

Abstract
The histologic presence of macrophages (tumor-associated macrophages, TAMs) and neutrophils (tumor-associated neutrophils, TANs) has been linked to poor clinical outcomes for solid tumors. The exact mechanism for this association with worsened prognosis is unclear. It has been theorized that TAMs are immunomodulated to an alternatively activated state and promote tumor progression. Similarly, TANs have been shown to promote angiogenesis and tumor detachment. TAMs and TANs were characterized for activation state and production of prometastatic mediators in an immunocompetent murine model of pancreatic adenocarcinoma. Specimens from liver metastases were evaluated by immunofluorescence and immunoblotting. TAMS have upregulated expression of CD206 and CD163 markers of alternative activation, (4.14 ± 0.55-fold and 7.36 ± 1.13-fold over control, respectively, P < 0.001) but do not have increased expression of classically activated macrophage markers CCR2 and CCR5. TAMs also express oncostatin M (OSM). We found that TANs, not TAMs, predominantly produce matrix metalloproteinase-9 (MMP-9) in this metastatic tumor microenvironment, while MMP-2 production is pan-tumoral. Moreover, increased expression of VEGF colocalized with TAMs as opposed to TANs. TAMs and TANs may act as distinct effector cells, with TAMs phenotypically exhibiting alternative activation and releasing OSM and VEGF. TANs are localized at the invasive front of the metastasis, where they colocalize with MMP-9. Improved understanding of these interactions may lead to targeted therapies for pancreas adenocarcinoma.
Keywords: macrophages, neutrophils, pancreatic cancer, tumor microenvironment, metastasis
solid tumors are made up of a variety of cells. Recent data suggest that tumor-associated immune cells (leukocytes, mononuclear, and polymorphonuclear) may be integral in tumor progression, as they have the ability to release mediators that facilitate growth and metastasis (10, 31). Approximately 80% of pathological reports for solid tumors demonstrate that increased numbers of tumor-associated macrophages (TAMs) are associated with poorer outcomes, while increased TAMs may be protective in 10% of malignancies (7, 31). Although the role of tumor-associated neutrophils (TANs) in tumor progression is unclear (24), there is evidence that increased TANs have a negative prognostic impact on clinical outcomes (4, 7, 10).
In their role as effector cells, macrophages move from a quiescent to an activated state. Activated macrophages are a heterogeneous group of cells, able to take on different physiological functions but are frequently referred to as “classically activated” or “alternatively activated” (25, 26). Classically activated macrophages (CAMs), or type I macrophages, result from the inflammatory signals IFN-γ and TNF-α, which enhance macrophage antigen presentation, intracellular pathogen cytotoxicity, and the production of nitric oxide (26). CCR2 and CCR5 are two receptors expressed in monocytes in the CAM-TH1 paradigm (28). In contrast, alternatively activated macrophages (AAMs), or type II macrophages, are induced by IL-4 and glucocorticoids, have limited cytotoxicity for intracellular pathogens and poor antigen presentation, and fail to produce nitrogen radicals (26). The mannose receptor (CD206) and CD163 are distinguished markers of alternatively activated macrophages (14, 34, 38). This alternative activation state produces signals involved in the synthesis and remodeling of the extracellular matrix and is integral in wound healing by promoting cell growth, inducing angiogenesis, and tissue repair (15, 26, 34). AAMs also actively participate in anti-inflammatory signals and tolerance induction (13), which may be a mechanism of tumor avoidance of host immunosurveillance. Tumor cells and their surrounding stroma express and secrete a multitude of cytokines and chemokines that recruit hematopoietic cells (23, 31) and may co-opt TAMs into an alternatively activated phenotype (13). This is felt to create a cycle that promotes further inflammatory cell recruitment, producing growth signals that promote migration, survival, and angiogenesis (25, 31).
Recently, TANs have been implicated as effector cells in the tumor microenvironment and are believed to be integral in the angiogenic switch in the early stages of cellular dysplasia and tumorigenesis in a malignant model of murine pancreatic neuroendocrine cancer (29). TANs have been shown to promote angiogenesis, increase cell detachment, and increase invasive capacity in an in vitro model of breast adenocarcinoma by the expression of oncostatin M (OSM) (33). OSM has the ability to induce cancer cells to produce VEGF and induce angiogenesis (11, 16, 33), and it is a potent chemoattractant for neutrophils (17). Neutrophils have been shown to be a primary early producer of OSM in inflammatory processes; however, macrophages are producers of OSM in chronic inflammatory conditions (17), as one might expect within a tumor microenvironment. Macrophages and neutrophils have the ability to produce MMP-2 and MMP-9, which can contribute to tumor progression and are upregulated in angiogenic lesions (6). MMP-9 also has the ability to regulate and liberate VEGF (6). Neutrophils are also key contributors of tumor MMP-9 (9). Our laboratory has recently shown an increase in TANs at the leading edge of tumors in a transgenic murine model of pancreatic adenocarcinoma (35). Like TAMs, TANs have the ability to secrete growth factors that may sustain tumorigenesis (12), as well as factors that have the potential to facilitate angiogenesis and metastasis, such as VEGF, MMPs, and other proteases (10, 19, 37), which break down the basement membrane, allowing tumor cells to invade surrounding tissue and spread distantly (35).
