) with the endogenous c-Abl gene deleted (Abl−/−) and the corresponding MEFs with a reconstituted c-Abl gene at a physiological level (Abl+) (Koleske et al., 1998; Plattner et al., 2003) were compared for the level of phospho-Y211 PCNA (Fig. 2D). Our previous study established that Y211 phosphorylation of PCNA is a target in cells expressing c-Abl. Consistently, treatment of Y211F peptide resulted in significant growth inhibition in these MEF cells, with the Abl-positive cells more sensitive than the Abl−/− MIG cells (Supplementary Figure 1). These results together demonstrate that c-Abl is an important factor for Y211 phosphorylation.
Figure 2. EGFR-independent Y211 phosphorylation in T47D and BT474 cells.

(A) Lapatinib inhibits phospho-Y211 in MDA-MB-468 but not in T47D and BT474 cells. Upper panels, breast cancer cell lines MDA-MB-468, BT474, and T47D were examined for the levels of Y211 phosphorylation of PCNA in the presence or absence of the EGFR inhibitor lapatinib (5 μM) for 40 hours. Cells were then lysed and phospho-Y211 PCNA was measured by immunoprecipitation with the anti-phospho-Y211 antibody, followed by Western analysis using a total PCNA antibody. Lower panels, the relative intensities of phospho-Y211 PCNA were quantitated using NIH ImageJ software and normalized to the levels of total PCNA. (B) Lapatinib effectively repressed activity of EGFR and ERBB2. Cells were treated with lapatinib 10 uM for 48 hours. Specific antibodies against total tyrosine phosphorylation migrated at ~180 KDa (Millipore), phospho-EGFR at Y1068 (p-EGFR), and phospho-ErbB2/HER2 at Y1221/1222 (p-HER2) were used to determine the phosphorylation levels by Western analysis of whole cell lysates.
(C) Low EGFR expression and high Ron expression in T47D and BT474 cells. The protein levels of EGFR, Ron, c-Abl, CrkL, phospho-CrkL (p-CrkL), and PCNA were measured by Western analysis. (D) Total cell lysates were isolated from MEF cells in which the endogenous c-Abl is deleted and stably re-introduced with a retroviral vector expressing c-Abl (Abl) or the green fluorescence protein (MIGR), and the level of Y211 phosphorylation was determined by immunoprecipitation and western analysis as described in (A).
We and others have shown that the Ron receptor tyrosine kinase regulates cell growth in breast cancer cells, including T47D (Maggiora et al., 1998; McClaine et al., 2010). To test the role of Ron signaling and c-Abl in phosphorylation of PCNA at Y211, T47D cells were infected with a lentiviral vector expressing a specific shRNA against c-Abl (shAbl) in the presence and absence of the Ron ligand HGFL. The results show that depleting c-Abl abrogated Y211 phosphorylation, whereas treatment with HGFL significantly stimulated Y211 phosphorylation (Figure 3). Importantly, HGFL failed to induce Y211 phosphorylation when c-Abl is depleted, indicating that c-Abl is required for HGFL-induced PCNA phosphorylation. This requirement of c-Abl for induction is specific to PCNA, since treatment with HGFL stimulates downstream MAPKs irrespective to the status of c-Abl.
Figure 3. HGFL stimulates Y211 phosphorylation through c-Abl.

T47D cells were starved for 24 hours, followed by stimulation with 80 ng/ml of HGFL for 30 minutes. Cells were then lysed and the levels of phospho-Y211 PCNA and the indicated proteins were analyzed by Western analysis using the corresponding specific antibodies.
The finding that HGFL stimulated Y207 phosphorylation of CrkL suggested that stimulation of the HGFL-Ron axis leads to c-Abl activation (Figure 3). To interrogate this possibility, we tested whether c-Abl and Ron interact. An immunocomplex of c-Abl was pulled down using an anti-c-Abl antibody by immunoprecipitation and found to contain co-immunoprecipitated Ron protein (Figure 4A). This result indicates that c-Abl and Ron form a complex. Interestingly, formation of the complex was abolished upon HGFL stimulation as shown by the diminished level of co-immunoprecipitated Ron with c-Abl, suggesting that Ron activation induces c-Abl dissociation from the receptor (Figure 4B). We therefore examined whether the alteration of Ron-Abl interaction can be translated to enhanced signaling of the Ron-Abl axis. Endogenous c-Abl was immunoprecipitated from T47D cells harboring a Ron-specific shRNA (T47D/shRon) or the control cells harboring a scrambled shRNA (T47D/shCtrL) and incubated with the recombinant substrate of c-Abl (GST-Crk). Ron depletion significantly suppressed the kinase activity of c-Abl as measured by the phosphorylation level of GST-Crk with autoradiography (Figure 4C). Consistent with the observation that HGFL stimulates endogenous CrkL phosphorylation (Figure 3), treatment with HGFL activated c-Abl kinase activity as measured by an immunocomplex kinase assay (Figure 4D). On the other hand, activation of c-Abl was dependent on the function of Ron since HGFL-mediated c-Abl activation was abolished in T47D/shRon but not T47D/shCtrl cells (Figure 4D). These results suggest that c-Abl is phosphorylated in association with Ron. This possibility was tested using an immunocomplex kinase assay in which the endogenous Ron protein was immunoprecipitated from T47D cells. The isolated Ron was incubated with c-Abl which was ectopically expressed in and immunoprecipitated from HEK293T cells in a kinase reaction containing [γ-32P]ATP (Fig. 4E). The isolated Ron, but not the control immunoprecipitate by IgG, was active to mediate c-Abl phosphorylation. These results together demonstrate a potential signaling pathway of HGFL-Ron-Abl-PCNA during cancer cell proliferation.
