


Abstract
CRISPR/Cas9 systems are a versatile tool for genome editing due to the highly efficient targeting of DNA sequences complementary to their RNA guide strands. However, it has been shown that RNA-guided Cas9 nuclease cleaves genomic DNA sequences containing mismatches to the guide strand. A better understanding of the CRISPR/Cas9 specificity is needed to minimize off-target cleavage in large mammalian genomes. Here we show that genomic sites could be cleaved by CRISPR/Cas9 systems when DNA sequences contain insertions (‘DNA bulge’) or deletions (‘RNA bulge’) compared to the RNA guide strand, and Cas9 nickases used for paired nicking can also tolerate bulges in one of the guide strands. Variants of single-guide RNAs (sgRNAs) for four endogenous loci were used as model systems, and their cleavage activities were quantified at different positions with 1- to 5-bp bulges. We further investigated 114 putative genomic off-target loci of 27 different sgRNAs and confirmed 15 off-target sites, each harboring a single-base bulge and one to three mismatches to the guide strand. Our results strongly indicate the need to perform comprehensive off-target analysis related to DNA and sgRNA bulges in addition to base mismatches, and suggest specific guidelines for reducing potential off-target cleavage.
INTRODUCTION
Advances with engineered nucleases allow high-efficiency, targeted gene editing in numerous organisms, primary cells and cell lines. Gene editing was used to create user-defined cells, model animals and gene-modified stem cells with novel characteristics that can be used for gene functional studies disease modeling and therapeutic applications. Clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins constitute a bacterial defense system that cleaves invading foreign nucleic acids (1–8). Chimeric single-guided RNAs (sgRNAs) based on CRISPR (9) have been engineered to direct the Cas9 nuclease to cleave complementary genomic sequences when followed by a 5′-NGG protospacer-adjacent motif (PAM) in eukaryotic cells (10–12). Since gene targeting by CRISPR/Cas9 is directed by base pairing, such that only the short 20-nt sequence of the sgRNA needs to be changed for different target sites, CRISPR/Cas systems enable simultaneous targeting of multiple deoxyribonucleic acid (DNA) sequences and robust gene modification (9–11,13–18).
Endogenous DNA sequences followed by a PAM sequence can be targeted for cleavage by designing a ∼20-nt sequence of the sgRNA complementary to the target. However, other sequences in the genome may also be cleaved non-specifically, and such off-target cleavage by CRISPR/Cas systems remains a major concern. Generally speaking, there is a partial match between the on- and off-target sites and the differences between the on- and off-target sequences can be grouped into three cases: (a) same length but with base mismatches; (b) off-target site has one or more bases missing (‘deletions’); (c) off-target site has one or more extra bases (‘insertions’). Recent studies have shown that CRISPR/Cas9 systems non-specifically cleave genomic DNA sequences containing base-pair mismatches (case a) generating off-target mutations in mammalian cells with considerable frequencies (19–24). Mismatches in the PAM sequence are less tolerated, although Cas9 also recognizes an alternative NAG PAM with low frequency (20,23,25). In addition, Cas9 off-target cleavage at a similar gene sequence with a base pair mismatch may lead to gross chromosomal deletions with high frequencies, as demonstrated by the deletion of the 7-kb sequence between two cleavage sites in HBB and HBD, respectively (22). These results indicate that, although Cas9 specificity extends past the 7–12 bp seed sequence (20,21), off-target effects may limit the applications of Cas9-mediated gene modification, especially in large mammalian genomes that contain multiple DNA sequences differing by only a few mismatches. A recent report revealed that 99.96% of the sites previously assumed to be unique Cas9 targets in human exons may have potential off-target sites containing a functional (NAG or NGG) PAM and one single-base mismatch compared with the on-target site (23).
In this work, we investigated the above-mentioned cases (b) and (c) of potential CRISPR/Cas9 off-target cleavage in human cells by systematically varying sgRNAs at different positions throughout the guide sequence to mimic insertions or deletions between off-target sequences and RNA guide strand. To avoid confusion, for single-base insertions, we use a ‘DNA bulge’ to represent the extra, unpaired base in the DNA sequence compared with the guide sequence. Similarly, for single-base deletions, we use a ‘RNA bulge’ to represent the extra, unpaired base in the guide sequence compared with the DNA sequence (Figure 1). Therefore, adding a base into the guide RNA would result in an RNA bulge, while removing a base in the guide strand can be used to model a DNA bulge. The cleavage activity of RNA-guided Cas9 at endogenous loci in HEK293T cells transfected with plasmids encoding Cas9 and sgRNA variants was quantified as the mutation rates induced by Non-Homologous End Joining (NHEJ). We found that off-target cleavage resulted from the sgRNA variants occurred with DNA bulge or sgRNA bulge at multiple positions in the guide strands, sometimes at levels comparable to or even higher than those of original sgRNAs. We further examined the Cas9-mediated mutagenesis at 114 potential off-target loci in the human genome carrying single-base DNA bulges or sgRNA bulges together with a range of base mismatches, and confirmed 15 off-target sites with mutation frequencies up to 45.5%. Our results clearly indicate the need to search for genomic sites with base-pair mismatches, insertions and deletions compared with the guide RNA sequence in analyzing CRISPR/Cas9 off-target activity and in designing RNA guide strands for targeting specific genomic sites.
Figure 1.
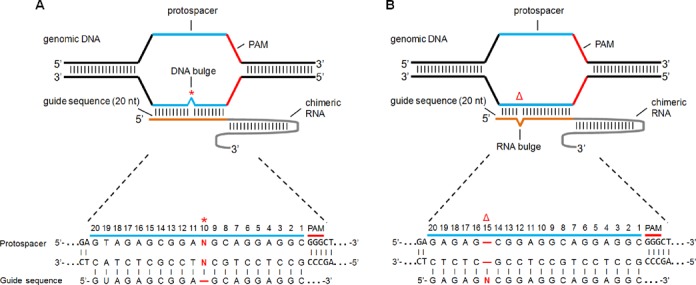
Schematic of CRISPR/Cas9 off-target sites with (A) 1-bp insertion (DNA bulge) or (B) 1-bp deletion (RNA bulge). The 20-nt guide sequence (orange line) in the sgRNA is shown with genomic target sequence (protospacer) containing single-base DNA bulge (red asterisk) or single-base sgRNA bulge (red Δ). The zoom-in sequences of protospacer and PAM are shown above the sgRNA guide sequence. Positions of nucleotides in the target are numbered 3′ to 5′ starting from the nucleotide next to PAM.