On the basis of these data, we hypothesize that tumor-associated inflammatory cells (leukocytes) display an alternatively activated phenotype and are equipped to act as effector cells by facilitating growth within metastasis by secreting growth factors OSM, VEGF, and enzymes MMP-2 and MMP-9. Moreover, the purpose of this study was to examine the characteristics of TANs and TAMs in the tumor microenvironment in an immunocompetent model of murine pancreatic adenocarcinoma (3).
MATERIALS AND METHODS
Chemicals and reagents.
RPMI 1640 medium, FBS, and 1% penicillin-streptomycin for cell culture, and wheat germ agglutinin were purchased from Invitrogen (Grand Island, NY, USA). HEPES-buffered saline solution, trypsin-EDTA, and trypsin-neutralizing solution were purchased from Lonza (Walkersville, MD). Primary antibodies against mouse CD206, CD163, CCR2, and CCR5 were purchased from Santa Cruz Biotechnology (San Diego, CA). Antibodies against mouse CCR2, MMP-2, and MMP-9 were also purchased from Abcam (Cambridge, MA). Antibodies against mouse PMN (clone 7/4) and macrophage (CD68) were from ABD Serotec (Oxford, UK). Anti-mouse OSM was purchased from R&D Systems (Minneapolis, MN), and anti-mouse VEGF:VEGF R2 complex-Gv39M was purchased from East Coast Biologics (New Berwick, ME). Anti-mouse GAPDH was purchased from Cell Signaling Technology (Boston, MA).
Murine pancreatic adenocarcinoma culture.
Pan02 cells, purchased from the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute (Frederick, MD), were cultured and maintained, as described previously (5). Cells were reconstituted from culture at a concentration of 5 × 106 cells/ml, to provide a tumor inoculation of 2.5 × 105 cells/50 μl.
Immunocompetent murine metastatic model of pancreatic adenocarcinoma.
Experiments were performed on four groups of five C57/BL6 mice (Jackson Laboratories, Bar Harbor, ME), aged 8–9 wk; five mice were used as the control group. All studies were performed under the guidelines of an approved protocol of the University of Colorado at Denver Institutional Animal Care and Use Committee (IACUC). After acclimation, mice underwent general anesthesia and were inoculated by a subcapsular splenic injection of 2.5 × 105 Pan02 cells (35), control mice did not receive tumor inoculation. Mice were clinically followed (2), weighed 3 times weekly, and killed 5 wk after injection of tumor cells or earlier if there were clinical deterioration per IACUC regulations. Necropsy was performed, and organs with gross metastatic disease were harvested for tissue analysis, preserving them in tissue-freezing media (Fischer Scientific, Pittsburgh, PA) for tissue histology, or snap-frozen for tissue homogenization and stored at −80°C. This model has been shown to consistently produce metastases to the liver, liver hilum, mesentery, and retroperitoneal basin (3). Using this model, we numbered visible liver lesions from 0 to 13 (5), measuring from 1 to 10 mm each.
Hematoxylin-and-eosin staining.
Liver sections from control mice not inoculated with Pan02 cells, as well as livers from mice with metastatic disease, were processed for hematoxylin-and-eosin (H&E) staining. Five-micrometer sections were stained and examined at ×40 magnification. Digital images were obtained using a Nikon Digital Sight DS-Fi1 microscope camera (Nikon Instruments, Melville, NY).
Immunofluorescent staining.
Tumors from liver specimens with gross metastatic disease, as well as liver specimens from control mice that were not inoculated with Pan02 cells, were examined for the presence of neutrophils, macrophages, and protein markers using immunofluorescent staining (IF). Sectioning and staining were performed in 5-μm serial sections of frozen liver segments. Slides were treated in a solution of 30% acetone and 70% methanol for 10 min, and dried at room temperature for 2 min. After three washes with PBS, they were fixed in 4% paraformaldehyde. Sections were washed in PBS and then blocked with 10% donkey serum for 30 min. They were then incubated overnight at 4°C in primary antibody, anti-mouse neutrophil (clone 7/4, dilution 1:500 in 1% BSA in PBS), or anti-mouse macrophage (CD68, dilution 1:500 in 1% BSA), costained with one of the following: anti-mouse VEGF:VEGF R2 complex −Gv39M (dilution 1:50 in 1% BSA), OSM (dilution 1:10 in 1% BSA), CD206 (dilution 1:10 in 1% BSA), CD163 (dilution 1:10 in 1% BSA), CCR2 (dilution 1:50 in 1% BSA), CCR5 (dilution 1:50 in 1% BSA), MMP-2 (dilution 1:200 in 1% BSA), or MMP-9 (dilution 1:200 in 1% BSA). They were then washed in PBS and incubated at room temperature in a dark environment with appropriate secondary antibodies of either a donkey anti-mouse CY3 IgG, donkey anti-goat CY3 IgG, or a donkey anti-rabbit CY3 IgG (all imaged on the red channel), Alexa Fluor 488-labeled donkey anti-rat IgG (Molecular Probes, Invitrogen; imaged on the green channel), and bisBenzimide H 33342 trihydrochloride (Sigma-Aldrich, St. Louis, MO; imaged on the blue channel) for 60 min. Slides were then washed with PBS, rinsed with distilled water, and air dried. They were mounted with anti-quenching medium and sealed with nail polish. Slides were then examined with a Leica DMRXA digital microscope (Leica Mikroskopie und Systeme, Wetzlar, Germany) and three random pictures from each slide were taken of both areas of tumor, as well as the nontumor surrounding liver parenchyma using SlideBook 2.6 software (Intelligent Imaging Innovations, Denver, CO) at ×40 magnification. Neutrophil and macrophage antibody signaling was calculated by SlideBook using the area of the stain signal for neutrophils and macrophages compared with the total area of the image at ×40 magnification. Data are presented as mean percentage of masked area ± SE of the mean.