Figure 4. Ron stimulates the tyrosine kinase activity of c-Abl.

(A) Interaction between endogenous Ron and c-Abl in T47D cells was demonstrated by immunoprecipitation with an anti-c-Abl antibody. After gel electrophoresis, co-precipitated Ron and c-Abl were detected using the corresponding antibodies. (B) T47D cells were treated with HGFL for 10 minutes. The endogenous c-Abl was immunoprecipitated and the levels of associated Ron receptor were determined with Western blotting (upper panel). The levels of Rona and c-Abl in the input were shown (lower panel). (C) Ron is required for c-Abl kinase activity. Upper panel, c-Abl was immunoprecipitated from cell lysates derived from pooled clones of T47D cells harboring a shRNA with scrambled sequence (T47D/shCtrl) or a shRNA against Ron (T47D/shRon). The immunocomplex was then incubated with GST-Crk fusion protein as the substrate in the presence of [γ-32P]ATP. Reaction mixtures were run on a SDS-PAGE gel, stained with Coomassie Blue for the GST-Crk protein, and autoradiographed to film. Bottom panel, expression of Ron and c-Abl in the lysates was determined by Western analysis. The experiment shown is representative of three experiments. Relative intensities of the phosphorylated GST-Crk are indicated with that of un-stimulated T47D/shCtrl cells set as 1. (D) HGFL activates c-Abl kinase activity through Ron. Upper panel, T47D/shCtrl and T47D/shRon cells were starved for 24 hours, followed by treatment with HGFL (80 ng/ml) for 30 minutes. Endogenous c-Abl was then immunoprecipitated and the kinase activity was measured as described in (B). Relative intensities of the phosphorylated GST-Crk are indicated with that of un-stimulated T47D/shCtrl cells set as 1. Bottom panel, expression of Ron and c-Abl in the lysates was determined by Western analysis. (E) HEK-293 cells were transfected with c-Abl cDNA and the expressed c-Abl was immunoprecipitated using an anti-c-Abl antibody (Calbiochem). The isolated c-Abl was then incubated with endogenous Ron protein immunoprecipitated from T47D cells in the presence of [γ-32P]ATP. Negative controls: the immunoprecipitate with IgG (left panel), and the kinase reaction without c-Abl (right panel).
Corroborating these observations, stimulation with HGFL induced Y211 phosphorylation of PCNA in serum-starved cells in a time-dependent manner, accompanied by increased levels of phospho-Y207 CrkL and MAPK phosphorylation (Figure 5A). Conversely, Ron depletion in cells cultured in serum-supplemented normal medium caused decreased Y211 phosphorylation of PCNA (Figure 5B). These results indicated that Y211 phosphorylation of PCNA is a downstream signaling event triggered by the activated Ron pathway. We have reported the use of a peptide-based strategy to target phospho-Y211 PCNA in which the synthetic peptide spanning the Y211 phosphorylation motif but with the site changed to phenylalanine (termed Y211F peptide) specifically inhibited endogenousY211 phosphorylation and inhibited cell proliferation (Wang et al., 2006; Zhao et al., 2012; Zhao et al., 2011a). Furthermore, we have previously demonstrated that Y211F peptide treatment conferred a more significant growth inhibition effect in cells expressing c-Abl than in the cells in which c-Abl was depleted (Zhao et al., 2012). This suggested that Y211 phosphorylation is an important component for cell proliferation in c-Abl-expressing cells. Following this line, we reasoned that if phospho-Y211 PCNA has an important contribution to the cell proliferation-promoting function of Ron, down-regulation of Y211 phosphorylation should have a more significant impact on proliferation in cells expressing Ron than cells lacking Ron. As anticipated, treatment with the Y211F peptide, but not the control scrambled peptide, inhibited cell growth (Figure 5C; comparing control and Y211F peptides). Importantly, knockdown of Ron reduced sensitivity to the peptide in comparison with control cells (Figure 5C), indicating that Y211 phosphorylation of PCNA induced by Ron plays an essential role in promoting cell proliferation.
Figure 5. Y211 phosphorylation is a critical growth signal downstream of the Ron pathway.

(A) HGFL stimulates PCNA Y211 phosphorylation. T47D cells were serum-starved for 24 hours, then stimulated with HGFL (80 ng/ml) for indicated periods of time. Cells were then lysed and phospho-Y211 PCNA was pulled down using the anti-phospho-Y211 PCNA antibody, followed by Western analysis using an anti-PCNA antibody. The result was quantitated using NIH ImageJ software with the value at time point 0 minutes set as 1. The expression levels of PCNA, CrkL, phospho-CrkL (p-CrkL), MAPK, phospho-MAPK (p-MAPK), and tubulin in the input lysates were determined. (B) Y211 phosphorylation of PCNA, as measured in (A), was inhibited in T47D/shRon cells in comparison with T47D/shCtrl cells. The levels of endogenous PCNA, Ron, and tubulin in the input lysates were determined by Western analysis. (C) Depleting Ron expression de-sensitized cells to inhibition of Y211 phosphorylation. T47D/shRon and T47D/shCtrl cells were treated with the Y211F peptide or the control peptide with scrambled sequence at 10 μM for 48 hours. The ratio between T47D/shCtrL and T47D/shAbl in each group was determined. **, p < 0.01.