MATERIALS AND METHODS
CRISPR/Cas9 plasmid assembly
DNA oligonucleotides containing a G followed by a 19-nt guide sequence (Supplementary Table S1) were kinased, annealed to create sticky ends and ligated into the pX330 plasmid that contains the +85 chimeric RNA under the U6 promoter and a Cas9 expression cassette under the CBh promoter (kindly provided by Dr Feng Zhang; it is also available at Addgene) (26). Variants of sgRNAs were constructed and tested with one or more nucleotides inserted or deleted (Supplementary Table S2). The annealed oligonucleotides have 4-bp overhangs that are compatible with the ends of BbsI-digested pX330 plasmid. Constructed plasmids were sequenced to confirm the guide strand region using the primer CRISPR_seq (5′-CGATACAAGGCTGTTAGAGAGATAATTGG-3′).
T7 endonuclease I (T7E1) mutation detection assay for measuring endogenous gene modification rates
The cleavage activity of RNA-guided Cas9 at endogenous loci was quantified based on the mutation rates resulting from the imperfect repair of double-stranded breaks by NHEJ. In a 24-well plate, 60 000 HEK293T cells per well were seeded and cultured in Dulbecco's Modified Eagle Medium (DMEM) media supplemented with 10% Fetal Bovine Serum (FBS) and 2 mM fresh l-glutamine, 24 h prior to transfection. Cells were transfected with 750 ng (sgRNA variants) or 1000 ng of CRISPR plasmids using 3.4 μl FuGene HD (Promega), following manufacturer's instructions. Each sgRNA plasmid was transfected as biological duplicates in two separate transfections. All subsequent steps, including the T7E1 assay were performed independently for the duplicates. A HEK293T-derived cell line containing stably integrated EGFP gene was used for sgRNAs targeted to the EGFP gene. This cell line was constructed by correcting the mutations in the EGFP gene in the cell line 293/A658 (27) (kindly provided by Dr Francesca Storici). The genomic DNA was harvested after 3 days using QuickExtract DNA extraction solution (Epicentre), as described in (28). T7E1 mutation detection assays were performed, as described previously (29) and the digestions separated on 2% agarose gels. The cleavage bands were quantified using ImageJ. The percentage of gene modification = 100 × (1 – (1 – fraction cleaved)0.5), as described (28). Unless otherwise stated, all polymerase chain reactions (PCRs) were performed using AccuPrime Taq DNA Polymerase High Fidelity (Life Technologies) following manufacturer's instructions for 40 cycles (94°C, 30 s; 60°C, 30 s; 68°C, 60 s) in a 50 μl reaction containing 1.5 μl of the cell lysate, 3% Dimethyl sulfoxide (DMSO) and 1.5 μl of each 10 μM target region amplification primer (Supplementary Table S3) or off-target region amplification primer (Supplementary Table S4).
Sanger sequencing of gene modifications resulted from Cas9
To validate the mutation rates measured by T7E1 assay, the PCR products used in the T7E1 assays were cloned into plasmid vectors using TOPO TA Cloning Kit for Sequencing (Life Technologies) or Zero Blunt TOPO PCR Cloning Kit (Life Technologies), following manufacturer's instructions. Plasmid DNAs were purified and Sanger sequenced using a M13F primer (5′-TGTAAAACGACGGCCAGT-3′).
Identification of off-target sites
Potential off-target sites in the human genome (hg19) were identified using TagScan (http://www.isrec.isb-sib.ch/tagger), a web tool providing genome searches for short sequences (30). Guide sequences containing single-base insertions (represented with an ‘N’ in the sequence) and single-base deletions at different positions were entered, followed by the PAM sequence ‘NGG’. We alternatively searched for off-target sites using the recently developed bioinformatics program COSMID that can identify potential off-target sites due to insertions and deletions between target DNA and guide RNA sequences (Cradick et al., submitted for publication). Primers were individually designed to amplify the genomic loci identified in the output.
Quantitative PCR to measure the expression levels of different guide RNAs
HEK 293T cells were transfected with 750 ng sgRNA variants, as described above. Each sgRNA was transfected as biological triplicates in three separate wells and processed independently. Total RNA was isolated from cells using the RNAeasy kit (Qiagen). Extracted RNA was reverse-transcribed using the iScript cDNA Synthesis (BioRad). The cDNA was amplified using the iTaq Universal SYBR Green Supermix (BioRad) and analyzed with quantitative PCR using specific primers that annealed at 60°C (Supplementary Table S3). Quantitative PCR was performed in technical triplicates for each cDNA sample from single transfected well. Relative mRNA expression was analyzed using an MX3005P (Agilent) and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. GAPDH expression remained relatively constant among treatments.
Relative mRNA expression of target genes was calculated with the ddCT method. All target genes were normalized to GAPDH in reactions performed in triplicate. Differences in CT values (ΔCT = CT gene of interest − CT GAPDH in experimental samples) were calculated for each target mRNA by subtracting the mean value of GAPDH. ΔCT values were subsequently normalized to the reference sample (mock transfected cells) to get ΔΔCT or ddCT (relative expression = 2−ΔΔCT).
Deep sequencing to determine activities at genomic loci
Genomic DNAs from mock and nuclease-treated cells that were prepared for T7E1 assays were used as templates for the first round of PCR using locus-specific primers that contained overhang adapter sequences to be used in the second PCR (Supplementary Tables S5 and S6). PCR reactions for each locus were performed independently for eight touchdown cycles in which annealing temperature was lowered by 1°C each cycle from 65 to 57°C, followed by 35 cycles with annealing temperature at 57°C. PCR products were purified using Agencourt AmPure XP (Beckman Coulter) following manufacturer's protocol. The second PCR amplification was performed for each individual amplicon from first PCR using primers containing the adapter sequences from the first PCR, P5/P7 adapters and sample barcodes in the reverse primers (Supplementary Table S5). PCR products were purified as in first PCR, pooled in an equimolar ratio, and subjected to 2 × 250 paired-end sequencing with an Illumina MiSeq.
Paired-end reads from MiSeq were filtered by an average Phred quality (Q score) greater than 20 and merged into a longer single read from each pair with a minimum overlap of 10 nucleotides. Alignments were performed using Borrows-Wheeler Aligner (BWA) for each barcode (31) and percentage of insertions and deletions containing bases within a ±10-bp window of the predicted cut sites were quantified. Error bounds for indel percentages are Wilson score intervals calculated using binom package for R statistical software (version 3.0.3) with a confidence level of 95% (32). To determine if each off-target indel percentage from a CRISPR-treated sample is significant compared to a mock-treated sample, a two-tailed P-value was calculated using Fisher's exact test.
