

Abstract
Drug-resistant tuberculosis is a growing threat to global public health. Recent efforts to understand the evolution of drug resistance have shown that changes in drug–target interactions are only the first step in a longer adaptive process. The emergence of transmissible drug-resistant Mycobacterium tuberculosis is the result of a multitude of additional genetic mutations, many of which interact, a phenomenon known as epistasis. The varied effects of these epistatic interactions include compensating for the reduction of the biological cost associated with the development of drug resistance, increasing the level of resistance, and possibly accommodating broader changes in the physiology of resistant bacteria. Knowledge of these processes and our ability to detect them as they happen informs the development of diagnostic tools and better control strategies. In particular, the use of whole genome sequencing combined with surveillance efforts in the field could provide a powerful instrument to prevent future epidemics of drug-resistant tuberculosis.
Introduction
The burden of tuberculosis (TB) caused by drug-resistant Mycobacterium tuberculosis is increasing. As a result, the standard treatment—directly observed therapy, short course (DOTS), composed of 2 months of a four-drug regimen (isoniazid [INH], rifampicin [RIF], pyrazinamide [PZA], and ethambutol [EMB]) followed by 4 months of treatment with RIF and INH—is failing in many settings. While any degree of drug resistance might worsen the prognosis of a TB patient [1, 2], there are two definitions of M. tuberculosis drug resistance that are particularly relevant in the clinic. The first refers to strains resistant to both INH and RIF and is termed multidrug-resistant (MDR) TB. Treatment of patients with MDR-TB takes up to 2 years, and currently relies on fluoroquinolones (FQ) and injectable aminoglycosides (AG) to compensate for the loss of two of the most potent drugs. The acquisition of resistance to these classes of antibiotics defines extensively drug-resistant (XDR) TB. XDR-TB requires even longer treatment with drugs that are much more costly and show limited efficacy and increased side effects. Thus, XDR-TB is often associated with poor treatment outcomes [3].
The global genetic diversity of drug-resistant M. tuberculosis [4–10] indicates that drug resistance evolved on multiple occasions in geographical hotspots characterized by a high incidence of TB and inappropriate drug treatment. The latter is mostly driven by a lack of resources resulting in two important failures: an inability to detect drug resistance and a systemic failure to deploy effective treatments [11–13]. The ongoing evolution of M. tuberculosis in these settings [14, 15], provides an excellent opportunity to explore the genetic determinants of drug resistance in this microbe.
Unlike most other bacterial pathogens, resistance plasmids and horizontal gene transfer play no role in the acquisition of drug resistance in M. tuberculosis. Moreover, efflux mechanisms appear to serve only as a ‘stepping stone’ to high-level resistance. They allow the bacteria to tolerate higher levels of drug but do not per se result in clinically relevant levels of resistance to multiple antibiotics in M. tuberculosis [14, 16–18]. Instead, the evolution of strains resistant to multiple antibiotics is driven by the sequential acquisition and accumulation of resistance-conferring mutations on the bacterial chromosome. These mutations primarily interfere with drug-target binding (e.g. RIF-rpoB [19], FQ-gyrA [20]), compromise prodrug activation (e.g. INH-katG [21], PA-824-fgd [22]), or cause over-expression of the target (INH/ETH—promoter region of inhA). The elucidation of the mechanisms of action for many antimycobacterials led to the identification of key determinants of resistance (see Zhang and Yew [23] and Almeida Da Silva and Palomino [24] for reviews), and the realization that the corresponding genetic mutations can be used as reliable molecular markers for drug susceptibility testing (DST) [25]. The application of this knowledge to the clinic has resulted in the development of diagnostic tools based on nucleic acid amplification (NAA) that overcome many of the shortcomings of phenotypic DST; these include a long turnaround time, outcome variability, and infrastructure requirements [26–29]. A comprehensive overview of these diagnostic tools has been given elsewhere [30]. Recently, one of these tools, known as Xpert MTB/RIF, has been the subject of a policy update published by the World Health Organization. Their recommendation for Xpert MTB/RIF to replace microscopy in HIV-positive patients, patients suspected of MDR-TB, and those suspected of TB meningitis follows its successful implementation in many countries [31, 32]. The instrument is designed to analyse sputum directly, hence bypassing the need for primary bacterial culture. It simultaneously tests for the presence of M. tuberculosis and RIF resistance. Even though this technology is having a positive impact on TB control by offering high sensitivity and reducing the time to TB diagnosis, the associated costs and infrastructural requirements (e.g. a constant power supply, machine maintenance) remain limiting for many high-burden countries [32, 33]. Hence, on-going efforts in product development focus on cheaper and simpler so-called ‘point-of-care’ diagnostics [34]. Nonetheless, DST without bacterial culturing will continue to require NAA. The choice of targets for NAA will determine the power of future diagnostics. It should be based on detailed knowledge of the relationship between strain genotype, drug resistance phenotype, and the patient treatment outcome in terms of relapse rate and treatment failure. Whole genome sequencing (WGS) of clinical drug-resistant strains of M. tuberculosis should be combined with the analysis of clinical information from patients infected by these strains to yield valuable new insights into the biology of drug resistance.
In this review, we first summarize key findings from recent WGS studies of mycobacterial drug resistance. We focus specifically on results that shed light on why the acquisition of individual drug resistance-conferring mutations is only part of the problem. We then use specific examples to illustrate how understanding more broadly the evolutionary mechanisms that drive drug resistance can inform the development of improved diagnostic tools as well as better strategies to preserve both existing and new treatment regimens.
New Genomic Insights into Mycobacterium tuberculosis Drug Resistance
The evolutionary path leading through drug resistance is strongly influenced by two factors: epistasis and bacterial fitness [35–38]. We define epistasis as a set of genetic interactions where the phenotypic effect of one mutation is determined by the presence of one or more other mutations. For example, resistant strains carrying the same resistance mutations vary in their capacity to transmit from patient to patient [39–42], showing that the strain genetic background can determine the course of evolution of drug resistance. Bacterial fitness, on the other hand, is a function of growth rate, virulence, and transmissibility [15, 43]. Any mutation that reduces it in relation to the wild-type strain is said to carry a ‘fitness cost’. The most immediate way to estimate the relative fitness is to measure the growth rate of bacteria in culture. On average, most drug-resistance mutations carry a fitness cost (see Fig. 3) [44] whose magnitude positively correlates with the frequency of different resistance mutations in the clinic; resistant strains with the smallest fitness defect are most abundant [45, 46]. Furthermore, the fitness of resistant mutants is not fixed; evolution is an ongoing process, and a comparison between clinical and laboratory strains carrying the same drug-resistance mutation shows that clinical strains often successfully bypass any fitness cost imposed by resistance [45]. The acquisition of such compensatory mutations is also an example of epistasis and is key in the evolution of transmissible drug-resistant strains that pose the greatest risk to public health [6, 10, 47–51]. Unfortunately, we do not currently have sufficient knowledge to predict epistatic interactions a priori, so we must rely on detecting them empirically by studying the genetics of drug resistance [52].