We previously showed that Y211 phosphorylation stabilized chromatin-bound PCNA, which in turn promoted cell proliferation (Wang et al., 2006), and that c-Abl functioned to enhance chromatin association of PCNA in cancer cells (Zhao et al., 2012). Identification of Y211 phosphorylation of PCNA as a downstream event of the HGFL-Ron-c-Abl pathway predicts that Ron activation should also enhance chromatin association of PCNA. Indeed, stimulation with HGFL increased chromatin-bound PCNA as shown by the increased level of PCNA resistant to Triton X-100 extraction, while there is no difference in the chromatin-unbound pool of PCNA, which is soluble with Triton X-100 extraction (Figure 6A). The observation that Ron induces c-Abl activation suggests that the Ron-induced chromatin association of PCNA relies on c-Abl. Indeed, the HGFL-induced PCNA chromatin association was abrogated by c-Abl depletion (Figure 6A), which also suppressed the proliferative effect of HGFL (Figure 6B). A similar effect was observed with pharmacological inhibition of c-Abl by its kinase inhibitor imatinib (STI571), which abrogated the proliferative effect of HGFL (Figure 6C). Like imatinib, treatment with the Y211 peptide blocked HGFL-induced cell proliferation (Figure 6C).
Figure 6. c-Abl is required for Ron-mediated cell growth.

(A) HGFL stimulated chromatin association of PCNA, which required c-Abl function. T47D/shAbl and T47D/shCtrl cells were starved for 24 hours, followed by stimulation with 80 ng/ml of HGFL for 30 minutes. The cells were then extracted with 5% of Triton X-100, and the levels of PCNA in the Triton-insoluble and Triton-soluble fractions were determined by Western analysis. Histone H3 and α-tubulin were used as the internal control for Triton-insoluble and Triton-soluble fractions, respectively. The results were quantitated using ImageJ software. (B) Silencing c-Abl mitigated HGFL-stimulated cell growth. T47D/shAbl and T47D/shCtrl cells were starved with 0.5% FBS medium for 24 hours, then treated with 80 ng/ml HGFL for 72 hours. Viable cells were assessed by a MTT assay and the data was plotted. ***, p<0.001. (C) Pharmacological inhibition of c-Abl or Y211 phosphorylation abrogated HGFL-induced cell growth. T47D cells were starved by maintaining in a medium containing 0.5% of serum for 24 hours, then subject to the indicated treatment for 72 hours (HGFL, 80 ng/ml, imatinib, 10 μM, peptides, 6 μM). Cell growth was assessed by MTT assay and the data was plotted. ***, p < 0.001.
These results predict a correlation between phospho-Y211 PCNA and Ron expression in primary breast cancer tumor tissues. To test this hypothesis, immunohistochemistry (IHC) analyses were performed using IHC-proven antibodies of Ron (O’Toole et al., 2006; Thobe et al., 2010), EGFR, PCNA, and phospho-Y211 PCNA (Wang et al., 2006) in a cohort of breast cancer tumor tissues (see Materials and Methods). As we previously reported, expression levels of EGFR in tumor tissues are strongly correlated with phospho-Y211 PCNA (n=118; p=0.0027). Importantly, we found that Ron expression is significantly correlated with Y211 phosphorylation of PCNA in the current cohort of tissues (n=162; p=0.048) (Table 1). No correlation was observed between total PCNA and EGFR (n=118; p=0.999) or total PCNA and Ron (n=162; p=0.416). These results indicate that both EGFR and Ron are associated with Y211 phosphorylation of PCNA in human breast cancer. EGFR overexpression is a frequent event in this cohort of samples. When examining the samples with low EGFR expression but positive phospho-Y211 PCNA (n=28), we found that more than half of them (n=16; 57%) were Ron-positive, indicating that Ron as well as EGFR is associated with Y211 phosphorylation of PCNA in human breast cancer. We then examined the correlation between Ron and phospho-Y211 PCNA with c-Abl. Due to sample availability, this was examined in another independent cohort of patients (see Materials and Methods). Consistent with the activity of c-Abl in catalyzing Y211 phosphorylation of PCNA, the levels of phospho-Y211 PCNA were significantly correlated with the level of c-Abl (n=81, p=0.0008; Table 2). However, significant correlation between Ron and cAbl was not found within this cohort of samples. This suggests that multiple other pathways contribute to the Abl-PCNA cascade. Indeed, among the tissues with relatively low levels of EGFR (H score lower than medium) and high levels of phospho-Y211 PCNA (H score higher than medium) and c-Abl (H score higher than medium) (n=24), 62.5% of these tissues have high Ron expression (H score above the medium). Representative IHC staining of tissues with positive expression of Ron, c-Abl, and phospho-Y211 PCNA are shown in Figure 7.
Table 1.