Immunoblotting.
Homogenates were prepared from liver segments of control mice that were not inoculated with tumor cells (control). Portions of livers from mice inoculated with Pan02 cells were assessed for areas of gross metastatic disease. Areas grossly free of metastatic disease (liver) were microdissected, and those with visible metastatic disease (tumor) were microdissected and cleared of visibly normal liver, and each were homogenized by placing samples into equal volumes of dissection buffer containing 10.3 M sucrose, 25 mM imidazole, 11 mM EDTA, and 1× protease inhibitor then titrated to a pH of 7.2. Samples were homogenized with a Tissuemizer (Tekmar, Cincinnati, OH) for ∼10 s, centrifuged at 21,000 g, and supernatants were collected. Samples for PAGE were prepared by adding equal volume of homogenate supernatant and 2× Laemmli sample buffer. Forty micrograms of total protein was loaded into each lane and fractioned on a 4–20% acrylamide gradient gel and transferred onto a nitrocellulose membrane. Membranes were incubated in 5% nonfat dry milk in PBS containing 0.1% Tween-20 (T-PBS) for 60 min at room temperature to block nonspecific binding. After washing with T-PBS, membranes were incubated overnight at 4°C with appropriate primary antibodies; VEGF:VEGF R2 complex −Gv39M (dilution 1:200 in 5% BSA in PBS), OSM (dilution 1:1,000 in 5% BSA), CD206 (dilution 1:200 in 5% BSA), CD163 (dilution 1:200 in 5% BSA), CCR2 (dilution 1:200 in 5% BSA), CCR5 (dilution 1:100 in 5% BSA), MMP-2 (dilution 1:1,000 in 5% BSA), MMP-9 (dilution 1:1,000 in 5% BSA), and GAPDH (dilution 1:1,000 in 5% BSA). Membranes were then washed with T-PBS and incubated with appropriate horseradish peroxidase-labeled secondary antibodies in 5% nonfat dry milk in T-PBS for 1 h at room temperature. Protein bands were developed using enhanced chemiluminescence technique. Membranes were exposed to Kodak X-Omat film. Densitometry was performed using the NIH software, ImageJ. A density ratio was created by dividing primary band density by density of the GAPDH band for each sample. Data were then normalized to the expression of control liver for each marker from seven experiments run for each antibody and are presented as the mean fold increases over control liver expression ± SE of the mean.
Statistical analysis.
Parametric statistics were used to analyze the data with the assumption of a normal distribution of activation state. ANOVA was applied with a Fisher exact, post hoc examination, P < 0.05 was considered significant.
RESULTS
H&E staining.
H&E evaluation of liver metastasis confirmed pancreatic adenocarcinoma, with pleomorphic tumor cells with high nuclear to cytoplasm ratio and heavy hematoxylin staining consistent with typical adenocarcinoma features (Fig. 1).
Fig. 1.
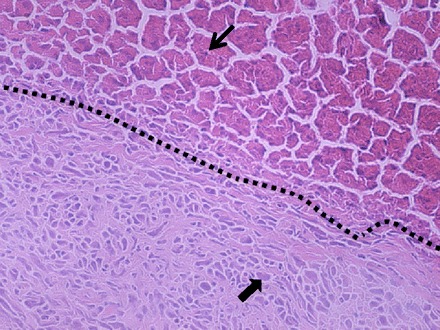
Representative hematoxylin-and-eosin stain of a metastatic lesion in the liver, with corresponding tumor-liver interface (black dashed line), with normal liver and hepatocytes (thin arrow) in the upper-right and tumor in the lower-left, with pleomorphic cells with high nuclear to cytoplasm ratio and heavy hematoxylin staining consistent with adenocarcinoma (thick arrow).
Tumor microenvironment TAMs and TANs.
Immunofluorescence (IF) performed on liver metastases of mice with either anti-CD-68 (macrophage) or anti-7/4 (neutrophil) is shown in Fig. 2. In animals that had undergone tumor inoculation, macrophages and neutrophils were found throughout the liver parenchyma in low amounts (Fig. 2, A and B). However, the amount of each of these inflammatory cells was significantly increased inside the metastases (Fig. 2, C and D). The masked signal percent of the total area for macrophages was 4.30% ± 0.55 in the liver parenchyma compared with 9.32% ± 0.82 in the tumor microenvironment (P < 0.001). For neutrophils, the signal was 0.95% ± 0.12 in the liver parenchyma compared with 5.24% ± 0.59 in the metastasis (P < 0.001). Additionally, the neutrophil infiltration within the tumor microenvironment appeared to be at a higher density along the leading edge (Fig. 2E). In livers from control mice that were not inoculated with tumor cells, there was a trend toward less macrophage area compared with liver parenchyma from tumor-bearing mice, 2.15% ± 0.26 vs. 4.30% ± 0.55, respectively (P = 0.06). There was also a trend toward decreased neutrophil area in control mouse liver compared with liver parenchyma from tumor-bearing mice, 0.51 ± 0.08 vs. 0.95% ± 0.12, respectively (P = 0.08).