Fig. 3.
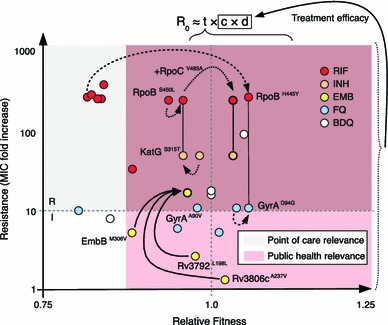
Evolutionary trajectories of epistasis-driven resistance. The relative fitness of strains carrying key drug-resistance mutations grown in the absence of drug was plotted against their contribution to MIC to illustrate the relationship between genotype and phenotype for important drug-resistance mutations. Lines connecting individual mutations denote strains carrying two mutations, while arrows denote estimates of the fitness of double/triple-mutants (three different types of arrows are used to illustrate different evolutionary trajectories). Reproductive number (R 0) defined as the number of secondary cases caused by an infected individual is roughly equal to the product of an organism’s transmissibility (t), number of contacts (c), and the duration of infection (d). Fitness estimates were summarized from Gagneux et al. [45] (RIF, M. tuberculosis), Borrell et al. [46] (FQ, Msm), Safi et al. [47] (EMB, M. tuberculosis), and Huitric et al. [85] (BDQ, M. tuberculosis). We were unable to find true relative fitness measurements for KatG S315T and GyrA A90V; these were estimated from Pym et al. [95] and Poissy et al. [96]. Fold increases in MIC shown are averages of values obtained from Sougakoff et al. [97–99], Pang et al. [97–99], and Anthony et al. [97–99] for RIF; Pym et al. [95] for INH; Safi et al. [47, 100, 101], Plinke et al. [47, 100, 101], and Starks et al. [47, 100, 101] for EMB; Aubry et al. [94, 102–105], Matrat et al. [94, 102–105], Cheng et al. [94, 102–105], Duong et al. [94, 102–105], and Malik et al. [94, 102–105] for FQ; and Huitric et al. [85] for BDQ. BDQ bedaquiline, EMB ethambutol, FQ fluoroquinolones, INH isoniazid, MIC minimum inhibitory concentration, RIF rifampicin
A number of recent studies used WGS to address the evolution of drug-resistant M. tuberculosis [7, 9, 51, 53, 54]. The authors of these studies used approaches based on phylogeny [7], molecular epidemiology [9, 51, 53], and mutation frequency analyses [54] to compare drug-susceptible and drug-resistant clinical strains of M. tuberculosis. The shared aim of these studies was the identification of bacterial genes under positive evolutionary selection by drug pressure. The detailed discussion of the merits and shortcomings of the analytical approaches used in these studies is beyond the scope of this review, but it is important to note that there is considerable overlap in the findings of these studies. In addition to known drug targets, all studies identified novel bacterial genes and intergenic regions whose function may be ancillary to drug-resistance mutations (see Fig. 1). In particular, several genes involved in lipid metabolism, cell wall homeostasis, purine metabolism, and transcriptional control appear to be positively selected in presence of anti-TB drugs [7, 54]. These studies represent an important step towards an improved understanding of drug resistance. However, with the exception of just a few mutations (e.g. ponA1, and the promoters of thyA and thyX [7, 54]), the actual role of these genes remains unclear, essentially offering an extended list of genes of interest that require further investigation. Consequently, it is currently not possible to evaluate the role of these genes in the development of future diagnostics or therapies.
Fig. 1.
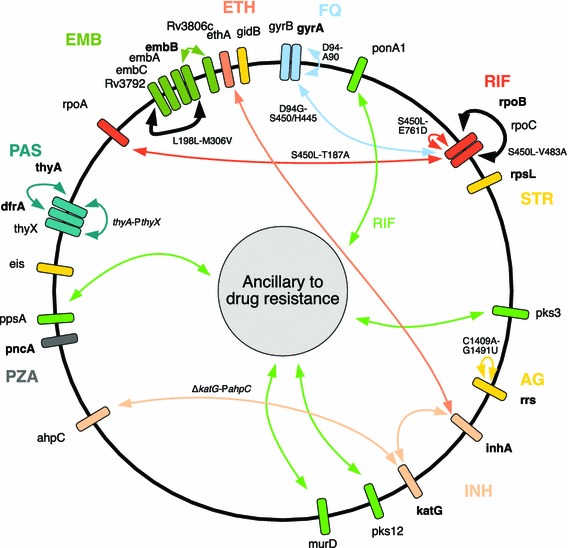
A web of epistasis mediates drug resistance in M. tuberculosis. Key genes in M. tuberculosis drug resistance have been plotted, taking into account their approximate position in the genome. Genes in bold are known to be directly involved in antibiotic resistance. Lines denote putative epistatic interactions; connecting genes involved in the physiology of a drug as well as more broad/indirect mechanisms referred to as ‘ancillary to drug resistance’. This categorization is meant to include factors mediating complex aspects of cell physiology, such as cell permeability and mutation-induced physiological changes. Bold lines connecting rpoB to rpoC and embB to Rv3972 refer to in vitro validated compensatory mechanisms. Specific mutation pairs using M. tuberculosis numbering are shown where known. Figure based on information from the following references: [6, 7, 24, 46, 47, 49, 51, 54, 91–94]. AG aminoglycosides, EMB ethambutol, ETH ethionamide, FQ fluoroquinolones, INH isoniazid, PAS para-aminosalicylic acid, PZA pyrazinamide, RIF rifampicin, STR streptomycin. Many ancillary genes were omitted for the sake of clarity—see original papers for a more comprehensive list [7, 54]
In addition to the above genes, there are a growing number of novel genes for which experimental evidence is available to support a role of epistasis in adaptation to resistance. These genes were identified through recent studies aimed at elucidating the evolutionary trajectory of drug-resistant M. tuberculosis in the clinic [6, 47] and include rpoC, which mediates the adaptation to RIF resistance, and Rv3806c, which appears to have a role in EMB resistance. RIF resistance is caused by mutations of the beta subunit of RNA polymerase encoded by rpoB. Mutated amino acids are normally involved in drug binding and are usually restricted within a short stretch of the protein termed the rifampicin resistance determining region (RRDR) [19]. Many of these mutations carry a fitness cost [45, 49] that appears to be negatively correlated with the activity of the mutant enzyme [55]. The importance of this aspect of RNA polymerase physiology is clearly illustrated by the fact that one of the most frequent resistance mutations observed in the clinic, S450L, (equivalent to S531L in Escherichia coli) also carries the lowest fitness cost [45]. Moreover, this mutation appears to be almost ubiquitous among MDR strains of M. tuberculosis [6, 9, 10, 48, 51], and perhaps more importantly, is shown to be strongly associated with the acquisition of compensatory mutations within RNA polymerase genes (rpoA, rpoB, and rpoC). This combination of mutations is strongly associated with improved transmissibility of strains as evidenced by clonal expansion of M. tuberculosis strains carrying these particular mutations in patient populations [9, 48, 51]. The role of these compensatory mutations (see Koch et al. [56] for a more comprehensive review) appears to be to restore wild-type function of mutant RNA polymerase, probably on the level of enzymatic activity [49, 50]. Alternatively, these compensatory mutations maybe also restore the baseline gene expression profile of cells. Specifically, rpoB mutants were shown to have a modified lipid profile [57] as well as a modified expression of many proteins involved in lipid metabolism, particularly phthiocerol dimycocerosates (PDIMs) [58]. Interestingly, lipid metabolism genes, including those for PDIM metabolism, seem to be under positive selection during the evolution of drug resistance [7, 54]. Because PDIMs and other mycobacterial lipids play an important role in virulence [59, 60], this argues that global physiological consequences of drug-resistance mutations could provide a contextual framework, within which the compensatory mechanisms mediated by mutations in ancillary genes can be explored [7, 51, 54].