RESULTS
Cas9 cleavage with sgRNA variants containing single-base DNA bulges
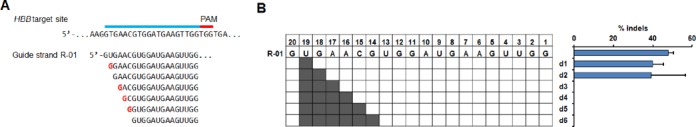
To determine if CRISPR/Cas9 systems tolerate genomic target sites containing single-base DNA bulges (Figure 1a), we used the sgRNA–DNA interfaces of two sgRNAs, R-01 and R-30, targeting the HBB and CCR5 genes, respectively as a model system (22). Systematically removing single nucleotides at all possible positions throughout the original 19-nt guide sequences of R-01 and R-30 resulted in single-base DNA bulges at their original HBB and CCR5 target sites that model single-base insertion at potential off-target sites in the genome (Figure 2A and B).
Figure 2.

Activity of sgRNA variants targeted to genomic loci containing single-base DNA bulges. A single nucleotide was deleted from the original sgRNA at all possible positions (red dashes) throughout the guide sequence for (A) sgRNA R-01 targeting HBB or (B) sgRNA R-30 targeting CCR5. Cleavage activity for the corresponding sgRNA variants measured by T7E1 assay in HEK293T cells at (C) the HBB site or (D) CCR5 site for the sgRNA variants in (A) and (B). Sequence of the original sgRNA is in the top row of the grid. Positions of the deleted nucleotides are highlighted for A (green), G (black), C (blue), or U (red) in the grid. Semi-transparent colors in two positions in the same sgRNA indicate that deletions can be interpreted at either of adjacent positions (also marked by ‘or’) due to identical nucleotides at both positions. The bar graph on the right shows cleavage activity aligned to the corresponding sgRNA variants using the same color scheme. Positions relative to PAM are labeled on the y-axis. The vertical dashed lines mark the activity levels of the original sgRNAs. Error bar, SEM (n = 2).
Cleavage of the genomic DNA in HEK293T cells was quantified using the T7E1 mutation detection assay. For both groups of sgRNA variants (generated from R-01 and R-30 respectively), single-base DNA bulges at certain positions in the DNA sequences were well tolerated (e.g. still had Cas9 induced cleavage), though variants of R-30 had higher cleavage activity at more locations (Figure 2C and D). For both groups, it was clear that Cas9 tolerated DNA bulges in target sites in three regions: seven bases from PAM, the 5′-end (PAM-distal) and the 3′-end (PAM-proximal). Specifically, "-1 nt" variants of R-01 induced Cas9 cleavage activity when a single-base DNA bulge is present at positions 1 or 2, 6 or 7, 18 and 19 of the target DNA sequence from the PAM (Figure 2C). Note that due to the presence of consecutive identical nucleotides at positions 1 and 2, 6 and 7, removing either one of the identical nucleotides in the sgRNA at these adjacent positions would give the same sequence and have the same sgRNA–DNA interface (their position is therefore marked as ‘or’ in Figure 2C and D). In contrast, "-1 nt" variants of R-30 induced variable cleavage activity at more positions throughout the guide sequence: positions 1, 2 or 3, 7, 8, 9 or 10, 11, 16, 17, 18 and 19 from the PAM (Figure 2D). Seven R-30 variants have activities comparable to or even higher than that of the original sgRNA. These variants correspond to DNA bulges at positions 1, 2 or 3, 8, 9 or 10, 11, 18 and 19 from the PAM (Figure 2D). Consistent with previous studies showing that the specificity of CRISPR/Cas9 systems is guide-strand and target-site dependent (19,20,22), the positions in R-01 sgRNA variants where DNA-bulges were tolerated are different from that in R-30 sgRNA variants. However, these positions seem to group in the 5′-end, middle and 3′-end regions of the target loci, as in both R-01 and R-30 sgRNA–DNA interfaces, single-base DNA-bulges at the following five positions seems to be tolerated: positions 1, 2, 7, 18 and 19. Although additional studies are needed to determine if these positions are common for different target sequences, single-base DNA-bulges at the target sites corresponding to these positions may be worth investigating when performing off-target analysis for CRISPR/Cas9 systems.
In certain cases, off-target sites with DNA bulges may also be interpreted as sequences having various base mismatches with guide sequence and/or PAM (Supplementary Figure S1). For example, the sgRNA–DNA interfaces corresponding to removing 5′-end bases in the guide sequences (positions 18 and 19 of the R-01 interface and 16–19 of the R-30 interface) can be viewed as having DNA bulges or having mismatches in the 5′-end region of sgRNA, which have been shown to be better tolerated compared to the 3′-end region (11,19,20). Therefore, the Cas9 cleavage activities induced by these guide strands may be interpreted as tolerance of base mismatches at the 5′-end of the guide RNA. In addition, the position-1 variant of R-30 results in a shift in the adjacent PAM from GGG to CGG (another canonical PAM), which could explain why the activity of this guide sequence variant was similar to the original R-30. However, off-target activities associated with most other DNA bulges for the R-01 and R-30 interfaces cannot be attributed to base mismatch tolerance, since a base removal in the sgRNAs (corresponding to a DNA bulge) could result in many base mismatches or mutation in the PAM sequence. For example, the cleavage activity induced by the R-01 variant at position 2/1 may be alternatively interpreted as Cas9 cleavage with a GTG PAM (Figure 2C and Supplementary Figure S1), which is highly unlikely according to previous studies (20,21). Further, a R-30 guide strand variant at position 11 would contain at least seven mismatches if modeled without a bulge. This guide strand resulted in a 1.8-fold higher cleavage activity compared to the original R-30 (Supplementary Figure S1 and Figure S2D), which cannot be readily explained by the high level of base mismatches (which should prohibit cleavage), and thus should be attributed to the tolerance of DNA bulges.
Cas9 cleavage with small sgRNA truncations
We further investigated if sgRNAs with small truncations at the 5′-end retain cleavage activity. One to six nucleotides were deleted from the 5′ end of R-01 except for the nucleotide at position 20, because the guanine here is required for the expression under the U6 promoter (Figure 3A). For these guide sequence truncations, we found that 1- to 2-bp 5′ truncations could still induce cleavage activities similar to the full-length sgRNA (Figure 3B).
Figure 3.
Activity for sgRNAs containing 5′-end truncations. (A) 1–6 bp truncations at the 5′ end of the guide sequence R-01 targeted to the HBB gene. (B) Activity for truncated sgRNAs. Truncated positions are highlighted in gray in the grid. Bar graph shows corresponding cleavage activity measured by T7E1 assay in HEK293T cells. Error bar, SEM (n = 2).
Cas9 cleavage with sgRNA variants containing single-base sgRNA bulges
In addition to Cas9 induced cleave at off-target sites with single-base DNA bulges, we further investigated if single-base sgRNA bulges (that model single-base deletions in DNA sequence) could induce Cas9 cleavage (Figure 1B). Again, using sgRNA–DNA interfaces R-01 and R-30 as model systems, we systematically added single nucleotides at positions throughout the original guide sequences, so that the interfaces with target sequences in HBB or CCR5 carries single-base sgRNA bulges (Figure 4). For some positions, the addition of single nucleotide A, C, G and U, respectively to the guide sequence was all tested to account for the effect of base identity. As above, HEK293T cells were transfected with plasmids of the Cas9 and sgRNA variants and the T7E1 mutation detection assay was used to measure the Cas9 cleavage activity.