Fig. 2.
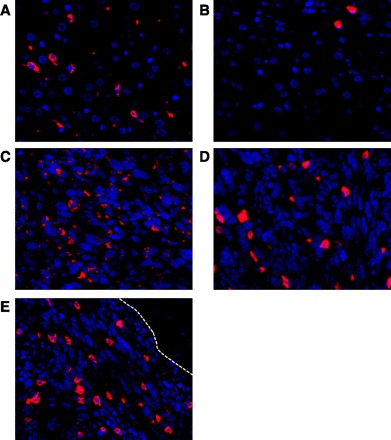
Immunofluorescence staining for macrophages and neutrophils in livers from mice with pancreatic adenocarcinoma. Blue signal in nuclear staining, images are ×40 amplification. A: low density of macrophages (red) is shown in the peritumor liver parenchyma. B: few neutrophils (red) are also present in the nontumor liver parenchyma. Macrophages (C) and neutrophils (D) are densely populated in the tumor microenvironment. E: neutrophil density is high at the leading edge of the tumor microenvironment (white broken line indicates tumor-liver interface).
Expression of alternatively activated markers.
In evaluating the metastatic tumor microenvironment for the expression of the AAM markers, immunoblotting on homogenates for CD206 and CD163 shows low expression of each in control livers and a small, but significant, increase of expression in the liver parenchyma from livers of tumor-bearing mice compared with expression from control livers, 1.40 ± 0.16-fold over control for CD206 (P = 0.018), and 2.58 ± 0.37-fold over control for CD163 (P < 0.001). Examination of metastases for alternatively activated macrophage markers shows increased expression of CD206 and CD163 compared with control livers and peritumor liver parenchyma, 4.14 ± 0.55-fold over control and 7.36 ± 1.13-fold over control, respectively (both P < 0.001 vs. control, Fig. 3, A and B). IF staining of the metastatic tumor microenvironment shows increased signaling of CD206 (Fig. 3, C–E) and CD163 (Fig. 3, F–H), and areas of the tumors with increased signaling of each AAM marker colocalized well with areas densely populated with TAMs. Results of staining for these factors in the liver parenchyma from tumor-bearing mice showed very little signaling of both CD206 and CD163 (data not shown).
Fig. 3.
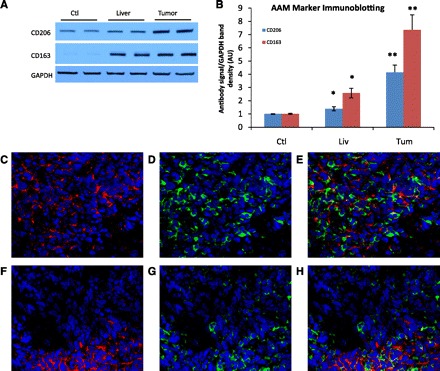
A: Representative immunoblotting of CD206 and CD163 from one experiment shows the increase of expression in homogenates from the tumor (lanes 5 and 6) compared with liver parenchyma from the same animal (lanes 3 and 4) or control mice (lanes 1 and 2). B: densitometry data normalized to control liver expression from seven experiments are represented. *P < 0.02 compared with control. **P < 0.001 compared with control. C–H: immunofluorescence staining for CD206 and CD163 with macrophages from metastatic tumors is shown. Images are ×40 magnification; blue denotes nuclear stain. C-CD206 expression (red) is elevated in the tumor microenvironment. D: macrophage staining (green) of the same image. E: composite image represents areas of overlap, in that areas of high CD206 signal inside the tumor microenvironment colocalize with areas of the tumor that have a denser population of macrophages. F–H: similar pattern in the signal of CD163 (red) that colocalizes with macrophages.
In evaluating the metastatic tumor microenvironment for the expression of the CAM marker CCR2, IF staining showed there was no specific signal in metastases or in the liver parenchyma of tumor-bearing mice and did not colocalize with macrophages (data not shown). Macrophages colocalizing with CCR2 were not identified in the liver tissue of tumor-bearing mice; they were, however, identified in populations of macrophages in the nontumor splenic parenchyma of the same, tumor-bearing mice, which was used as a positive control to confirm antibody function. These results were confirmed by immunoblotting, showing low expression in homogenates of livers of control mice without cancer, liver parenchyma of tumor-bearing mice, and slightly decreased expression among the metastases compared with the liver parenchyma. CCR5 expression within homogenates of liver metastases was decreased 0.32 ± 0.09-fold compared with control (P < 0.001, data not shown). Similar to CCR2, specific CCR5 signaling by IF staining was not identified among macrophages in the metastases or in the macrophages in the liver parenchyma from tumor-bearing mice (data not shown). Selective signaling of CCR5 colocalizing with populations of macrophages in nontumor spleen from the same mice was again used as a positive control. In addition, IF staining further showed that neither CAM nor AAM markers colocalized with TANs (data not shown).