A recent set of experiments reported by Safi et al. [47] demonstrate the effect of epistasis in the progressive increase of minimal inhibitory concentration (MIC) for a drug. Focusing their study on EMB, they observed that acquisition of high-level resistance to EMB is a multistep process. In addition to the most frequently isolated resistance mutant—embB M306V—they identified nonsynonymous mutations in Rv3806c and a synonymous mutation in Rv3792 as important contributors to EMB resistance in vitro. They proceeded to show that Rv3806c is involved in modulating the availability of EmbB substrates, while the synonymous single nucleotide polymorphism in Rv3792 apparently stabilized embC RNA, leading to a de facto over-expression of the gene, resulting in reduced susceptibility to EMB [47].
Finally, epistasis is not limited to the physiology of a single drug. Investigations into the interaction between mutations resulting in resistance to disparate drugs have suggested that positive epistasis may drive multidrug resistance [61]. Our group recently published a report showing that specific resistance mutations in rpoB and gyrA can compensate for each other’s fitness defects, to the point that some strains carrying both mutations are fitter than either single mutant [46]. Moreover, the particular combinations of mutations conferring resistance to RIF and ofloxacin (OFX) associated with the highest overall fitness appear to be positively selected in high-burden settings.
The examples listed here are not designed to offer a comprehensive overview of all reported examples of epistasis in drug-resistant M. tuberculosis; we have tried to sketch a more complete picture in Fig. 1. Instead, they were chosen to illustrate important concepts brought to light by recent studies: in the first place, based on current data, it appears that compensatory mutations occur most frequently in strains that already harbor the least costly mutation [48]. Second, epistatic interactions occur between specific mutations [46, 47, 49], and in some cases these can be mutually exclusive; for example, a strain harboring an rpoA mutation does not then also acquire an rpoC or additional rpoB mutations [6, 50, 51, 62]. Continued exposure to a drug seems to impose constraints on evolution that facilitate the acquisition of compensatory mutations in strains that are already resistant [36, 50]. Once generated, these strains are more likely to be transmitted than strains carrying the resistance mutation alone [48, 51]. A further consequence of continued drug exposure is the stepwise accumulation of mutations that result in an increased level of resistance to a drug [47]; a factor that is already influencing INH resistance in XDR strains [63] and may contribute to higher levels of resistance to FQ [64, 65]. Moreover, epistasis can occur between drug-resistance mutations [46, 61, 66], implicating individual resistance determinants as drivers of polydrug resistance. Combined, these observations clearly indicate that continued inappropriate treatment, in part caused by misdiagnosis of resistance, drives the evolution of more transmissible, increasingly drug-resistant strains [10, 51].
A further set of factors, not considered hitherto, is involved in the generation of protein heterogeneity in the cell. The primary source of this is mutations, and mutation rates were shown to vary between different phylogenetic lineages of M. tuberculosis. The Beijing family of strains in particular seems to have a higher mutation rate [67–69]. The exact consequences of this remain to be determined, but differences in mutation rate have been used to explain the correlation between this genotype with a higher rate of drug-resistance acquisition. Mathematical models estimate that Beijing strains are 22-fold more likely to produce MDR strains [68]. Another intriguing possibility was recently put forward by Javid et al. [70], who argue that protein variability driven by errors in the central information-processing pathway (DNA-RNA-protein) may provide a phenotypic stepping-stone to resistance akin perhaps to other ancillary mechanisms shown in Fig. 1. While intellectually appealing, the clinical relevance of such a scenario remains to be substantiated.
Implications for Diagnosis and Therapy
The translational potential of the knowledge gained from the above studies lies in the implementation of molecular tools in TB control (see Table 1 for a summary). Wells et al. [71] offer an excellent review of current and future needs for the clinical management of TB, addressing diagnostics, surveillance, and programmatic approaches. It is useful to take advantage of their conceptual framework to discuss the impact of the themes that emerge from evolutionary studies. They place capabilities available to TB control programs onto a continuum that spans from community-based physicians to supranational reference laboratories (see Fig. 2). Accordingly, the available resources combined with the specific needs of each level will dictate the contribution that molecular approaches can offer.
Table 1.
Implications of understanding evolutionary mechanisms of drug resistance for tuberculosis control
| Experimental observation | Physiological consequence | Implications |
|---|---|---|
| Resistant strains gain compensatory mutations that change the basic physiology, e.g. RIF-rpoC | Increased transmissibility, increased propensity to acquire additional resistance |
Focus surveillance on mutations that correlate with transmissible, highly resistant strains Use spent biosamples to establish wide catchment area for WGS-based surveillance |
| Continued exposure to RIF directs evolution towards the acquisition of compensatory mutations | More rapid evolution of compensatory mutations | Use molecular tools to probe the genotype of strains and stop administering ineffective drugs immediately |
| Epistasis exists between drug-resistance mutations for a single drug: e.g. EMB, FQ, INH, RIF | Multi-step acquisition of high level of drug resistance | Define clinical breakpoint concentrations based on specific susceptibility profiles for a drug |
| Positive epistasis between rpoB and gyrA mutations | Strains with specific combinations of resistance mutations (e.g. gyrA D94G and rpoB H445Y) are fitter than the wild type |
Drug regimens containing both RIF and FQ may fail more quickly. Assess current clinical trials for evidence Evaluate if the order in which drugs are administered might enhance negative epistatic interactions |
| Some mutations conferring resistance to FQ and bedaquiline have no fitness costs attached | No need to compensate for specific resistance mutations |
Enforce appropriate administration of drugs to avoid adding these antibiotics to failing regimens or use as monotherapy Explore the spectrum of resistance mutations, identify low cost and frequent mutations before releasing the drug into the market |
| Mutation rates vary between M. tuberculosis lineages | Beijing family strains acquire resistance to INH and RIF at a higher rate and are 22 times more likely to develop into MDR than the laboratory-adapted strain | Increase frequency of phylogeny-based surveillance and focus monitoring on areas with high rates of Beijing strains |
EMB ethambutol, FQ fluoroquinolones, INH isoniazid, RIF rifampicin, WGS whole genome sequencing
Fig. 2.