Figure 4.
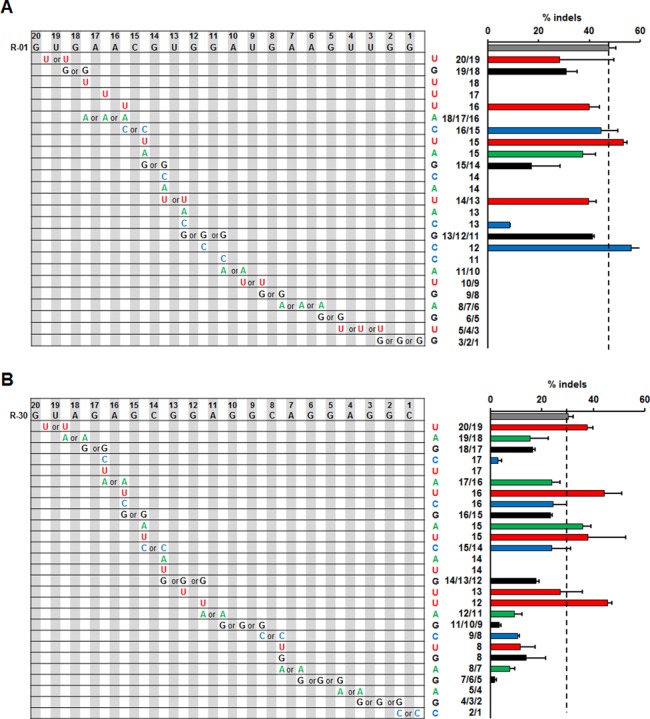
Activity of sgRNA variants targeted to genomic loci containing single-base sgRNA bulges. (A and B) Activity of Cas9 at (A)HBB target site and (B)CCR5 target site carrying single-base sgRNA bulges associated with different variants of the original sgRNAs (A) R-01 and (B) R-30. Single nucleotide, A (green), G (black), C (blue), or U (red), was inserted into the original sgRNA throughout the guide sequence. Sequence of the original sgRNA is in the top row of the grid. Positions of the original guide sequence are shaded in gray, while the inserted positions are white. Due to identical nucleotides at adjacent positions, some inserted nucleotides can be in multiple positions (marked by ‘or’). Bar graphs on the right show corresponding cleavage activities quantified by T7E1 assay in HEK293T cells, with the same color scheme for different inserted nucleotides. Positions relative to PAM and the single nucleotides added are labeled on the y-axis. Error bar, SEM (n = 2).
We found that sgRNA bulges in the R-30 sgRNA–DNA interface were better tolerated compared to those of R-01. In contrast to the tolerances of DNA bulges adjacent to the PAM, sgRNA bulges close to the PAM prohibited cleavage (Figure 4). For the R-01 interface, single-base sgRNA bulges between each of the 11 PAM-proximal guide-strand nucleotides resulted in no detectable activity (Figure 4A). Single-base sgRNA bulges of the four nucleotides closest to the PAM in R-30 also eliminated T7E1 activity (Figure 4B). The sgRNA bulges 3′ to the position 11 in R-30 resulted in reduced cleavage activities (Figure 4B). The lack of activity with PAM-proximal sgRNA bulges in R-01 and low levels of activity with PAM-proximal sgRNA bulges in R-30 are consistent with the reduced mismatch tolerance in the ‘seed sequence’ reported in previous studies (9,11,33). Nucleotides additions in sgRNA sometimes created consecutive identical nucleotides, such as adding a G before or after position 14 of R-01 or before or after position 15 of R-30. These sgRNA variants model a G-bulge that can be at either position in the sgRNA (Figure 4A). We found that in many cases sgRNA bulges with a single U gave rise to high nuclease activities. Among all sgRNA variants with activities higher than the original sgRNAs, ∼71% (5/7) were targeted to the loci with a U-bulge. Overall, single-base sgRNA bulges induced higher Cas9 cleavage activities at many more positions than that with single-base DNA bulges. This is not surprising since RNA molecules are more flexible than DNA molecules, thus having smaller binding energy penalty with single-base RNA bulges, resulting in a higher tolerance (34).
RNA–DNA interfaces with single-base RNA bulges can also be viewed as sequences with various mismatches in the guide sequence and PAM (Supplementary Figure S2). Specifically, sgRNA bulges at the 5′-end of guide RNA sequences (e.g. U+20/19 for R-01 and R-30 interfaces) can be alternatively viewed as having one to a few base mismatches with the 3′-end of DNA sequences (Supplementary Figure S2), which are often tolerated, similar to deletions of 1–2 bp at the 5′ end of guide strands (Figure 3). SgRNA bulges close to the 3′-end of guide sequence can be alternatively viewed as having base mismatches in the 3′-end region, including those at the third base of PAM (R-30 variants) (the last six variants in Supplementary Figure S2). Among all sgRNA variants with considerable activities (Supplementary Figure S2), most of them could not be explained by tolerance of base mismatches, since they would contain more than five mismatches or change in the third base of PAM, which was shown to abolish cleavage activity (20).
The effect of GC (guanine-cytosine) content of sgRNAs on the tolerance of single-base sgRNA bulges
As revealed in our study, the specificity profile (location and level of off-target cleavage) of R-01 variants is substantially different from that of R-30 variants. R-30, which showed a higher level of tolerance to DNA and RNA bulges than R-01, has a GC content of 70%, whereas R-01 has a GC content of 50%. We hypothesized that the GC content of guide strands R-01 and R-30 played a significant role in causing this difference. To investigate this hypothesis, we tested two additional sets of guide strands targeted to HBB and CCR5 genes, respectively, with different GC contents compared to R-01 and R-30 (Figure 5A). Specifically, R-08 has a moderately higher GC content compared to R-01 (65% compared to 50%), whereas the GC content of R-25 is half of that of R-30 (35% compared to 70%). Cas9 induced cleavage with sgRNA variants of R-08 and R-25 was individually tested to quantify the bulge tolerance in HEK 293T cells.
Figure 5.