Evaluation of MMP-9 colocalization with tumor-associated neutrophils.
Figure 4, A and B show MMP-9 is highly expressed in homogenates from livers with tumors compared with control liver expression, 36.69 ± 6.69-fold over control in the tumor (P < 0.001) and 9.90 ± 3.28-fold over control in the surrounding liver parenchyma (P = 0.019). In evaluating the tumor microenvironment by IF for MMP-9, focal areas inside the tumor of very high MMP-9 signaling colocalizes to a high degree with TANs (Fig. 4, C–E). Additionally, high signaling of MMP-9 colocalized with the high density of TANs at the leading edge of the tumor (Fig. 4F). There does not appear to be any evidence of colocalization of MMP-9 with macrophages within the tumor (Fig. 4G), or in the peritumor liver parenchyma (data not shown).
Fig. 4.
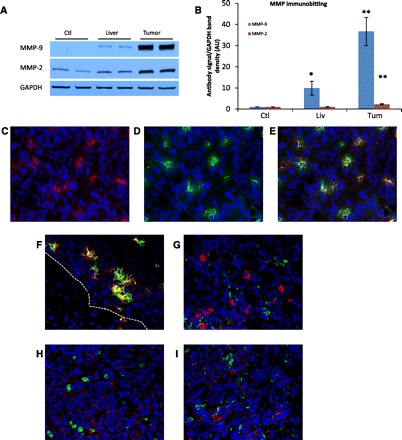
Tumor expression of matrix metalloproteinase-2 (MMP-2) and MMP-9. A representative immunoblotting for both MMPs from one experiment reveal slightly increased expression in the homogenates from liver parenchyma of tumor-bearing mice for MMP-9 vs. control livers, and very high expression of both MMPs in the tumors. GAPDH loading control (see Fig. 3) was reused as an appropriate control for analytes. B: densitometry data normalized to control liver expression from seven experiments are represented, *P ≤ 0.03, **P < 0.001 compared with control. Immunofluorescence of MMPs from the metastases are shown in C–I; images are ×40 amplification, blue denotes nuclear stain. C: MMP-9 (red) stains at low levels in the tumor with areas of focal high signaling. D: neutrophil stain (green) correlates with focal areas of increased signal of MMP-9. E: composite image shows highly specific colocalization of neutrophils and MMP-9. F: high density of MMP-9 signaling colocalizing with neutrophils are shown at the tumor leading edge (white broken line indicates tumor-liver interface, tumor upper right, liver lower left). G: tumors stained for MMP-9 and macrophages (green) do not show the same spatial colocalization. Staining of MMP-2 (red) in tumors does not colocalize well with neutrophils (green, H) or macrophages (green, I).
Metastatic tumor MMP-2 expression among homogenates was also upregulated in a similar manner as MMP-9, 2.53 ± 0.39-fold over control liver expression (P < 0.001) but was not increased in the liver parenchyma from tumor-bearing mice, 0.95 ± 0.11 fold compared with control (Fig. 4, A and B). However, IF staining of MMP-2 within the tumor microenvironment did not selectively colocalize with either TAMs or TANs (Fig. 4, H and I).
Tumor-associated macrophages and OSM and VEGF colocalization.
Immunoblotting of OSM in the homogenates from livers of tumor-bearing mice resulted in small but significantly increased expression of OSM compared with control liver expression, 1.51 ± 0.14-fold (P = 0.004), and markedly increased expression in the metastases, 3.54 ± 0.39-fold (P < 0.001, Fig. 5, A and B). IF staining of OSM and macrophages in the metastatic tumor microenvironment demonstrated colocalization of OSM with TAMs (Fig. 5C). OSM signal in the tumors did not colocalize well with TANs (Fig. 5D).
Fig. 5.
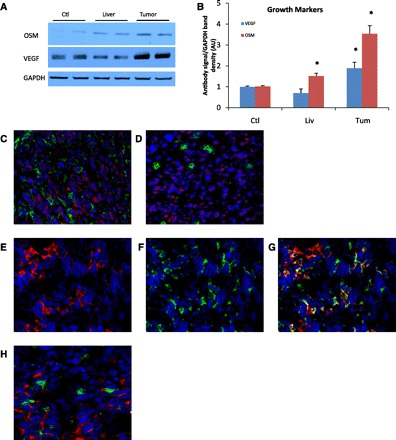
Tumor expression of oncostatin M (OSM) and VEGF. A: representative immunoblotting of OSM and VEGF from one experiment show increased expression of both in homogenates from the tumors. VEGF expression is nonsignificantly decreased in the liver parenchyma from malignant mice compared with control livers. GAPDH loading control (see Fig. 3), was reused as appropriate control for analytes. B: densitometry data normalized to control liver expression from seven experiments are represented. *P < 0.01 compared with control. Immunofluorescence of tumors is shown in C–F, images are ×40 amplification; blue signal denotes nuclear stain. C: increased signal of OSM (red) in the tumor colocalizes with macrophages (green). D: staining of OSM in the tumor does not colocalize well with neutrophils (green). E–G: staining for VEGF (red, E) signal in the tumor and macrophages (green, F) shows colocalization in the combined image (G). H: tumoral VEGF signal does not reveal colocalization with neutrophils (green).