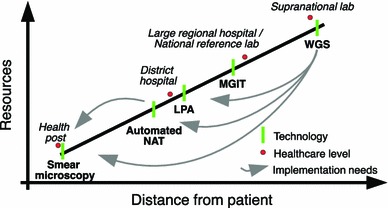
The healthcare continuum. A diagrammatic representation of the current healthcare continuum as described in Wells et al. [71], showing different healthcare levels with their distance from the patient. The resources dimension encompasses a breadth of parameters, from access to infrastructure and apparatus, to technical proficiency of staff and financial resources that are available. Different diagnostic and DST technologies are shown as bars with the arrows indicating the levels at which we would ideally deploy them in the future. WGS whole genome sequencing, LPA line probe assay, MGIT mycobacterial growth indicator tube: phenotypic DST using the Bactec MGIT 960 instrument, automated NAT automated nucleic acid amplification technology
In the clinic, the main goal is rapid and effective treatment of infected individuals. This relies on quick and reliable case detection, and, given the need for speed, is likely to depend on molecular diagnostics in the future [71]. In this setting, focusing on drug-resistance mutations offers high sensitivity and specificity for key first-line drugs [25]. Including compensatory mutations (e.g. in rpoC or rpoA, discussed above) as diagnostic markers is unlikely to provide additional clinical benefit, because mutations in rpoB already display a high sensitivity and specificity for detecting MDR-TB in individual patients [33, 72]. In contrast, a key area in which evolutionary lessons could be brought to bear in a significant manner is surveillance. Consider the reproductive number of a pathogen (R 0, see Fig. 3). Two dimensions are paramount to the outcome of transmission cycles; one is pathogen transmissibility (t) and the other is time to effective treatment (c × d). Strain fitness plays a role in the former, while drug resistance considerably influences the latter. It is in the interest of public health to identify and eradicate transmissible drug-resistant strains quickly. We are currently failing on this front—in 2012, fewer than one-quarter of estimated MDR-TB cases were detected, and only 23 % of those had DST results reported for PZA and FQ [73]. To this end, there is scope for the implementation of ‘molecular diagnostics for public health’. Screening for the emergence of compensatory mutations in a population, perhaps by focusing on re-treatment cases [74] to pinpoint areas with evolved highly transmissible strains as they emerge [48, 51], would aid the public health official to prioritize resource allocation and interrupt the spread of these strains. Examples of such measures include targeted delivery of individualized, albeit expensive treatments, resource-intensive active case finding, and, in extreme cases, isolation [75, 76]. In line with the described differences in mutation rates between M. tuberculosis lineages [67, 68], the public health official may include phylogenetic markers to focus monitoring on areas with a high incidence of Beijing strains. Performing surveillance based on high-resistance mutations alone may be insufficient. Mutations conferring intermediate resistance, namely, below that of empirically determined resistance breakpoints, play a potentially pivotal role in the spread of resistance [47]. We have used the ‘fitness-MIC space’ to illustrate these concepts in Fig. 3. It is also important to stress at this point, that strains harboring mutations conferring low-level resistance can often be treated effectively with existing drugs [77], either by increasing the dosage of the same drug, or by using alternative drugs from the same class, as is the case of FQ [78]. Conversely, the use of ineffective drugs should be stopped immediately to avoid directing the evolution of a strain [36, 50] towards a more transmissible phenotype.
In addition to surveillance, knowledge of epistasis should be applied to the design and deployment of future drug regimens. Given the genetic interaction between mutations in rpoB and gyrA [46], it may be dangerous to administer RIF and FQ simultaneously, and should perhaps be avoided. A number of current clinical trials are testing an iteration of such a combination [79], and their outcomes should be carefully scrutinized by using WGS for evidence of epistasis-driven drug resistance [74, 80–82]. Protecting new drug classes is equally important. Bedaquiline is the first new antimycobacterial to be approved by the US FDA for over 40 years [83, 84]. Given the availability of bedaquiline resistance mutations with no fitness cost, or even a fitness benefit (see Fig. 3) [85], it is key that the guidelines for the administration of the drug are adhered to [86, 87], especially in light of the fact that no standardized regimen exists for it yet. Most importantly, bedaquiline should not be used to rescue a failing regimen, and should always be administered with other effective drugs to minimize the emergence of resistance.
Conclusion
Consensus is growing that biological parameters such as the frequency of resistance mutations, the occurrence of low- or no-cost drug-resistance mutations, epistasis between resistance mutations, as well as host genetics should be considered when deciding on future treatment protocols [88]. Efficient inclusion of all of these factors for drug-resistance surveillance requires broader implementation of WGS. The World Health Organization is currently analyzing the results of a surveillance effort looking for underlying resistance to PZA and FQ where phenotypic DST was paired with DNA sequencing (Zignol, unpublished). The timing of the study is crucial in view of the fact that many of the new treatment regimens under investigation rely on these two drugs, and underlying resistance may severely compromise their success. Applying WGS to clinical trials can also contribute important information on the success of a new treatment [81], as well as sorely needed data for new-in-class antibiotics such as PA-824, delamanide, bedaquiline, and SQ109 [89]. Together, this knowledge should be used to build an accurate picture of how the genetic makeup of a strain ultimately determines the success of treatment.
