
Figure 1.
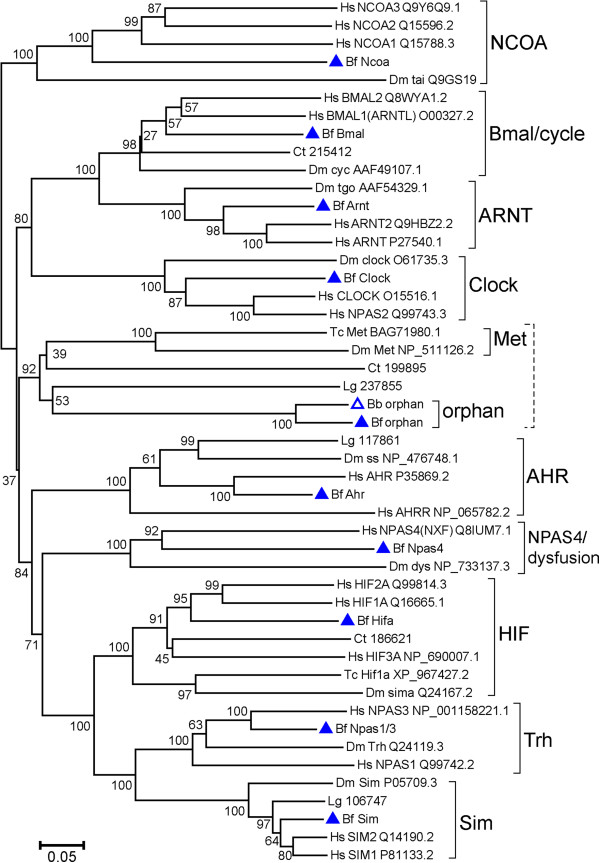
Phylogenetic analysis of all bHLH-PAS protein families with neighbor-joining method. The tree is a neighbor-joining bootstrap consensus tree based on a concatenated alignment of bHLH, PAS A, and PAS B domains. The rooting should be considered as arbitrary. Bootstrap support values (as percentages) from 1,000 replicates of each branch are shown. Branchiostoma floridae proteins are labeled with filled blue triangles. The BbbHLHPAS-orphan (Bb orphan) from B. belcheri is labeled with an open blue triangle. Insect methoprene-tolerant (Met) proteins and spiralian predicted proteins were included because these sequences had high scores when we used the BfbHLHPAS-orphan sequence to perform BLASTp searches. Spiralian sequences are labeled as abbreviations (Ct for Capitella teleta and Lg for Lottia gigantea) + protein ID on the Joint Genome Institute genome browser. This tree shows that nine amphioxus proteins are grouped into well-known bHLH-PAS families with high bootstrap support (≥98%). Two amphioxus ‘orphan’ proteins, two insect Met proteins, and two spiralian predicted proteins (Ct199895 and Lg237855) form a cluster with a 92% bootstrap support. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the p-distance method and units used are the number of amino acid differences per site. The analysis involved 48 amino acid sequences. All ambiguous positions were removed for each sequence pair. There were a total of 258 positions in the final dataset. Bb, Branchiostoma belcheri; Bf, Branchiostoma floridae; Ct, Capitella teleta; Dm, Drosophila melanogaster; Hs, Homo sapiens; Lg, Lottia gigantea; Tc, Tribolium castaneum.