Correlation between Ron and phospho-PCNA in breast cancer tissues*
| Phospho PCNA | Total PCNA | ||||||
|---|---|---|---|---|---|---|---|
| high | low | high | low | ||||
| Ron | high | 41 | 44 | P = 0.048 | 63 | 43 | P = 0.416 |
| low | 38 | 30 | 24 | 32 | |||
The number of tissue samples in each category is shown in the table with the indicated P value determined by Fisher’s test. Dichotomization of Ron expression was based on the median IHC score (=150). For total PCNA and phospho-PCNA, the average IHC scores are used (PCNA=199; phospho-PCNA=155).
Table 2.
Correlation between c-Abl and phospho-PCNA in breast cancer tissues*
| Phospho PCNA | ||||
|---|---|---|---|---|
| high | low | |||
| c-Abl | high | 41 | 18 | P = 0.0008 |
| low | 10 | 22 | ||
The number of tissue samples in each category is shown in the table with the indicated P value determined by Fisher’s test. Dichotomization of c-Abl and phospho-PCNA expression was based on the median IHC scores (c-Abl=280; phospho-PCNA=90).
Figure 7. Expression of Ron and phospho-Y211 PCNA in primary breast cancer tissues.
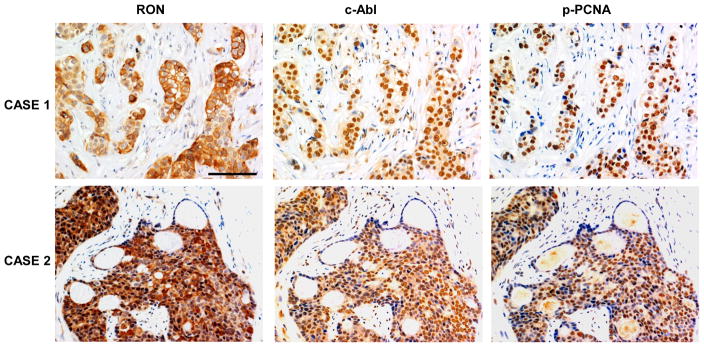
Ron, c-Abl, and phospho-Y211 PCNA in the tumor tissues were assessed by IHC using specific antibodies as described in the Methods section. Representative staining in two pairs of consecutive tissue sections is shown. The tissues were selected from a tumor microarray purchased from US Biomax (BRC1021).
DISSCUSSION
The current study identifies c-Abl as a non-receptor tyrosine kinase that is capable of catalyzing Y211 phosphorylation of PCNA in breast cancer cells. We further demonstrate that this pathway is regulated by the receptor tyrosine kinase Ron in EGFR-negative cells. Thus, Y211 phosphorylation of PCNA can be mediated by different kinases in different cell contexts to foster cell proliferation.
We previously showed that the SH2 domain of c-Abl interacts with the phosphorylated Y211 motif of PCNA and that targeting the phosphorylation resulted in inhibition of breast cancer growth (Zhao et al., 2012). In the current study, we are able to establish a new pathway in which the Ron receptor tyrosine kinase activates c-Abl which in turn catalyzes Y211 phosphorylation of PCNA. This mechanism may augment the intensity and speed of the signaling mediated by c-Abl. Augmented interaction with the substrate upon phosphorylation is a common mechanism adopted by the c-Abl kinase. Examples include c-Jun (Barila et al., 2000), RNA polymerase II (Duyster et al., 1995), and p73 (Tsai & Yuan, 2003). For PCNA, this mechanism can provide a unique advantage given its topology as a trimeric ring in which a phosphorylated PCNA unit at Y211 can serve as the docking site of c-Abl that in turn catalyzes phosphorylation on other PCNA units in the same trimer. In addition, binding of c-Abl to the phosphorylated Y211 of PCNA through its SH2 domain may lend an opportunity for the c-Abl kinase to regulate other proteins associated with the PCNA sliding clamp.
In addition, our findings are consistent with the notion that multiple pathways are activated by Ron signaling, such as MAPK, which is not dependent on c-Abl, and Y211 phosphorylation of PCNA, which requires c-Abl function. This is the first report which has shown that c-Abl is regulated by Ron. It is interesting that stimulation with HGFL resulted in decreased levels of the Ron-Abl complex. Although the underlying mechanism remains to be determined, this phenomenon is consistent with an enzyme-substrate relationship between the activated Ron and c-Abl in which the catalyzed substrate is dissociated from the enzyme. It is conceivable that the activated c-Abl undergoes conformation changes which facilitate its translocation and association with other downstream binding partners. c-Abl has also been shown to regulate receptor tyrosine kinases by phosphorylating the receptors (Srinivasan et al., 2009; Tanos & Pendergast, 2006). Thus the molecular outcome subsequent to the interaction between c-Abl and membrane receptors can be complex. Molecular characterization of the interaction between by Ron and c-Abl in response to HGFL will be necessary to further understand the mechanism of crosstalk between Ron and c-Abl. In addition to molecular evidence derived from breast cancer cell lines, significant correlation between Ron and phospho-Y211 PCNA as well as phospho-Y211 PCNA and c-Abl was also established in archived primary tumor tissues. Although correlation between Ron and c-Abl was not detected among the relatively small number of tissue samples in this study, Ron positivity was enriched in samples which express relatively low levels of EGFR and high levels of c-Abl and phospho-Y211 PCNA.