Due to high costs and logistical requirements, WGS technology might not find its way into many of the most affected and often resource-poor countries in the short term. However, applied at a supranational or national level with a wide-spread community-based catchment area, WGS would allow high-throughput analysis of known but also unknown mutations. Healthcare officials would thus obtain surveillance data and essential information for the development of new diagnostic tests adjusted to the prevailing resistance pattern [39, 40, 42, 90]. For example, one could imagine an approach in which used sputum-microscopy slides from primary TB-diagnostic centers are recycled as a source of M. tuberculosis DNA for pooled DNA sequencing to measure the frequency of drug-resistance alleles in a particular patient population. Similarly, spent DST samples, such as mycobacterial growth indicator tubes (MGIT) and Loewenstein–Jensen slopes, would provide an excellent source of DNA for WGS-based surveillance. The application of WGS and evolutionary principles to drug resistance in M. tuberculosis has been furthering our understanding of the challenges faced in the clinic as well as contributing key data for developing tools and strategies to control drug-resistant TB. As new treatment regimens containing new drugs are implemented, we will have to establish the spectrum of epidemiologically relevant mutations as soon as possible. This will help us track the emergence of strains resistant to these drugs in real-time, thereby prolonging the life span of the new regimens—a fundamental concern given how precious new drugs are.
Acknowledgments
The authors thank the other members of our group for the stimulating discussions. Our work is supported by the Swiss National Science Foundation (grant PP00P3_150750), the National Institutes of Health (AI090928), The European Research Council (309540-EVODRTB), and SystemsX.ch (2013/154). Drs Trauner, Borrell, Reither, and Gagneux have no conflicts of interest to declare.
References
- 1.Lew W, Pai M, Oxlade O, Martin D, Menzies D. Initial drug resistance and tuberculosis treatment outcomes: systematic review and meta-analysis. Ann Intern Med. 2008;149:123–134. doi: 10.7326/0003-4819-149-2-200807150-00008. [DOI] [PubMed] [Google Scholar]
- 2.Winston CA, Mitruka K. Treatment duration for patients with drug-resistant tuberculosis, United States. Emerg Infect Dis. 2012;18:1201–1202. doi: 10.3201/eid1807.120261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pietersen E, Ignatius E, Streicher EM, Mastrapa B, Padanilam X, Pooran A, et al. Long-term outcomes of patients with extensively drug-resistant tuberculosis in South Africa: a cohort study. Lancet. 2014;383:1230–1239. doi: 10.1016/S0140-6736(13)62675-6. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Gao X, Luo T, Wu J, Sun G, Liu Q, et al. Association of gyrA/B mutations and resistance levels to fluoroquinolones in clinical isolates of Mycobacterium tuberculosis. Emerg Microbiol Infect. 2014;3:e19. doi: 10.1038/emi.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ioerger TR, Feng Y, Chen X, Dobos KM, Victor TC, Streicher EM, et al. The non-clonality of drug resistance in Beijing-genotype isolates of Mycobacterium tuberculosis from the Western Cape of South Africa. BMC Genomics. 2010;11:670. doi: 10.1186/1471-2164-11-670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, et al. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet. 2011;44:106–110. doi: 10.1038/ng.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farhat MR, Shapiro BJ, Kieser KJ, Sultana R, Jacobson KR, Victor TC, et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat Genet. 2013;45:1183–1189. doi: 10.1038/ng.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hazbon MH, Motiwala AS, Cavatore M, Brimacombe M, Whittam TS, Alland D. Convergent evolutionary analysis identifies significant mutations in drug resistance targets of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2008;52:3369–3376. doi: 10.1128/AAC.00309-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lanzas F, Karakousis PC, Sacchettini JC, Ioerger TR. Multidrug-resistant tuberculosis in Panama is driven by clonal expansion of a multidrug-resistant Mycobacterium tuberculosis strain related to the KZN extensively drug-resistant M. tuberculosis strain from South Africa. J Clin Microbiol. 2013;51:3277–3285. doi: 10.1128/JCM.01122-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller B, Chihota VN, Pillay M, Klopper M, Streicher EM, Coetzee G, et al. Programmatically selected multidrug-resistant strains drive the emergence of extensively drug-resistant tuberculosis in South Africa (Karakousis PC, editor). PLoS One 2013;8:e70919. [DOI] [PMC free article] [PubMed]
- 11.Gillespie SH. Evolution of drug resistance in Mycobacterium tuberculosis: clinical and molecular perspective. Antimicrob Agents Chemother. 2002;46:267–274. doi: 10.1128/AAC.46.2.267-274.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, van Soolingen D, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet. 2010;375:1830–1843. doi: 10.1016/S0140-6736(10)60410-2. [DOI] [PubMed] [Google Scholar]
- 13.de J Sosa A, Byarugaba DK, Amabile C, Hsueh P-R, Kariuki S, Okeke IN. Antimicrobial resistance in developing countries. Springer, New York; 2009.
- 14.Borrell S, Gagneux S. Strain diversity, epistasis and the evolution of drug resistance in Mycobacterium tuberculosis. Clin Microbiol Infect. 2011;17:815–820. doi: 10.1111/j.1469-0691.2011.03556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Müller B, Borrell S, Rose G, Gagneux S. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 2013;29:160–169. doi: 10.1016/j.tig.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X-Z, Nikaido H. Efflux-mediated drug resistance in bacteria. Drugs. 2009;69:1555–1623. doi: 10.2165/11317030-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machado D, Couto I, Perdigão J, Rodrigues L, Portugal I, Baptista P, et al. Contribution of efflux to the emergence of isoniazid and multidrug resistance in Mycobacterium tuberculosis (Mokrousov I, editor). PLoS One 2012;7:e34538. [DOI] [PMC free article] [PubMed]