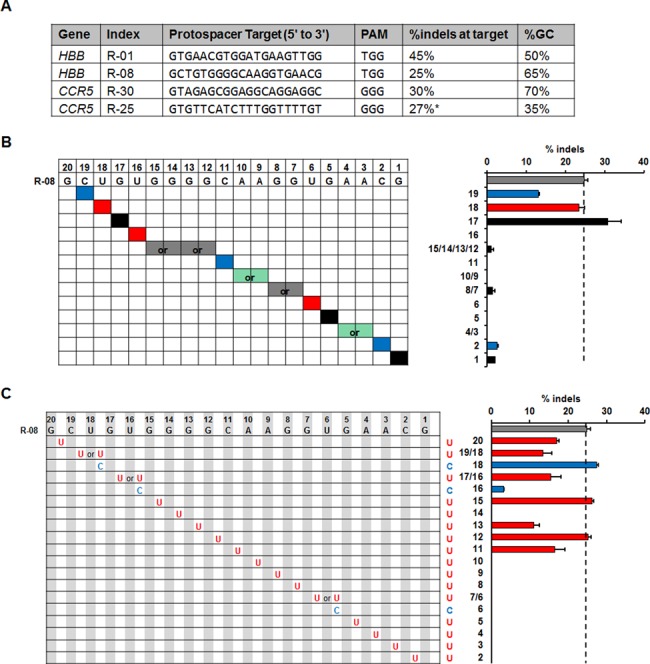
Activity of sgRNA variants with bulges targeted to genomic loci with different GC contents. (A) Target sites, cleavage activities (% indels by T7E1 assay) and GC contents of different guide strands targeted to HBB and CCR5 genes. *Cleavage activity of R-25 is from reference (22). (B and C) T7E1 activity of R-08 variants targeted to HBB genomic loci with (B) single-base DNA bulges or (C) single-base sgRNA bulges. Color schemes and labels are similar to Figures 2 and 4. Error bar, SEM (n = 2).
For the guide strand R-25, which contains a low percentage of GC, we found that all R-25 variants tested showed non-detectable activities using the T7E1 assay (Supplementary Table S2). In contrast, for R-08 variants with bulges throughout the guide sequence, we observed cleavage activities at more positions compared with R-01 (Figure 5B and C). These results of bulge tolerance for variants of R-08 and R-25 support our GC dependence hypothesis.
Cas9 cleavage with sgRNA variants containing 2- to 5-bp bulges
In addition to single-base bulges between sgRNA and target sequence, it is important to determine if bulges longer than 1 bp can also be tolerated by the CRISPR/Cas9 systems. Consequently, the tolerance of 2- to 5-bp bulges was tested at locations where single-base bulges were well tolerated. For sgRNA bulges, we added two to five U's 15- or 12-bp upstream of PAM into the guide sequences of R-01 and R-30, respectively. To generate DNA bulges, we deleted two bases from the guide sequences of R-01 and R-30 (Figure 6A). Strikingly, we found that sgRNA variants forming 2-, 3- and 4-bp RNA bulges induced cleavage activities as determined by the T7E1 assay in HEK 293T cells (Figure 6B). Since sgRNA variants forming 2-bp DNA bulges did not show any detectable activity, we did not test longer DNA bulges. Our findings that sgRNA bulges of >2-bp are better tolerated than DNA bulges of similar size are consistent with the higher cleavage activities by guide strands with 1-bp sgRNA bulges compared to those with 1-bp DNA bulges as shown in Figures 2 and 4.
Figure 6.
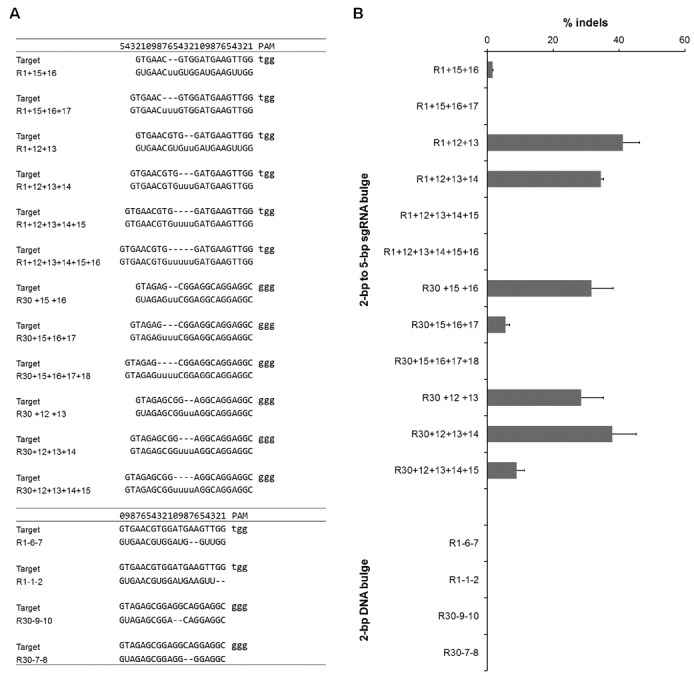
Activity of sgRNA variants with 2-bp DNA or 2- to 5-bp sgRNA bulges. Guide strands with 2- to 5-bp addition are labeled with ‘+’ and positions of the added bases and guide strands with 2-bp deletion are labeled with ‘−’ and positions of the deleted bases. (A) Sequences comparison of guide RNAs and target sites, with position numbers on top. (B) Bar graph showing cleavage activities of these sgRNA variants quantified by T7E1 assay in HEK293T cells. Error bar, SEM (n = 2).
Cleavage by paired Cas9 nickases with sgRNA variants containing single-base bulges
Paired Cas9 nickases (Cas9n) were recently developed to generate DNA double-strand breaks by inducing two closely spaced single-strand nicks using an appropriately designed pair of guide RNAs (23,35). This strategy may lower the off-target cleavage, as double stranded breaks (DSBs) could occur only when both guide RNAs of the pair induced two nicks adjacent to each other at roughly the same time. Here we tested if paired Cas9n systems can tolerate bulges by using one bulge-forming guide variant paired with a perfectly matched guide strand. Specifically, four variants of R-01 showing high activities with Cas9 were paired with R-02, including R1 U+14/13 and R1 C+12 to test sgRNA bulges and R1 −7/6 and R1 −2/1 to test DNA bulges. Each paired sgRNAs created a 34-bp 5′ overhang in the HBB gene (Figure 7A) (22), and the Cas9n cleavage activities were determined by the T7E1 assay. We found that both sgRNA and DNA bulges were also well tolerated in the Cas9n system (Figure 7B). The paired Cas9 nickases with single sgRNA bulges showed activities comparable to Cas9 system having one bulge in R0–1; however, for DNA bulges, the activities of paired Cas9 nickases were >2-fold higher than that of Cas9.
Figure 7.