EGFR has been demonstrated to be able to stimulate c-Abl (Jones et al., 2006; Plattner et al., 1999; Srinivasan & Plattner, 2006) and EGFR signaling may also lead to Y211 phosphorylation of PCNA through c-Abl, in addition to the direct mechanism in which nucleus-localized EGFR interacts with PCNA and catalyzes the phosphorylation event (Wang et al., 2006). PCNA Y211 phosphorylation may be a point of convergence for proliferation signaling governed by EGFR and Ron, since both have significant roles in breast cancer (Figure 8). Multiple EGFR-specific small molecules and antibodies have been developed as therapeutic agents in the past decades. However, their impact on breast cancer as single treatments has been disappointing. Thus, development of effective combined therapeutic approaches are desperately needed (Mendelsohn & Baselga, 2003). Our observation suggests that combined treatment of EGFR and the Ron-Abl axis is a promising strategy to target breast cancer. Combining the EGFR-ERBB2 dual inhibitor lapatinib and the c-Abl inhibitor imatinib resulted in synergistic efficacy in growth inhibition of breast cancer cells (Lo et al., 2011).
Figure 8. Schematic illustration of the Y211 phosphorylation pathways.

EGFR has been shown to be able to directly phosphorylate PCNA at Y211. The current study demonstrates a new pathway in which stimulation of the Ron receptor induces Y211 phosphorylation through activation of c-Abl which subsequently catalyzes phosphorylation of PCNA at Y211. Given the fact that Ron, EGFR, and c-Abl are important targets in cancer therapy, combined inhibition of phospho-Y211 PCNA may be an efficient approach to enhance sensitivity and help overcome drug resistance.
It remains possible that other kinases may also involved in Y211 phosphorylation of PCNA. Further understanding the signaling mechanisms underlying PCNA phosphorylation can allow this event to be used not only as a prognostic marker for disease development, but also as a therapeutic target of combination treatment in multiple cancer types and to help overcome the frequent challenge of drug resistance in cancer therapy.
METHODS AND REGENTS
Cell culture and antibodies
T47D, BT474, and MDA-MB-468 cell lines were purchased from American Type Culture Collection. T47D/shAbl and T47D/shCtrl cells were generated as described previously (Zhao et al., 2010). The Abl+ and Abl−/− mouse embryonic fibroblasts (MEFs) were derived from double knockout of the mouse Abl and Arg genes, followed by reintroduction of Abl with a retroviral vector (Abl+) or a control vector (Abl−/−) as described previously (Koleske et al., 1998; Plattner et al., 2003). All cells were grown in DMEM/F12 (1:1) with 10% fetal bovine serum. The following antibodies were purchased: α-tubulin (from Sigma); c-Abl (from EMD for immunoblotting analysis; from Abcam for IHC); EGFR, CrkL, phospho-EGFR (Y1068), phospho-HER2 (Y1221/1222), phospho-Y207-CrkL (from Cell Signaling); phospho-PCNA (Bethyl); PCNA, Ron, IgG (from Santa Cruz), Ron/MSRP (from R&D Systems). Lapatinib and imatinib were purchased from LC Laboratories.
Cell growth analysis
Measurement of cell growth was based on the reduction of the tetrazolium salt MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide as described previously (Furukawa et al., 1991). Briefly, 1000 to 3000 cells per well were plated in 96-well plates, in triplicates. The assay was initiated by adding 20 μl of MTT reagent (5 mg/ml in PBS) to the 100 μl culture and incubated for 4 hours. 100 μl of detergent (20% SDS, 50% N,N-dimethylformamide, pH 4.7) was then added to lyse the cells for overnight. Absorbance at 570 nm was recorded. The experiments were repeated at least three times.
Kinase assay
To examine PCNA as the substrate of c-Abl, 40 μl of kinase reaction mix contained 50–100 ng of purified c-Abl protein (Upstate), and 0.2–0.8 μg of GST-PCNA fusion protein in reaction buffer (10 mM Tris HCl, pH 7.5; 10 mM MgCl2; 1mM DTT; 10 μM cold ATP) was set up. Kinase reactions were initiated by adding 25–50 μCi of [γ-32P]ATP. After incubating 30 minutes at room temperature, reactions were stopped by adding 10 μl of 5X loading buffer. 32P incorporation into GST–PCNA was quantitated using a PhosphorImager (Molecular Dynamics). Endogenous c-Abl kinase activities were determined by immunocomplex kinase assay in which the endogenous c-Abl was immunoprecipitated from serum-starved cells and then mixed with recombinant GST-Crk substrate in the presence of [γ-32P]ATP as reported previously (Plattner et al., 1999; Plattner et al., 2004). For kinase activity of Ron, endogenous Ron was immunoprecipitated from 100 μg of T47D total lysate by a Ron/MSRP-specific antibody (R&D Systems) or a control IgG control. The immunoprecipitate was incubated with immunoprecipitate of c-Abl from 100 μg of the lysate of HEK-293T cells which had been transiently transfected with 10 μg of c-Abl cDNA.