- 18.Schmalstieg AM, Srivastava S, Belkaya S, Deshpande D, Meek C, Leff R, et al. The antibiotic resistance arrow of time: efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob Agents Chemother. 2012;56:4806–4815. doi: 10.1128/AAC.05546-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, et al. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 2001;104:901–912. doi: 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- 20.Barnard FM, Maxwell A. Interaction between DNA gyrase and quinolones: effects of alanine mutations at GyrA subunit residues Ser83 and Asp87. Antimicrob Agents Chemother. 2001;45:1994–2000. doi: 10.1128/AAC.45.7.1994-2000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouse DA, DeVito JA, Li Z, Byer H, Morris SL. Site-directed mutagenesis of the katG gene of Mycobacterium tuberculosis: effects on catalase-peroxidase activities and isoniazid resistance. Mol Microbiol. 1996;22:583–592. doi: 10.1046/j.1365-2958.1996.00133.x. [DOI] [PubMed] [Google Scholar]
- 22.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Yew WW. Mechanisms of drug resistance in Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 2009;13:1320–1330. [PubMed] [Google Scholar]
- 24.Almeida Da Silva PE, Palomino JC. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: classical and new drugs. J Antimicrob Chemother. 2011;66:1417–1430. doi: 10.1093/jac/dkr173. [DOI] [PubMed] [Google Scholar]
- 25.Campbell PJ, Morlock GP, Sikes RD, Dalton TL, Metchock B, Starks AM, et al. Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2011;55:2032–2041. doi: 10.1128/AAC.01550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SJ. Drug-susceptibility testing in tuberculosis: methods and reliability of results. Eur Resp J. 2005;25:564–569. doi: 10.1183/09031936.05.00111304. [DOI] [PubMed] [Google Scholar]
- 27.Lange C, Mori T. Advances in the diagnosis of tuberculosis. Respirology. 2010;15:220–240. doi: 10.1111/j.1440-1843.2009.01692.x. [DOI] [PubMed] [Google Scholar]
- 28.Banu S, Rahman SMM, Khan MSR, Ferdous SS, Ahmed S, Gratz J, et al. Discordance across several methods for drug susceptibility testing of drug-resistant Mycobacterium tuberculosis isolates in a single laboratory. J Clin Microbiol. 2013;52:156–163. doi: 10.1128/JCM.02378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Deun A, Wright A, Zignol M, Weyer K, Rieder HL. Drug susceptibility testing proficiency in the network of supranational tuberculosis reference laboratories. Int J Tuberc Lung Dis. 2011;15:116–124. [PubMed] [Google Scholar]
- 30.Laurenzo D, Mousa SA. Mechanisms of drug resistance in Mycobacterium tuberculosis and current status of rapid molecular diagnostic testing. Acta Tropica. 2011;119:5–10. doi: 10.1016/j.actatropica.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Helb D, Jones M, Story E, Boehme C, Wallace E, Ho K, et al. Rapid detection of Mycobacterium tuberculosis and rifampin resistance by use of on-demand, near-patient technology. J Clin Microbiol. 2010;48:229–237. doi: 10.1128/JCM.01463-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.World Health Organization. Automated real-time nucleic acid amplification technology for rapid and simultaneous detection of tuberculosis and rifampicin resistance: Xpert MTB/RIF assay for the diagnosis of pulmonary and extrapulmonary TB in adults and children (internet), 2nd edn. World Health Organisation; 2013, pp. 1–97. Report no. WHO/HTM/TB/2013.16. http://apps.who.int/iris/bitstream/10665/112472/1/9789241506335_eng.pdf?ua=1. [PubMed]
- 33.Lawn SD, Mwaba P, Bates M, Piatek A, Alexander H, Marais BJ, et al. Advances in tuberculosis diagnostics: the Xpert MTB/RIF assay and future prospects for a point-of-care test. Lancet Infect Dis. 2013;13:349–361. doi: 10.1016/S1473-3099(13)70008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.World Health Organization . Pathways to better diagnostics for tuberculosis: a blueprint for the development of TB diagnostics by the new diagnostics working group of the Stop TB Partnership. Geneva: World Health Organization; 2009. [Google Scholar]
- 35.Weinreich DM, Delaney NF, DePristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312:111–114. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 36.Gong LI, Suchard MA, Bloom JD. Stability-mediated epistasis constrains the evolution of an influenza protein. eLife. 2013;2:e00631-1.
- 37.Thomas VL, McReynolds AC, Shoichet BK. Structural bases for stability-function tradeoffs in antibiotic resistance. J Mol Biol. 2010;396:47–59. doi: 10.1016/j.jmb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lozovsky ER, Chookajorn T, Brown KM, Imwong M, Shaw PJ, Kamchonwongpaisan S, et al. Stepwise acquisition of pyrimethamine resistance in the malaria parasite. Proc Natl Acad Sci USA. 2009;106:12025–12030. doi: 10.1073/pnas.0905922106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fenner L, Egger M, Bodmer T, Altpeter E, Zwahlen M, Jaton K, et al. Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56:3047–3053. doi: 10.1128/AAC.06460-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker LV, Brown TJ, Maxwell O, Gibson AL, Fang Z, Yates MD, et al. Molecular analysis of isoniazid-resistant Mycobacterium tuberculosis isolates from England and Wales reveals the phylogenetic significance of the ahpC-46A polymorphism. Antimicrob Agents Chemother. 2005;49:1455–1464. doi: 10.1128/AAC.49.4.1455-1464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Doorn HR, de Haas PEW, Kremer K, Vandenbroucke-Grauls CMJE, Borgdorff MW, van Soolingen D. Public health impact of isoniazid-resistant Mycobacterium tuberculosis strains with a mutation at amino-acid position 315 of katG: a decade of experience in The Netherlands. Clin Microbiol Infect. 2006;12:769–775. doi: 10.1111/j.1469-0691.2006.01495.x. [DOI] [PubMed] [Google Scholar]
- 42.Gagneux S, Burgos MV, DeRiemer K, Encisco A, Muñoz S, Hopewell PC, et al. Impact of bacterial genetics on the transmission of isoniazid-resistant Mycobacterium tuberculosis. PLoS Pathol. 2006;2:e61. doi: 10.1371/journal.ppat.0020061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borrell S, Gagneux S. Infectiousness, reproductive fitness and evolution of drug-resistant Mycobacterium tuberculosis. Int J Tuberc Lung Dis. 2009;13:1456–1466. [PubMed] [Google Scholar]