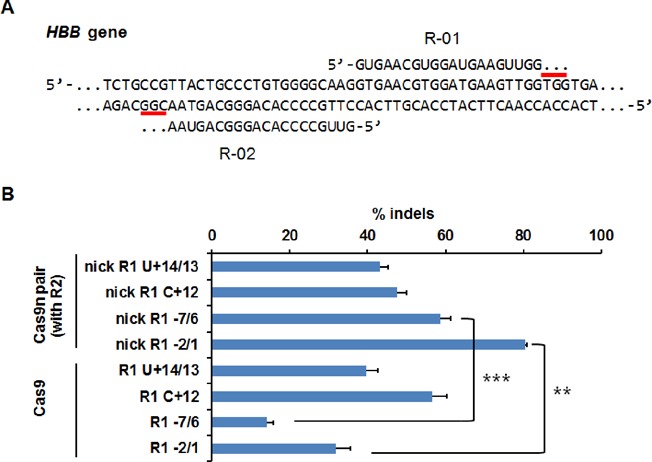
Paired Cas9 nickases with one bulge-containing sgRNA effectively cleave genomic DNA. (A) Human HBB gene targeted by Cas9 nickases (Cas9n) with paired guide strands R-01 and R-02. PAMs are indicated with grey bars. (B) T7E1 activities of Cas9n with R-01 bulge-variants paired with R-02, compared with original Cas9 activities of the R-01 bulge-variants as in Figures 2 and 4. Error bar, SEM (n = 2). Asterisks indicate P-values from a two-tailed independent two-sample t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
Cas9 cleavage at genomic loci with both base mismatches and DNA or sgRNA bulges
To gain a better understanding of CRISPR/Cas9 off-target activity, we examined 27 different sgRNAs targeting six different genes (Supplementary Table S1), seven targeted HBB, two for EGFP, five for CCR5, seven for ERCC5, four for TARDBP and two for HPRT1, respectively. We performed off-target analyses of these sgRNAs by searching the human genome for potential off-target sites and found that for the sgRNAs searched, single-base DNA or sgRNA bulges were not located without mismatches in the human genome. Therefore, for each sgRNA, we selected a subset of the potential sites with one to three mismatches and avoided mismatches close to the PAM as much as possible. All of these sgRNAs efficiently induced mutations at their intended target loci in human HEK293T cells, as measured by the T7E1 assay (Supplementary Figure S3). Using the T7E1 assay, we initially investigated 18 potential off-target sites containing target-site insertions and 62 containing deletions (Supplementary Table S4).
Two sgRNAs targeted to CCR5 and ERCC5, respectively, also induced cleavage at two off-target sites each bearing one DNA bulge and one mismatch (Figure 8A and B). For R-30, the identified off-target site R-30 Off-4 contains a single-base DNA bulge at position 5, 6 or 7 and a base mismatch at position 14. The off-target gene modification rate determined by T7E1 is 9%, almost one third of the 30% on-target activity at the CCR5 gene (Figure 8A). For an R-31 off-target site with a single-base DNA bulge at position 2 and a mismatch at position 20, the off-target gene modification rate determined by T7E1 was 3%, compared to 60% on-target activity at the ERCC5 gene (Figure 8B). Due to the high frequency of small indels (insertions and deletions) that result from repair of Cas9 induced cleavage, which may be poorly detected by the T7E1 assay, we verified the mutagenesis at these off-target sites using Sanger sequencing (Figure 8C and D). For both off-target sites, the mutation frequencies quantified by Sanger sequencing are higher than those by T7E1, which is consistent with a previous study (22). We did not observe any off-target cleavage for the 62 sites tested with both sgRNA bulge and base mismatch, although in our model systems with sgRNA bulges only, high cleavage activities were observed (Figure 4). This discrepancy suggests that sites forming sgRNA bulges may be less tolerant to additional base mismatches and vice versa.
Figure 8.
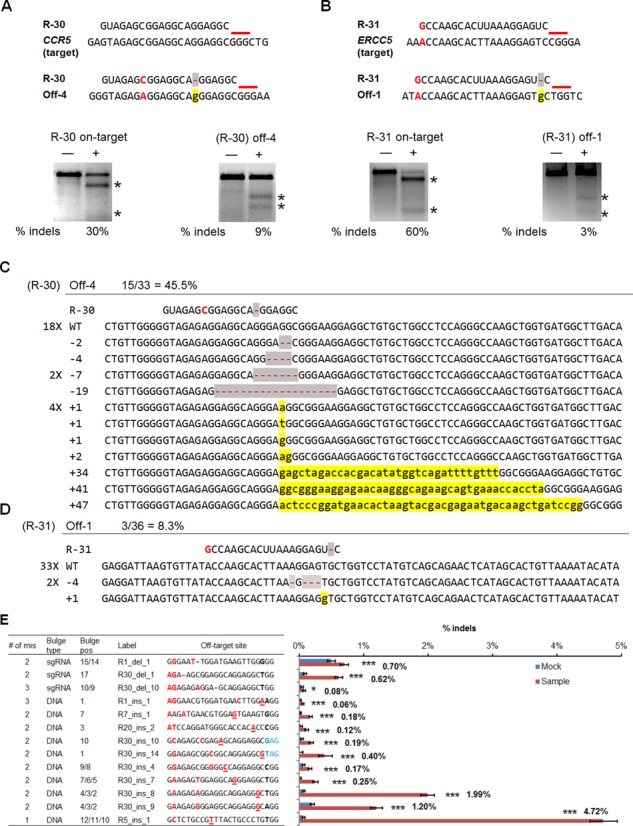
Activities of CRISPR/Cas9 nucleases at genomic target sites and at off-target sites with single-base DNA bulges coupled with mismatches. (A and B) On-target and off-target cleavage activities for (A) sgRNAs R-30 targeted to CCR5 gene, and (B) R-31 target to ERCC5 gene. Upper: target sequences (CCR5 and ERCC5) and off-target sequences (Off-4 and Off-1) with mismatch (red) and DNA bulge (shaded in yellow) shown next to the sgRNA (R-30 and R-31) tested. Red lines indicate the PAM. Bottom: Cleavage activities at the target sites and off-target sites measured by T7E1 assay in HEK293T cells. ‘−’ and ‘+’ denote samples treated without and with nuclease, respectively. Numbers below the lanes indicate average percentages of gene modification (n = 2). Asterisks indicate specific T7E1 cleavage products. (C and D) Sanger sequencing reads of amplified off-target sites aligned to the wild-type genomic sequence and sgRNAs for (C) R-30 and (D) R-31. The occurrence of each sequence is indicated to the left of the alignment, if greater than one. Unmodified reads are indicated by ‘WT’. Deletions are marked in gray and insertions marked in yellow. (E) Significant activities analyzed by deep sequencing at genomic off-target loci containing bulges coupled with mismatches and in some cases alternative NAG-PAM. Only bulge-containing off-target loci determined to have P-values less than 0.05 are shown. Table on the left shows numbers of mismatches at off-target loci in addition to bulge (no. of mis), bulge types, positions of bulges from PAM (bulge pos), labels for the loci as in Supplementary Table S6 and sequences of off-target sites including PAMs. In these off-target genomic sequences, mismatches are marked by red, deleted base compared to sgRNA marked as ‘−’ (sgRNA bulge), inserted base compared to sgRNA marked as underlined red letters (DNA bulge), NAG-PAMs are marked by blue. Bar graph on the right indicates indel percentages quantified for mock (blue) and treated samples (red) with sgRNAs at off-target loci shown in the table to the left. Error bars, Wilson intervals (see ‘Materials and Methods’ section). *P ≤ 0.05, ***P ≤ 0.001 as determined by Fisher's exact test. The % indel values of treated samples are also indicated.