The expression of VEGF is modestly decreased in homogenates of the liver parenchyma of mice with tumors compared with control liver expression, 0.70 ± 0.20-fold compared with control (P = 0.14, not significant), and is increased in the metastases 1.88 ± 0.29-fold over control (P = 0.006, Fig. 5, A and B). IF staining of VEGF from metastatic tumors showed colocalization with TAMs (Fig. 5, E–G), with some tumor surface signal as well. VEGF staining within tumors does not reveal colocalization with TANs (Fig. 5H).
Cumulative results of colocalization studies of TAMs and TANs with markers of activation and growth mediators are summarized in Fig. 6.
Fig. 6.

Colocalization results of alternatively activated markers (CD206 and CD163), classically activated markers (CCR2 and CCR5), and growth mediators (MMP-9, MMP-2, VEGF, and OSM) with tumor-associated macrophages (TAMs) and tumor-associated neutrophils (TANs) (+ denotes positive colocalization, ++ denotes strongly positive colocalization, and − denotes no identifiable colocalization). TAMs colocalized to alternatively activated markers and did not show colocalization to either classical activation marker. TANs did not colocalize with either classical or alternative activation markers. Colocalization of growth mediators suggest polarization of expression, with TANs specifically colocalizing with MMP-9, and TAMs colocalizing with OSM and VEGF.
DISCUSSION
It is generally believed that the immune system provides surveillance and protection against the development of malignancy; however, several studies have suggested that tumors modulate immune cell function to facilitate growth and metastasis (10, 31). TAMs and TANs are uniquely equipped to provide growth factors, angiogenic factors, and enzymes capable of breaking down the extracellular matrix and facilitating the metastatic process (36). Using a model of metastases that creates a hepatic dominant metastatic pattern, as observed in human pancreatic adenocarcinoma (35), we have demonstrated that TAMs express a “procancer” alternatively activated phenotype, as evidenced by a marked increase in expression of CD206 and CD163 in the hepatic metastatic tumor microenvironment. These data demonstrate that TAMs in the tumor microenvironment are AAMs in an in vivo model, which corroborates prior hypothesis papers (10, 31) and in vitro studies (39). Slightly increased expression of AAM markers in the liver parenchyma of tumor-bearing mice may be due to the presence of micrometastases within the grossly normal liver segments or may represent global changes caused by a circulating tumor volume, which allow “fertile soil” for metastasis (30). In our study, we did not observe a relationship between tumor burden and expression of alternative activation. Concomitantly, markers in the CAM-TH1 paradigm, which would be expected in immunosurveillance, were difficult to detect among macrophages within metastases. In this model, TAMs colocalize with VEGF and OSM, which may be indicative of their role in the production of each factor. TANs appear well equipped to act as effector cells via the production of MMP-9, which has been shown to promote angiogenesis and extracellular matrix degradation. We found TANs, not TAMs, are strongly colocalized with MMP-9 and aggregate along the leading edge of the tumor at the invasive front (Fig. 5F). These results suggest a polarity of roles between TAMs and TANs, where TAMs appear to supply growth and angiogenic factors that may recruit hematopoietic cells, while TANs coalesce on the tumor invasion front, where they can act as effector cells for invasion and metastasis.
We postulate that TAMs recruit neutrophils via expression of OSM. OSM is a known, potent chemoattractant for neutrophils (17). It has been shown that macrophages express OSM in chronic inflammatory conditions, which one would expect in a developed metastatic focus (17). Contrary to prior in vitro studies, we did not demonstrate that TANs were colocalized with OSM (33). It is possible that expression of OSM in the tumor microenvironment may mirror that of other known inflammatory conditions (17). We postulate that TANs may express OSM in the early phases of metastasis development, but TAMs predominate in expression chronically in established metastases. Additionally, in our metastatic model, the increased expression of VEGF colocalized with TAMs, suggesting in metastatic foci that TAMs produce VEGF as opposed to TANs, as has been suggested in models of primary neuroendocrine tumor progression (29).
With this preclinical model, we are able to control for many variables, such as tumor burden and genetic variability that make clinical research difficult (3). Pancreatic adenocarcinoma continues to be a disease of high morbidity and mortality (1, 20), with a propensity of metastases, even with small primary tumors (21). Pancreatic adenocarcinoma has generally been resistant to chemotherapy and radiotherapy (22), thus, the need to investigate therapeutic targets and elucidate the mechanism of tumor progression. As increased infiltrating inflammatory cells have been associated with worse outcomes in many types of cancer (10, 31), understanding the functional relationship between tumor cells and the surrounding stroma may lead to novel therapies. This study reveals that TAMs and TANs have the ability to potentiate metastasis by stimulating and sustaining growth, angiogenesis, and promoting invasiveness by breakdown of the extracellular matrix. There are some limitations to this model, which should be noted. In this set of experiments, we have intentionally injected Pan02 cells into the subcapsule of the spleen to expedite the development of hepatic metastases, presumably via the ability of tumors to access the splenic vein and move into the portal circulation. Our laboratory group has significant experience with a similar orthotopic model of pancreas cancer growth (3). However, in this work, we noted significant morbidity in the animals due to peritoneal metastases, particularly from bowel obstruction, and it was our intent to more specifically study hepatic metastases. It should be noted that the immunocompetent orthotopic murine model that we previously employed, involved direct injection of Pan02 cells into the tail of the pancreas. Clinically, patients with pancreatic carcinoma in this location often present with disseminated peritoneal disease, as opposed to patients with cancers in the head of the pancreas, where likely because of the proximity to the portal vein, they present with periportal lymph node metastases and hepatic metastases.