Immunohistochemical staining
Correlation among pY211-PCNA and Ron was conducted using consecutive sections of tissue microarrays (TMA) were purchased from the Cooperative Breast Cancer Tissue Resource (CBCTR) of the National Cancer Institute (NCI). To examine the correlation between Ron and pY211-PCNA with c-Abl, TMA purchased from US Biomax (BRC1021) were used. IHC was performed as previously described (Wang et al., 2006). In brief, paraffin was removed by heating slides at 60° C for 1 hour, extracted twice with xylene for 10 minutes, followed by a series of ethanol solutions. Antigen retrieval was conducted by heating in a microwave (90° ~ 100° C) in citrate buffer (10 mM, pH 6.0) for 30 minutes. Endogenous peroxidase was quenched with 3% H2O2 in methanol. For immunostaining, slides were blocked in 5% goat serum in TBST (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 0.1% Tween 20) for one hour. IHC staining and the scoring of pY211-PCNA and total PCNA (Wang et al., 2006), Ron (O’Toole et al., 2006; Thobe et al., 2010), c-Abl (Zhao et al., 2010), and EGFR (Wang et al., 2006) were conducted following procedures optimized and described previously by our group. IHC staining was scored by the histological scoring method (H score), as defined by the product of the percent positivity and the staining intensity. To test the correlation between different markers, IHC scores were dichotomized as high or low. In the study described in Table 1, Ron staining dichotomization was based on the medium H score. For other markers, the average H scores are used. In the study described in Table 2, the medium scores were used for dichotomization for all markers. P values were determined by Fisher’s test. Differences with p ≤ 0.05 were considered statistically significant.
Western analysis
Cell lysates of the treated cells were isolated by incubation with NETN buffer (150 mM NaCl, 1 mM EDTA pH 8.0, 20 mM Tris pH 8.0, 0.5% NP-40, phosphatase inhibitors consisting of 25 mM NaF and 2 mM Na3VO4, and the protease inhibitors 20 μl/ml aprotinin and 0.1 M PMSF). Cell lysates were separated on acrylamide gels, transferred to a PVDF membrane (Bio-Rad), and probed with the indicated antibodies. Bands were visualized by a chemiluminescence-based detection method (Fisher/Pierce) that used a horseradish peroxidase-conjugated secondary antibody.
Supplementary Material
Acknowledgments
We thank Drs. David Plas and Daniel Starczynowski for the help in discussion and reagents. The authors also like to thank Dr. Belinda Peace and Glenn Doerman for assistance in editing the manuscript and preparing the graphics, and Jerylin Gray for technical assistance. This work was supported in part by the Susan G. Komen Breast Cancer Research Award KG080540 (S.-C. W.), the Department of Defense Prostate Cancer Research Program New Investigator Award PC073951 (S.-C. W.), the VA Merit Award 1001BX000803 (S.E.W.), the Center for Clinical and Translational Science and Training (CCTST) of University of Cincinnati and Cincinnati Children’s Hospital (S.-C. W.), and the Elsa Pardee Foundation (S.-C. W.). This project was also supported in part by PHS Grant P30 DK078392 and CA125379 (S.E.W.).
References
- Agami R, Blandino G, Oren M, Shaul Y. Nature. 1999;399:809–13. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- Andoniou CE, Thien CB, Langdon WY. Oncogene. 1996;12:1981–9. [PubMed] [Google Scholar]
- Ayyagari R, Impellizzeri KJ, Yoder BL, Gary SL, Burgers PM. Mol Cell Biol. 1995;15:4420–4429. doi: 10.1128/mcb.15.8.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MA, Hetherington L, Curry B, Aitken RJ. Developmental Biology. 2009;333:57–66. doi: 10.1016/j.ydbio.2009.06.022. [DOI] [PubMed] [Google Scholar]
- Barila D, Mangano R, Gonfloni S, Kretzschmar J, Moro M, Bohmann D, Superti-Furga G. EMBO J. 2000;19:273–81. doi: 10.1093/emboj/19.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskaran R, Escobar SR, Wang JYJ. Cell Growth Differ. 1999;10:387–396. [PubMed] [Google Scholar]
- Baskaran R, Wood LD, Whitaker LL, Canman CE, Morgan SE, Xu Y, Barlow C, Baltimore D, Wynshaw-Boris A, Kastan MB, Wang JY. Nature. 1997;387:516–9. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- Duyster J, Baskaran R, Wang JY. Proc Natl Acad Sci U S A. 1995;92:1555–9. doi: 10.1073/pnas.92.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers J, Theil AF, Baldeyron C, van Cappellen WA, Houtsmuller AB, Kanaar R, Vermeulen W. Mol Cell Biol. 2005;25:9350–9. doi: 10.1128/MCB.25.21.9350-9359.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follenzi A, Bakovic S, Gual P, Stella MC, Longati P, Comoglio PM. Oncogene. 2000;19:3041–9. doi: 10.1038/sj.onc.1203620. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Kubota T, Suto A, Takahara T, Yamaguchi H, Takeuchi T, Kase S, Kodaira S, Ishibiki K, Kitajima M. J Surg Oncol. 1991;48:188–93. doi: 10.1002/jso.2930480310. [DOI] [PubMed] [Google Scholar]