Two genomic off-target sites for guide strand R-30, Off-4 and Off-5, have identical target sequences (Supplementary Table S4), but were cleaved at different rates. Specifically, R-30 Off-4 had a cleavage rate of 9%, while the cleavage at Off-5 was undetectable with the T7E1 assay (Supplementary Figure S4). Sanger sequencing revealed a 45.5% mutation rate at the R-30 Off-4 locus (Figure 8C), compared to a 4.2% mutation rate at R-30 Off-5 (Supplementary Figure S4). Since R-30 Off-4 and R-30 Off-5 sites have identical sequences, our results clearly suggest that off-target cleavage of Cas9 nuclease is very dependent on genomic context (22). Further investigation of these two sites using the ENCODE annotation from UCSC genome browser (36,37) revealed that R-30 Off-4, which had high off-target activity, targeted a site within 400 bp of the 3′ end of a long non-coding RNA (RP4-756H11.3) and 12 kb of the protein-coding gene RABGEF. Analysis of the ENCODE data for chromatin structure in normal human embryonic kidney cells (NHEK) cells, the cell type of origin for the HEK293 cells used in this study shows Off-4 to be within 3 kb of a strong enhancer (marked by H3K27Ac and H3K4me1) and a strong DNAse1 hypersensitive site, suggestive of an open chromatin structure. In contrast, R-30 Off-5, which had low activity, targeted a site in a 162-kb intergenic region between the WBSCR28 and ELN genes that is marked by the more heterochromatic H3K27me3, and hence may be less accessible for Cas9 induced cleavage (Supplementary Figure S5). Taken together, these data strongly suggest that differences in the local chromatin structure may underlie the observed differences in cleavage efficiency between Off-4 and Off-5.
We further performed deep sequencing at 55 putative off-target sites corresponding to single-base sgRNA bulges and 21 sites corresponding to single-base DNA bulges. The sites were amplified from genomic DNA harvested from HEK 293T cells transfected with Cas9 and sgRNAs (Supplementary Table S6). The 55 sites with sgRNA bulges contain 35 sites tested in the preliminary T7E1 assay, and the 21 sites with DNA bulges include seven sites tested in the T7E1 assay. Putative bulge-forming loci containing one to three PAM-distal mismatches were chosen, since we did not find sites associated with a bulge without any base mismatch. We also selected some of the bulge-forming sites with a high level of sequence similarity, but containing an alternative NAG-PAM. For comparison, the deep sequencing also investigated 16 on-target sites of the sgRNAs tested. Each locus was sequenced from mock-transfected cells as control.
We identified additional 13 bulge-forming off-target sites with significant cleavage activities resulted from CRISPR/Cas9 systems compared to the mock-transfected samples (Figure 8E). We found that the number of genomic off-target cleavage sites associated with sgRNA bulges was relatively small (some of these cases are indistinguishable from a few mismatches at 5′ end), but there was considerable activity at genomic sites with DNA bulges coupled with one to three additional base mismatches, even with an alternative NAG-PAM. Similar results showing more off-target effect with DNA bulges plus mismatches compared to sgRNA bulges plus mismatches were observed in the preliminary T7E1 assay (Figure 8A and B). The positions of these tolerated DNA bulges are 1–3 and 7–10 bp from PAM, consistent with the results from the model systems using sgRNA variants. The majority of the sites with off-target activities detected, as shown in Figure 8A, B and E are associated with the sgRNA R-30, which has a high GC content (70%). Other sgRNAs that resulted in off-target cleavage at bulge-forming loci have GC content ≥50%.
DISCUSSION
Although CRISPR/Cas9 systems can efficiently induce gene modification in many organisms, recent studies revealed that off-target cleavage may occur in mammalian cells with up to five-base mismatches between the short ∼20-nt guide RNA and DNA sequences (19–22). Here we show that CRISPR/Cas9 systems can have off-target cleavage when DNA sequences have an extra base (DNA bulge) or a missing base (sgRNA bulge) at various locations compared with the corresponding RNA guide strand. Importantly, our results revealed that, sgRNA bulges of up to 4-bp could be tolerated by CRISPR/Cas9 systems (Figure 6). The correlation between cleavage activity and the position of DNA bulge or sgRNA bulge relative to the PAM appears to be loci and sequence dependent when comparing the specificity profiles of guide sequences R-01 and R-30.
Our results suggest the need to perform comprehensive off-target analysis by considering cleavage due to DNA and sgRNA bulges in addition to base mismatches. We believe that the following design guidelines will help reduce potential off-target effects of CRISPR/Cas9 systems: (i) conservatively choose target sequences with relatively low GC contents (e.g. ≤35%), (ii) avoid target sequences (with either NGG- and NAG-PAM) with ≤3 mismatches that form DNA bulges at 5′ end, 3′ ends or around 7–10 bp from PAM and (iii) if possible, avoid potential sgRNA bulges further than 12 bp from PAM. To aid the rational design of sgRNAs for an intended DNA cleavage site, as well as experimental determination of off-target activity, a robust bioinformatic tool that incorporates these design guidelines and ranking potential off-target sites is desired, and more extensive studies of off-target cleavage by CRISPR/Cas9 systems may be needed concerning the dependence of off-target activity on the type (base mismatch, DNA bulge, sgRNA bulge), location and length of sequence differences.
We found that different specificity profiles of R-01 and R-30 guide sequences (and variants) are not due to different expression levels of the sgRNAs. Quantitative PCR of inactive R-01 variants and active R-30 variants indicated similar sgRNA expression levels (Supplementary Figure S6). We believe that high GC-content, which makes the RNA/DNA hybrids more stable (39), may be responsible for increased tolerance of DNA bulges and sgRNA bulges. Consistent with our hypothesis, guide strand R-30 (70% GC) showed the highest tolerance to sgRNA and DNA bulges among the four guide strands we tested (R-01, R-08, R-25 and R-30), while guide strand R-25 (35% GC) does not seem to tolerate any bulges. Guide sequences showing bulge-related off-target activity in Figure 8 all have GC contents ≥50%, which further confirms that it is important to consider DNA-bulges for sgRNAs with high GC content, even with up to three base mismatches, when investigating off-target effects.