- 44.Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol. 2010;8:260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 45.Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJM. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science. 2006;312:1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 46.Borrell S, Teo Y, Giardina F, Streicher EM, Klopper M, Feldmann J, et al. Epistasis between antibiotic resistance mutations drives the evolution of extensively drug-resistant tuberculosis. Evol Med Public Health. 2013;2013:65–74. doi: 10.1093/emph/eot003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Safi H, Lingaraju S, Amin A, Kim S, Jones M, Holmes M, et al. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-β-d-arabinose biosynthetic and utilization pathway genes. Nat Genet. 2013;45:1190–1197. doi: 10.1038/ng.2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Vos M, Muller B, Borrell S, Black PA, van Helden PD, Warren RM, et al. Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob Agents Chemother. 2013;57:827–832. doi: 10.1128/AAC.01541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song T, Park Y, Shamputa IC, Seo S, Lee SY, Jeon H-S, et al. Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β’ subunit of RNA polymerase. Mol Microbiol. 2014;91:1106–1119. doi: 10.1111/mmi.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brandis G, Hughes D. Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates. J Antimicrob Chemother. 2013;68:2493–2497. doi: 10.1093/jac/dkt224. [DOI] [PubMed] [Google Scholar]
- 51.Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. 2014;46:279–286. doi: 10.1038/ng.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lehner B. Molecular mechanisms of epistasis within and between genes. Trends Genet. 2011;27:323–331. doi: 10.1016/j.tig.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 53.Casali N, Nikolayevskyy V, Balabanova Y, Ignatyeva O, Kontsevaya I, Harris SR, et al. Microevolution of extensively drug-resistant tuberculosis in Russia. Genome Res. 2012;22:735–745. doi: 10.1101/gr.128678.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang H, Li D, Zhao L, Fleming J, Lin N, Wang T, et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat Genet. 2013;45:1255–1260. doi: 10.1038/ng.2735. [DOI] [PubMed] [Google Scholar]
- 55.Reynolds MG. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics. 2000;156:1471–1481. doi: 10.1093/genetics/156.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koch A, Mizrahi V, Warner DF. The impact of drug resistance on Mycobacterium tuberculosis physiology: what can we learn from rifampicin? Emerg Microb Infect. 2014;3:e17. doi: 10.1038/emi.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.du Preez I, Loots DT. Altered fatty acid metabolism due to rifampicin-resistance conferring mutations in the rpoB gene of Mycobacterium tuberculosis: mapping the potential of pharmaco-metabolomics for global health and personalized medicine. OMICS. 2012;16:596–603. doi: 10.1089/omi.2012.0028. [DOI] [PubMed] [Google Scholar]
- 58.Bisson GP, Mehaffy C, Broeckling C, Prenni J, Rifat D, Lun DS, et al. Upregulation of the phthiocerol dimycocerosate biosynthetic pathway by rifampin-resistant, rpoB mutant Mycobacterium tuberculosis. J Bacteriol. 2012;194:6441–6452. doi: 10.1128/JB.01013-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cox JS, Chen B, McNeil M, Jacobs WR. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 60.Kirksey MA, Tischler AD, Siméone R, Hisert KB, Uplekar S, Guilhot C, et al. Spontaneous phthiocerol dimycocerosate-deficient variants of Mycobacterium tuberculosis are susceptible to gamma interferon-mediated immunity. Infect Immun. 2011;79:2829–2838. doi: 10.1128/IAI.00097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Trindade S, Sousa A, Xavier KB, Dionisio F, Ferreira MG, Gordo I. Positive epistasis drives the acquisition of multidrug resistance (Zhang J, editor). PLoS Genet. 2009;5:e1000578. [DOI] [PMC free article] [PubMed]
- 62.Brandis G, Wrande M, Liljas L, Hughes D. Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol Microbiol. 2012;85:142–151. doi: 10.1111/j.1365-2958.2012.08099.x. [DOI] [PubMed] [Google Scholar]
- 63.Muller B, Streicher EM, Hoek KGP, Tait M, Trollip A, Bosman ME, et al. inhA promoter mutations: a gateway to extensively drug-resistant tuberculosis in South Africa? Int J Tuberc Lung Dis. 2011;15:344–351. [PubMed] [Google Scholar]
- 64.Baker S, Duy PT, Nga TVT, Dung TTN, Phat VV, Chau TT, et al. Fitness benefits in fluoroquinolone-resistant Salmonella typhi in the absence of antimicrobial pressure. eLife. 2013;2:e01229-9. [DOI] [PMC free article] [PubMed]
- 65.Zhao X, Drlica K. Restricting the selection of antibiotic-resistant mutants: a general strategy derived from fluoroquinolone studies. Clin Infect Dis. 2001;33(Suppl 3):S147–S156. doi: 10.1086/321841. [DOI] [PubMed] [Google Scholar]
- 66.Bergval I, Kwok B, Schuitema A, Kremer K, van Soolingen D, Klatser P, et al. Pre-existing isoniazid resistance, but not the genotype of Mycobacterium tuberculosis drives rifampicin resistance codon preference in vitro. PLoS One. 2012;7:e29108. doi: 10.1371/journal.pone.0029108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2006;103:2869–2873. doi: 10.1073/pnas.0511240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013;45:784–790. doi: 10.1038/ng.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGrath M, Gey van Pittius NC, van Helden PD, Warren RM, Warner DF. Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J Antimicrob Chemother. 2014;69(2):292–302. [DOI] [PubMed]
- 70.Javid B, Sorrentino F, Toosky M, Zheng W, Pinkham JT, Jain N, et al. Mycobacterial mistranslation is necessary and sufficient for rifampicin phenotypic resistance. Proc Natl Acad Sci USA. 2014;111:1132–1137. doi: 10.1073/pnas.1317580111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wells WA, Boehme CC, Cobelens FG, Daniels C, Dowdy D, Gardiner E, et al. Alignment of new tuberculosis drug regimens and drug susceptibility testing: a framework for action. Lancet Infect Dis. 2013;13:449–458. doi: 10.1016/S1473-3099(13)70025-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Denkinger CM, Pai M, Dowdy DW. Do we need to detect isoniazid resistance in addition to rifampicin resistance in diagnostic tests for tuberculosis? (DeRiemer K, editor) PLoS One. 2014;9:e84197. doi: 10.1371/journal.pone.0084197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.WHO. Global tuberculosis report 2013. 2013, pp. 1–303. http://www.who.int/tb/publications/global_report/en/index.html. Accessed 10 Feb 2013.