- Gong J, Costanzo A, Yang HQ, Melino G, Kaelin WG, Levrero M, Wang JYJ. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- Iwama A, Yamaguchi N, Suda T. EMBO J. 1996;15:5866–75. [PMC free article] [PubMed] [Google Scholar]
- Jones RB, Gordus A, Krall JA, MacBeath G. Nature. 2006;439:168–74. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- Kelman Z. Oncogene. 1997;14:629–40. doi: 10.1038/sj.onc.1200886. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Yuan ZM, Weichselbaum R, Kufe D. Oncogene. 1998;17:3309–18. doi: 10.1038/sj.onc.1202571. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Rajasekaran B, Brown KD. J Biol Chem. 2007;282:32021–31. doi: 10.1074/jbc.M701451200. [DOI] [PubMed] [Google Scholar]
- Koleske AJ, Gifford AM, Scott ML, Nee M, Bronson RT, Miczek KA, Baltimore D. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- Komatsu K, Wharton W, Hang H, Wu C, Singh S, Lieberman HB, Pledger WJ, Wang HG. Oncogene. 2000;19:5291–7. doi: 10.1038/sj.onc.1203901. [DOI] [PubMed] [Google Scholar]
- Lee WY, Chen HHW, Chow NH, Su WC, Lin PW, Guo HR. Clin Cancer Res. 2005;11:2222–2228. doi: 10.1158/1078-0432.CCR-04-1761. [DOI] [PubMed] [Google Scholar]
- Lehmann AR. Exp Cell Res. 2006;312:2673–2676. doi: 10.1016/j.yexcr.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW. J Clin Invest. 2007;117:1370–80. doi: 10.1172/JCI30866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonis MA, Thobe MN, Waltz SE. Future Oncol. 2007;3:441–448. doi: 10.2217/14796694.3.4.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levav-Cohen Y, Goldberg Z, Zuckerman V, Grossman T, Haupt S, Haupt Y. Biochemical and Biophysical Research Communications. 2005;331:737–749. doi: 10.1016/j.bbrc.2005.03.152. [DOI] [PubMed] [Google Scholar]
- Lin J, Arlinghaus R. Oncogene. 2008;27:4385–4391. doi: 10.1038/onc.2008.86. [DOI] [PubMed] [Google Scholar]
- Lin J, Sun T, Ji L, Deng W, Roth J, Minna J, Arlinghaus R. Oncogene. 2007;26:6989–96. doi: 10.1038/sj.onc.1210500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YH, Ho PC, Zhao H, Wang SC. Anticancer Res. 2011;31:789–95. [PubMed] [Google Scholar]
- Maga G, Hubscher U. J Cell Sci. 2003;116:3051–60. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- Maggiora P, Marchio S, Stella MC, Giai M, Belfiore A, De Bortoli M, Di Renzo MF, Costantino A, Sismondi P, Comoglio PM. Oncogene. 1998;16:2927–33. doi: 10.1038/sj.onc.1201812. [DOI] [PubMed] [Google Scholar]
- Mantovani F, Piazza S, Gostissa M, Strano S, Zacchi P, Mantovani R, Blandino G, Del Sal G. Mol Cell. 2004;14:625–36. doi: 10.1016/j.molcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Marti TM, Kunz C, Fleck O. J Cell Physiol. 2002;191:28–41. doi: 10.1002/jcp.10077. [DOI] [PubMed] [Google Scholar]
- Matunis MJ. Mol Cell. 2002;10:441–442. doi: 10.1016/s1097-2765(02)00653-6. [DOI] [PubMed] [Google Scholar]
- McClaine RJ, Marshall AM, Wagh PK, Waltz S. Neoplasia. 2010;12:650–658. doi: 10.1593/neo.10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J. J Clin Oncol. 2003;21:2787–99. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- Moldovan GL, Pfander B, Jentsch S. Cell. 2007;129:665–79. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Nehme A, Baskaran R, Aebi S, Fink D, Nebel S, Cenni B, Wang JYJ, Howell SB, Christen RD. Cancer Res. 1997;57:3253–3257. [PubMed] [Google Scholar]
- O’Toole JM, Rabenau KE, Burns K, Lu D, Mangalampalli V, Balderes P, Covino N, Bassi R, Prewett M, Gottfredsen KJ, Thobe MN, Cheng Y, Li Y, Hicklin DJ, Zhu Z, Waltz SE, Hayman MJ, Ludwig DL, Pereira DS. Cancer Res. 2006;66:9162–9170. doi: 10.1158/0008-5472.CAN-06-0283. [DOI] [PubMed] [Google Scholar]
- Paunesku T, Mittal S, Protic M, Oryhon J, Korolev SV, Joachimiak A, Woloschak GE. Int J Radiat Biol. 2001;77:1007–21. doi: 10.1080/09553000110069335. [DOI] [PubMed] [Google Scholar]
- Peace BE, Hughes MJ, Degen SJ, Waltz SE. Oncogene. 2001;20:6142–51. doi: 10.1038/sj.onc.1204836. [DOI] [PubMed] [Google Scholar]
- Peace BE, Toney-Earley K, Collins MH, Waltz SE. Cancer Res. 2005;65:1285–1293. doi: 10.1158/0008-5472.CAN-03-3580. [DOI] [PubMed] [Google Scholar]
- Plattner R, Irvin BJ, Guo S, Blackburn K, Kazlauskas A, Abraham RT, York JD, Pendergast AM. Nat Cell Biol. 2003;5:309–19. doi: 10.1038/ncb949. [DOI] [PubMed] [Google Scholar]