As shown in Supplementary Figures S1 and S2, bulges in the PAM distal or PAM proximal regions can reflect either mismatch tolerance or RNA/DNA bulge tolerance. In a bioinformatics search considering base mismatches only, some of the potential off-target sites identified may overlap with a search considering bulges. Although in both scenarios the mismatch and bulge-containing sites should be tested for off-target cleavage, a better understanding of the bulge tolerance as well as the difference in the mechanisms underlying these two scenarios is needed. A recent study revealed that a Cas9 ortholog from Streptococcus thermophilus has a PAM located 2 bps downstream of the protospacer (38). Thus, the cleavage resulting from the variant R-01 -2/1 (Supplementary Figure S1) may reflect the tolerance of a linker between the target sequence and PAM instead of a DNA-bulge. On the other hand, Cas9 cleavage with RNA or DNA bulges in the middle of the target sequence may reflect only the bulge tolerance.
An interesting finding from this study is that sgRNA variants with bulges had different indel spectra than sgRNA without bulges (Supplementary Figure S7). We quantified indel spectra for original sgRNAs R-01 and R-30, as well as sgRNA variants R1 −7/6, R1 C+12, R30 −11 and R30 U+12, using deep sequencing with around 104 reads for each sample. Bulge-forming sgRNA variants showed higher ratios of larger deletions (Δ10 or Δ7), whereas the original sgRNAs without bulges generate mostly 1-bp insertions. This effect is more prominent for variants forming sgRNA bulges (R1 C+12 and R30 U+12). Bulge-forming sgRNA variants may be more effective than regular sgRNAs in creating larger deletions that might be preferred in certain applications, such as targeted disruption of genomic elements.
Recently, paired Cas9 nickases have been shown to increase target specificity of CRISPR/Cas9 systems. However, only off-target activity associated with single guide RNAs were investigated (23,35), and the effect of cooperative nicking at potential off-target sites with sequence similarity to a pair of guide RNAs has not been characterized. We showed that Cas9n is able to cleave efficiently at target sites despite a single-base bulge in one of the paired guide RNAs. The results of this work provide some insight into off-target cleavage of the paired Cas9 nickases, since nicking of opposite DNA strands is likely to be independent events and the knowledge of bulge tolerance at the sgRNA–DNA interface would be applicable to off-target cleavage of Cas9 nickases.
Recent studies on the specificity of CRISPR/Cas9 systems revealed that a broad range of partial matches between sgRNA and DNA sequences could induce off-target cleavage (19–22), which may limit the choice of sgRNA designs. While the use of existing bioinformatic tools based on base mismatches is certainly useful for predicting the most likely potential off-target sites, it might miss some important sites, since there would be too many base mismatches if bulges were not allowed to form in the middle of a target sequence, so the potential off-target sites with bulges are not likely to be included in the output of these search tools. Therefore, based on our results, it is necessary to search partially matched sequences including base mismatches, deletions and insertions and their combinations in identifying off-target sites. Since there might be a large number of potential off-target sites due to the many partially matched sequences, and the effect of sgRNA–DNA sequence differences on off-target cleavage is target-site and genome-context dependent, experimentally determining the true off-target activities is necessary, including the use of deep sequencing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr Feng Zhang for providing the Cas9 expression plasmid and Drs Francesca Storici and Matthew Porteus for providing the cell line derived from 293/A658. We would like to acknowledge the help on sequencing and bioinformatics analysis from Brian Krueger and Joshua Bridgers at Center for Human Genome Variation at Duke University and Greg Doho and R. Ben Islett at the Emory Integrated Genomics Core.
FUNDING
National Institutes of Health (Nanomedicine Development Center Award) [PN2EY018244 to G.B.]. Funding for open access charge: National Institutes of Health Award PN2EY018244 to G.B.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bolotin A., Quinquis B., Sorokin A., Ehrlich S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 2.Horvath P., Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 3.Marraffini L.A., Sontheimer E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010;11:181–190. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garneau J.E., Dupuis M., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadán A.H., Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 5.Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009;139:945–956. doi: 10.1016/j.cell.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makarova K.S., Grishin N.V., Shabalina S.A., Wolf Y.I., Koonin E.V. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 8.Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mali P., Esvelt K.M., Church G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H., Wang H., Shivalila C.S., Cheng A.W., Shi L., Jaenisch R. One-Step generation of mice carrying reporter and conditional alleles by CRISPR/cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie K., Yang Y. RNA-guided genome editing in plants using a CRISPR-Cas system. Mol Plant. 2013;6 doi: 10.1093/mp/sst119. [DOI] [PubMed] [Google Scholar]
- 15.Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R., Joung J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho S.W., Kim S., Kim J.M., Kim J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 17.Li D., Qiu Z., Shao Y., Chen Y., Guan Y., Liu M., Li Y., Gao N., Wang L., Lu X., et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:681–683. doi: 10.1038/nbt.2661. [DOI] [PubMed] [Google Scholar]
- 18.Shan Q., Wang Y., Li J., Zhang Y., Chen K., Liang Z., Zhang K., Liu J., Xi J.J., Qiu J.L., et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:686–688. doi: 10.1038/nbt.2650. [DOI] [PubMed] [Google Scholar]
- 19.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cradick T.J., Fine E.J., Antico C.J., Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho S.W., Kim S., Kim Y., Kweon J., Kim H.S., Bae S., Kim J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang W., Bikard D., Cox D., Zhang F., Marraffini L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of the RNA-guided Cas9 nuclease. Nat Biotechnol. 2013;31 doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porteus M.H., Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 28.Guschin D.Y., Waite A.J., Katibah G.E., Miller J.C., Holmes M.C., Rebar E.J. A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- 29.Reyon D., Tsai S.Q., Khayter C., Foden J.A., Sander J.D., Joung J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012;30:460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iseli C., Ambrosini G., Bucher P., Jongeneel C.V. Indexing strategies for rapid searches of short words in genome sequences. PLoS One. 2007;2:e579. doi: 10.1371/journal.pone.0000579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.R Core Team. R Foundation for Statistical Computing. Vienna, Austria: 2013. [Google Scholar]
- 33.Sapranauskas R., Gasiunas G., Fremaux C., Barrangou R., Horvath P., Siksnys V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011;39:9275–9282. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. Molecular Biology of the Cell. New York: Garland Science; 2007. [Google Scholar]
- 35.Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., et al. Double nicking by RNA-Guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenbloom K.R., Sloan C.A., Malladi V.S., Dreszer T.R., Learned K., Kirkup V.M., Wong M.C., Maddren M., Fang R., Heitner S.G., et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen H., Choi J., Bailey S. Cut site selection by the two nuclease domains of the Cas9 RNA-guided endonuclease. J. Biol. Chem. 2014 doi: 10.1074/jbc.M113.539726. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugimoto N., Nakano S., Katoh M., Matsumura A., Nakamuta H., Ohmichi T., Yoneyama M., Sasaki M. Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry. 1995;34:11211–11216. doi: 10.1021/bi00035a029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.