- 74.Clark TG, Mallard K, Coll F, Preston M, Assefa S, Harris D, et al. Elucidating emergence and transmission of multidrug-resistant tuberculosis in treatment experienced patients by whole genome sequencing (Metcalfe JZ, editor). PLoS One. 2013;8:e83012. [DOI] [PMC free article] [PubMed]
- 75.Nardell E, Dharmadhikari A. Turning off the spigot: reducing drug-resistant tuberculosis transmission in resource-limited settings. Int J Tuberc Lung Dis. 2010;14:1233–1243. [PMC free article] [PubMed] [Google Scholar]
- 76.Harries AD, Maher D, Nunn P. Practical and affordable measures for the protection of health care workers from tuberculosis in low-income countries. Bull World Health Organ. 1997;75:477–489. [PMC free article] [PubMed] [Google Scholar]
- 77.Böttger EC, Springer B. Tuberculosis: drug resistance, fitness, and strategies for global control. Eur J Pediatr. 2007;167:141–148. doi: 10.1007/s00431-007-0606-9. [DOI] [PubMed] [Google Scholar]
- 78.McGrath M, Gey van Pittius NC, Sirgel FA, Van Helden PD, Warren RM. Moxifloxacin retains antimycobacterial activity in the presence of gyrA mutations. Antimicrob Agents Chemother. 2014;58(5):2912–5. [DOI] [PMC free article] [PubMed]
- 79.Zumla A, Nahid P, Cole ST. Advances in the development of new tuberculosis drugs and treatment regimens. Nature Rev Drug Discov. 2013;12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 80.Bloom JD, Gong LI, Baltimore D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science. 2010;328:1272–1275. doi: 10.1126/science.1187816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bryant JM, Harris SR, Parkhill J, Dawson R, Diacon AH, van Helden P, et al. Whole-genome sequencing to establish relapse or re-infection with Mycobacterium tuberculosis: a retrospective observational study. Lancet Resp Dis. 2013; doi:10.1016/S2213-2600(13)70231-5. Accessed 10 Jan 2014. [DOI] [PMC free article] [PubMed]
- 82.Köser CU, Bryant JM, Becq J, Török ME, Ellington MJ, Marti-Renom MA, et al. Whole-genome sequencing for rapid susceptibility testing of M. tuberculosis. N Engl J Med. 2013;369:290–292. doi: 10.1056/NEJMc1215305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andries K. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 84.Diacon AH, Dawson R, von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, et al. 14-day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: a randomised trial. Lancet. 2012;380:986–993. doi: 10.1016/S0140-6736(12)61080-0. [DOI] [PubMed] [Google Scholar]
- 85.Huitric E, Verhasselt P, Koul A, Andries K, Hoffner S, Andersson DI. Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrob Agents Chemother. 2010;54:1022–1028. doi: 10.1128/AAC.01611-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Centers for Disease Control and Prevention. Provisional CDC guidelines for the use and safety monitoring of bedaquiline fumarate (Sirturo) for the treatment of multidrug-resistant tuberculosis. MMWR Recomm Rep. 2013: 1–12. [PubMed]
- 87.Kakkar AK, Dahiya N. Bedaquiline for the treatment of resistant tuberculosis: Promises and pitfalls. Tuberculosis. 2014; 10.1016/j.tube.2014.04.001. Accessed 10 May 2014. [DOI] [PubMed]
- 88.Martínez JL, Baquero F, Andersson DI. Beyond serial passages: new methods for predicting the emergence of resistance to novel antibiotics. Curr Opin Pharmacol. 2011;11:439–445. doi: 10.1016/j.coph.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 89.Ginsberg AM. Drugs in development for tuberculosis. Drugs. 2010;70:2201–2214. doi: 10.2165/11538170-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 90.Homolka S, Meyer CG, Hillemann D, Owusu-Dabo E, Adjei O, Horstmann RD, et al. Unequal distribution of resistance-conferring mutations among Mycobacterium tuberculosis and Mycobacterium africanum strains from Ghana. Int J Med Microbiol. 2010;300:489–495. doi: 10.1016/j.ijmm.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 91.Shcherbakov D, Akbergenov R, Matt T, Sander P, Andersson DI, Böttger EC. Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol Microbiol. 2010;77:830–840. doi: 10.1111/j.1365-2958.2010.07218.x. [DOI] [PubMed] [Google Scholar]
- 92.Sherman DR, Mdluli K, Hickey MJ, Arain TM, Morris SL, Barry CE, et al. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science. 1996;272:1641–1643. doi: 10.1126/science.272.5268.1641. [DOI] [PubMed] [Google Scholar]
- 93.Zheng J, Rubin EJ, Bifani P, Mathys V, Lim V, Au M, et al. Para-aminosalicylic acid is a prodrug targeting dihydrofolate reductase in Mycobacterium tuberculosis. J Biol Chem. 2013;288:23447–23456. doi: 10.1074/jbc.M113.475798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Aubry A, Veziris N, Cambau E, Truffot-Pernot C, Jarlier V, Fisher LM. Novel gyrase mutations in quinolone-resistant and -hypersusceptible clinical isolates of Mycobacterium tuberculosis: functional analysis of mutant enzymes. Antimicro Agents Chemother. 2006;50:104–112. doi: 10.1128/AAC.50.1.104-112.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pym AS, Saint-Joanis B, Cole ST. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect Immun. 2002;70:4955–4960. doi: 10.1128/IAI.70.9.4955-4960.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Poissy J, Aubry A, Fernandez C, Lott MC, Chauffour A, Jarlier V, et al. Should moxifloxacin be used for the treatment of extensively drug-resistant tuberculosis? An answer from a murine model. Antimicrob Agents Chemother. 2010;54:4765–4771. doi: 10.1128/AAC.00968-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sougakoff W, Rodrigue M, Truffot-Pernot C, Renard M, Durin N, Szpytma M, et al. Use of a high-density DNA probe array for detecting mutations involved in rifampicin resistance in Mycobacterium tuberculosis. Clin Microbiol Infect. 2004;10:289–294. doi: 10.1111/j.1198-743X.2004.00889.x. [DOI] [PubMed] [Google Scholar]
- 98.Pang Y, Lu J, Wang Y, Song Y, Wang S, Zhao Y. Study of the rifampin monoresistance mechanism in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2013;57:893–900. doi: 10.1128/AAC.01024-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Anthony RM, Schuitema ARJ, Bergval IL, Brown TJ, Oskam L, Klatser PR. Acquisition of rifabutin resistance by a rifampicin resistant mutant of Mycobacterium tuberculosis involves an unusual spectrum of mutations and elevated frequency. Ann Clin Microb Antimicrob. 2005;4:9. doi: 10.1186/1476-0711-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Plinke C, Walter K, Aly S, Ehlers S, Niemann S. Mycobacterium tuberculosis embB codon 306 mutations confer moderately increased resistance to ethambutol in vitro and in vivo. Antimicrob Agents Chemother. 2011;55:2891–2896. doi: 10.1128/AAC.00007-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Starks AM, Gumusboga A, Plikaytis BB, Shinnick TM, Posey JE. Mutations at embB codon 306 are an important molecular indicator of ethambutol resistance in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2009;53:1061–1066. doi: 10.1128/AAC.01357-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matrat S, Aubry A, Mayer C, Jarlier V, Cambau E. Mutagenesis in the alpha3alpha4 GyrA helix and in the Toprim domain of GyrB refines the contribution of Mycobacterium tuberculosis DNA gyrase to intrinsic resistance to quinolones. Antimicrob Agents Chemother. 2008;52:2909–2914. doi: 10.1128/AAC.01380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cheng AFB, Yew WW, Chan EWC, Chin ML, Hui MMM, Chan RCY. Multiplex PCR amplimer conformation analysis for rapid detection of gyrA mutations in fluoroquinolone-resistant Mycobacterium tuberculosis clinical isolates. Antimicrob Agents Chemother. 2004;48:596–601. doi: 10.1128/AAC.48.2.596-601.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Duong DA, Nguyen THD, Nguyen TNL, Dai VH, Dang TMH, Vo SK, et al. Beijing genotype of Mycobacterium tuberculosis is significantly associated with high-level fluoroquinolone resistance in Vietnam. Antimicrob Agents Chemother. 2009;53:4835–4839. doi: 10.1128/AAC.00541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Malik S, Willby M, Sikes D, Tsodikov OV, Posey JE. New insights into fluoroquinolone resistance in Mycobacterium tuberculosis: functional genetic analysis of gyrA and gyrB mutations. PLoS One. 2012;7:e39754. doi: 10.1371/journal.pone.0039754. [DOI] [PMC free article] [PubMed] [Google Scholar]