- Plattner R, Kadlec L, DeMali KA, Kazlauskas A, Pendergast AM. Genes Dev. 1999;13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R, Koleske AJ, Kazlauskas A, Pendergast AM. Mol Cell Biol. 2004;24:2573–83. doi: 10.1128/MCB.24.6.2573-2583.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafman T, Khanna KK, Kedar P, Spring K, Kozlov S, Yen T, Hobson K, Gatei M, Zhang N, Watters D, Egerton M, Shiloh Y, Kharbanda S, Kufe D, Lavin MF. Nature. 1997;387:520–3. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- Shaul Y, Ben-Yehoyada M. Cell Res. 2005;15:33–5. doi: 10.1038/sj.cr.7290261. [DOI] [PubMed] [Google Scholar]
- Sirvent A, Boureux A, Simon V, Leroy C, Roche S. Oncogene. 2007;26:7313–23. doi: 10.1038/sj.onc.1210543. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Kaetzel DM, Plattner R. Cell Signal. 2009;21:1143–1150. doi: 10.1016/j.cellsig.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan D, Plattner R. Cancer Res. 2006;66:5648–5655. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Sims JT, Plattner R. Oncogene. 2008;27:1095–105. doi: 10.1038/sj.onc.1210714. [DOI] [PubMed] [Google Scholar]
- Stuart JR, Gonzalez FH, Kawai H, Yuan ZM. J Biol Chem. 2006;281:31290–31297. doi: 10.1074/jbc.M602389200. [DOI] [PubMed] [Google Scholar]
- Taagepera S, McDonald D, Loeb JE, Whitaker LL, McElroy AK, Wang JY, Hope TJ. Proc Natl Acad Sci U S A. 1998;95:7457–62. doi: 10.1073/pnas.95.13.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanos B, Pendergast AM. J Biol Chem. 2006;281:32714–32723. doi: 10.1074/jbc.M603126200. [DOI] [PubMed] [Google Scholar]
- Thobe MN, Gurusamy D, Pathrose P, Waltz SE. Oncogene. 2010;29:214–226. doi: 10.1038/onc.2009.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong T, Sun G, Doorly M, Wang JYJ, Schwartz MA. Proceedings of the National Academy of Sciences. 2003;100:10281–10286. doi: 10.1073/pnas.1635435100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KKC, Yuan ZM. Cancer Res. 2003;63:3418–3424. [PubMed] [Google Scholar]
- Ulrich HD. Cell Cycle. 2004;3:15–8. [PubMed] [Google Scholar]
- Wagh PK, Peace BE, Waltz SE. Advances in Cancer Research. Vol. 100. Academic Press; 2008. pp. 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Ki SW. Biochem Soc Trans. 2001;29:666–73. doi: 10.1042/0300-5127:0290666. [DOI] [PubMed] [Google Scholar]
- Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R, Hung MC. Nat Cell Biol. 2006;8:1359–1368. doi: 10.1038/ncb1501. [DOI] [PubMed] [Google Scholar]
- Welm AL, Sneddon JB, Taylor C, Nuyten DSA, van de Vijver MJ, Hasegawa BH, Bishop JM. Proc Natl Acad Sci U S A. 2007;104:7570–7575. doi: 10.1073/pnas.0702095104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodring PJ, Hunter T, Wang JYJ. J Cell Sci. 2003;116:2613–2626. doi: 10.1242/jcs.00622. [DOI] [PubMed] [Google Scholar]
- Yi W, Lee TH, Tompkins JD, Zhu F, Wu X, Her C. Cancer Res. 2006;66:151–158. doi: 10.1158/0008-5472.CAN-05-3019. [DOI] [PubMed] [Google Scholar]
- Yogalingam G, Pendergast AM. J Biol Chem. 2008;283:35941–53. doi: 10.1074/jbc.M804543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. Nat Cell Biol. 2005;7:278–285. doi: 10.1038/ncb1228. [DOI] [PubMed] [Google Scholar]
- Yuan ZM, Huang Y, Ishiko T, Kharbanda S, Weichselbaum R, Kufe D. PNAS. 1997;94:1437–1440. doi: 10.1073/pnas.94.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZM, Huang Y, Whang Y, Sawyers C, Weichselbaum R, Kharbanda S, Kufe D. Nature. 1996;382:272–274. doi: 10.1038/382272a0. [DOI] [PubMed] [Google Scholar]
- Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang Y, Lu H, Kharbanda S, Weichselbaum R, Kufe D. Nature. 1999;399:814–817. doi: 10.1038/21704. [DOI] [PubMed] [Google Scholar]
- Zhao H, Ho PC, Lo YH, Espejo A, Bedford MT, Hung MC, Wang SC. PLoS ONE. 2012;7:e29416. doi: 10.1371/journal.pone.0029416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Lo YH, Ma L, Waltz SE, Gray JK, Hung MC, Wang SC. Molecular Cancer Therapeutics. 2011a;10:29–36. doi: 10.1158/1535-7163.MCT-10-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Lo YH, Yu L, Wang SC. Mol Carcinog. 2011b;50:383–389. doi: 10.1002/mc.20721. [DOI] [PubMed] [Google Scholar]
- Zhao H, Ou-Yang F, Chen IF, Hou MF, Yuan SS, Chang HL, Lee YC, Plattner R, Waltz SE, Ho SM, Sims J, Wang SC. Neoplasia. 2010;12:214–23. doi: 10.1593/neo.91576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinser GM, Leonis MA, Toney K, Pathrose P, Thobe M, Kader SA, Peace BE, Beauman SR, Collins MH, Waltz SE. Cancer Res. 2006;66:11967–11974. doi: 10.1158/0008-5472.CAN-06-2473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.