Further, we have intentionally focused on hepatic metastases in this preclinical model as local and regional control of pancreatic adenocarcinoma can often be achieved surgically. Despite this surgical control, patients are still at extremely high risk to develop and succumb to hepatic metastases (8).
From a practical standpoint, hepatic metastases are more easily identified and have borders that lend themselves to facile, histologic and IF examination compared with retroperitoneal metastases. Pancreas cancer progression at nonhepatic locations has a tendency to produce a strong cicatrix reaction, making borders difficult to define (32). It is our expectation that similar and important microenvironmental findings are present in alternate locations, but these are more difficult to study.
Perspectives and Significance
Although the role of TAMs within the tumor microenvironment has been postulated, this work clearly demonstrates the alternatively activated macrophage paradigm (31) within a metastatic focus of pancreatic carcinoma. We hypothesize that this activation state is driven by the genotypically abnormal tumor cells, which affect the location and function of the metastatic events (18). Within this paradigm, the TAMs play a “managerial” role as they are well equipped to promote the recruitment of effector cells, neutrophils (TANs), that act via matrix metalloproteases at the invasive front of the metastasis to promote further tumor progression.
Further, these findings suggest that hepatic directed therapies that manipulate the tumor microenvironment and more specifically target the activation state of TAMs are a cogent strategy in treating pancreatic cancer metastases to the liver. Considering that hepatic metastases derive their blood supply exclusively from the hepatic artery, the use of hepatic arterial infusion pumps has the potential to improve survival and may reduce the morbidity of traditional systemic chemotherapies (27).
GRANTS
This work was supported, in part, by the American Cancer Society, Grant MSRG-09-034-01-CCE.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.D.B., J.H.L., L.A., and C.C.B. performed experiments; D.D.B., X.M., J.H.L., and C.C.S. analyzed data; D.D.B., X.M., D.A.F., E.E.M., J.H.L., C.C.S., and C.C.B. interpreted results of experiments; D.D.B., J.H.L., and L.A. prepared figures; D.D.B. drafted manuscript; D.D.B., X.M., D.A.F., C.C.S., and C.C.B. edited and revised manuscript; D.D.B. and C.C.B. approved final version of manuscript; X.M., D.A.F., E.E.M., C.C.S., and C.C.B. conception and design of research.
REFERENCES
- 1. American Cancer Society. Surveillance and Health Policy research, 2010. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-026210.pdf, accessed Oct 2010
- 3. Barnett CC, Jr, Beck AW, Holloway SE, Kelher M, Schluterman MK, Brekken RA, Fleming JB, Silliman CC. Intravenous delivery of the plasma fraction of stored packed red cells promotes pancreatic cancer growth in immunocompetent mice. Cancer 116: 3862–3874, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B, Bernaudin JF, Mayaud C, Milleron B, Baud L, Cadranel J. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol 152: 83–92, 1998 [PMC free article] [PubMed] [Google Scholar]
- 5. Benson DD, Kelher MR, Meng X, Fullerton DA, Lee JH, Silliman CC, Barnett CC., Jr Gender-specific transfusion affects tumor-associated neutrophil: macrophage rations in murine pancreatic adenocarcinoma. J Gastrointest Surg 14: 1560–1565, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloprotease-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Bio 2: 737–744, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumor progression: implications for new anticancer therapies. J Pathol 196: 254–265, 2002 [DOI] [PubMed] [Google Scholar]
- 8. Breslin TM, Hess KR, Harbison DB, Jean ME, Cleary KR, Dackiw AP, Wolff RA, Abrbruzzese Janjan NA, Crane CH, Vauthey JN, Lee JE, Pisters PWT, Evans DB. Neoadjuvant chemoradiotherapy for adenocarcinoma of the pancreas: treatment variables and survival duration. Ann Surg Oncol 8: 123–132, 2000 [DOI] [PubMed] [Google Scholar]
- 9. Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103: 481–490, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coussens LM, Werb Z. Inflammation and cancer. Nature 420: 860–867, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cross A, Edwards SW, Bucknall RC, Moots RJ. Secretion of oncostatin M by neutrophils in rheumatoid arthritis. Arthritis Rheum 50: 1430–1436, 2004 [DOI] [PubMed] [Google Scholar]
- 12. Di Carlo E, Forni G, Musiani P. Neutrophils in the antitumoral response. Chem Immunol Allergy 83: 182–203, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Elgert KD, Alleva DG, Mullins DW. Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol 64: 275–290, 1998 [DOI] [PubMed] [Google Scholar]
- 14. Goerdt S, Orfanos C. Other functions, other genes: Alternative activation of antigen-presenting cells. Immunity 10: 137–142, 1999 [DOI] [PubMed] [Google Scholar]
- 15. Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, Pearce EJ, Wynn TA. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: Granulomatous pathology is shaped by the pattern of l-arginine metabolism. J Immunol 167: 6533–6544, 2001 [DOI] [PubMed] [Google Scholar]
- 16. Holzer RG, Ryan RE, Tommach M, Schlekeway E, Jorcyk CI. Oncostatin M stimulates the detachment of a reservoir of invasive mammary carcinoma cells: role of cyclooxygenase-2. Clin Exp Metastasis 21: 167–176, 2004 [DOI] [PubMed] [Google Scholar]
- 17. Hurst SM, McLoughlin RM, Monslow J, Owens S, Morgan L, Fuller GM, Topley N, Jones SA. Secretion of oncostatin M by infiltrating neutrophils: regulation of IL-6 and chemokine expression in human mesothelial cells. J Immunol 169: 5244–5251, 2002 [DOI] [PubMed] [Google Scholar]
- 18. Iacobuzio-Donahue CA, Yachida S. The pathology and genetics of metastatic pancreas cancer. Arch Pathol Lab Med 133: 413–422, 2009 [DOI] [PubMed] [Google Scholar]
- 19. Iwatsuki K, Kumara E, Yoshimine E, Nakagawa H, Sato M, Hayakawa T. Elastase expression by infiltrating neutrophils in gliomas. Neurol Res 22: 465–468, 2000 [DOI] [PubMed] [Google Scholar]
- 19a. Jackson Laboratories. Animal Resources Body Weight Study of 1993. JAX Notes 457: Spring, 1994 [Google Scholar]
- 20. Jemal A, Siegal R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics 2009. CA Cancer J Clin 59: 225–249, 2009 [DOI] [PubMed] [Google Scholar]
- 21. Kern S, Hruban R, Hollingsworth MA, Brand R, Adrian TE, Jaffee E, Tempero MA. A white paper: the product of a pancreas cancer think tank. Cancer Res 61: 4923–4932, 2001 [PubMed] [Google Scholar]
- 22. Kornmann M, Beger HG, Link KH. Chemosensitivity testing and test-directed chemotherapy in human pancreatic cancer. Recent Results Cancer Res 161: 180–195, 2003 [DOI] [PubMed] [Google Scholar]
- 23. Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia 7: 177–189, 2002 [DOI] [PubMed] [Google Scholar]
- 24. Lin EY, Pollard JW. Role of infiltrated leukocytes in tumor growth and spread. Br J Cancer 90: 2053–2058, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23: 549–555, 2002 [DOI] [PubMed] [Google Scholar]
- 26. Mosser DM. The many faces of macrophage activation. J Leukoc Biol 73: 209–212, 2003 [DOI] [PubMed] [Google Scholar]
- 27. Murata S, Tajima H, Abe Y, Komada Y, Fukunaga T, Nakazawa K, Kumazaki T. Transcatheter management for multiple liver tumors after hepatic artery obstruction following reservoir placement. Hepatogastroenterology 52: 852–856, 2005 [PubMed] [Google Scholar]
- 28. Murdoch C, Finn A. Chemokine receptors and their role in inflammation and infectious diseases. Blood 95: 3032–43, 2000 [PubMed] [Google Scholar]
- 29. Nozawa H, Chui C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA 103: 12493–12498, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paget S. The distribution of secondary growths in cancer of the breast. Lancet 1: 571–573, 1889 [PubMed] [Google Scholar]
- 31. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4: 71–78, 2004 [DOI] [PubMed] [Google Scholar]
- 32. Puolakkainen PA, Brekken RA, Muneer S, Sage H. Enhanced growth of pancreatic tumors in SPARC-null mice is associated with decreased deposition of extracellular matrix and reduced tumor cell deposition. Mol Cancer Res 2: 215–224, 2004 [PubMed] [Google Scholar]
- 33. Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL. Breast cancer cells stimulate neutrophils to produce oncostatin M: Potential implications for tumor progression. Cancer Res 65: 8896–8904, 2005 [DOI] [PubMed] [Google Scholar]
- 34. Raes G, De Baetselier P, Noel W, Beschin A, Brombacher F, Hassanzadeh GG. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J Leukoc Biol 71: 597–602, 2002 [PubMed] [Google Scholar]
- 35. Roland CL, Dineen SP, Toombs JE, Carbon JG, Smith CW, Brekken RA, Barnett CC., Jr Tumor-derived intercellular adhesion molecule-1 mediates tumor-associated leukocyte infiltration in orthotopic pancreas xenografts. Exp Biol Med (Maywood) 235: 263–70, 2010 [DOI] [PubMed] [Google Scholar]
- 36. Roland CL, Harken AH, Sarr MG, Barnett CC., Jr ICAM-1expression determines malignant potential of cancer. Surgery 141: 705–707, 2007 [DOI] [PubMed] [Google Scholar]
- 37. Scapini P, Nesi L, Morini M, Tanghetti H, Belleri M, Noonan D, Presta M, Albini A, Cassatella MA. Generation of biologically active angiostatin kringle 1–3 by activated human neutrophils. J Immunol 168: 5798–5804, 2002 [DOI] [PubMed] [Google Scholar]
- 38. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J Exp Med 176: 287–292, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang B, Wang J, Gao J, Gao Y, Chen X, Wang B, Gao J, Rao Z, Chen Z. Alternatively activated RAW264.7 macrophages enhance tumor lymphangiogenesis in mouse lung adenocarcinoma. J Cell Biochem 107: 134–143, 2009 [DOI] [PubMed] [Google Scholar]