

Abstract
The DNA deaminase AID initiates somatic hypermutation (SHM) and class switch recombination (CSR) by deaminating cytidines to uridines at variable region (V) genes and switch (S) regions. The mechanism by which AID is recruited to V genes and S region DNA is poorly understood. Here we have employed the CH12 B lymphoma line to demonstrate that while S regions can efficiently recruit AID and undergo mutations and deletions, AID neither binds to nor mutates the V gene, thus clearly demonstrating intra-immunoglobulin locus specificity. Depletion of the RNA-binding protein Ptpb2, previously shown to promote recruitment of AID to S regions, enables stable association of AID with the V gene. Surprisingly, AID binding to the V gene does not induce SHM. These results unmask a striking lack of correlation between AID binding and its mutator activity, providing evidence for the presence of factors required downstream of AID binding to effect SHM. Furthermore, our findings suggest that S regions are preferred targets for AID and, aided by Ptbp2, act as “sinks” to sequester AID activity from other genomic regions.
Introduction
Activation induced cytidine deaminase (AID) is essential for somatic hypermutation (SHM) and class switch recombination (CSR) (1, 2). During SHM, AID deaminates deoxycytidines (dCs) to deoxyuridines (dUs) at the variable region exons (V gene) of the immunoglobulin (Ig) heavy (Igh) and light chains (3). Engagement of base excision repair (BER) and mismatch repair (MMR) pathways along with DNA synthesis by error-prone DNA polymerases at the dU:dG mismatch mutates the V genes at a high rate (~10−2–10−3 mutations per bp per generation) leading to selection of B cells with increased antigen affinity (4). CSR exchanges the initially expressed Igh constant region Cμ for an alternative set of downstream CH exons (or genes) such as Cγ, Cε, or Cα, altering the B cell expression from IgM to a secondary antibody isotype (IgG, IgE, IgA) with distinct effector function (5). CSR is a deletional-recombination reaction that is initiated by AID-mediated deamination of transcribed, repetitive switch (S) region DNA elements that precede each CH gene (5). End-joining of DSBs between two distinct S regions deletes the intervening DNA as an extrachromosomal circle and juxtaposes a new CH gene downstream of the rearranged VDJ segment. Thus CSR allows for the generation of Ig molecules with the same affinity for antigen, but with new effector function.
AID is a general mutator and can mutate and induce DSBs at many non-Ig genes (6–11). In fact, aberrant AID activity at oncogenes is a major contributing factor in the ontogeny of a large number of mature B cell lymphomas (12). Despite the ability of AID to target non-Ig genes, the V genes and S region DNA serve as major AID targets, with the efficiency of AID association at the Ig loci several fold higher than at non-Ig genes (7, 8). In addition to specificity of AID for the Ig loci, there is evidence for intra-Ig locus specificity, as B cells undergoing CSR in culture do not mutate their variable regions (13, 14). Thus, mechanisms must exist to actively recruit AID to V genes and S regions during SHM and CSR, respectively. Several factors, including Spt5, Ptbp2, RNA exosome subunits and 14-3-3 adapter proteins, have been implicated in the recruitment of AID to S regions (7, 15–17), though the precise role of these proteins in CSR is yet to be fully elucidated. The mechanism by which AID is specifically recruited to V genes is even more enigmatic. Unlike S regions that are unique in their G:C richness and in their ability to form RNA:DNA hybrid structures (R-loops) upon transcription (18, 19), V genes do not present a recognizable primary or predicted secondary structure that could explain specificity for AID binding. The RGYW (R=A/G, Y=C/T, W=A/T) tetranucleotide does serve as an SHM hot-spot motif and E2A-transcription factor binding sites promote SHM (6, 20); however, the ubiquitous nature of these sequences at almost all transcribed genes fails to explain AID specificity.
We have previously identified polypyrimidine tract binding protein 2 (Ptbp2) as an AID interactor (15). Depletion of Ptbp2 significantly impaired CSR due to a defect in the recruitment of AID to S regions. Here we use the B lymphoma cell line CH12 to show that when AID recruitment to S regions is impaired through Ptbp2 depletion, association of AID with the expressed V gene is remarkably promoted. Surprisingly, despite the binding of AID to V genes, SHM is not induced. Therefore, AID binding does not correlate with mutation activity, suggesting that SHM requires specific factors and/or subversion of DNA repair pathways that operate downstream of AID binding.
Materials and Methods
Cell culture and protein analysis
CH12 cells (21) were stimulated at a density of 0.25 × 106 cells/ml for 96 hours with CIT, which consisted of anti-CD40 antibody (1 μg/ml; HM40-3; eBioscience), IL-4 (12.5 μg/ml; R&D Systems; 404-ML) and TGF-β1 (0.1 ng/ml; 240-B; R&D Systems). IgA+ cells were generated from CIT-stimulated CH12 cells by negative selection with anti-IgM MicroBeads (Miltenyl Biotec), followed by positive selection using anti-IgA biotin antibody (eBioscience) and Streptavidin MicroBeads (Miltenyl Biotec). The predominantly IgA+ cell population was subcloned by serial dilution.
Flow cytometry and western blotting
Cells were stained with IgM-PECy7 (R6-60.2; BD Biosciences) and IgA-FITC (C10-3; BD Biosciences), acquired on a LSR II (BD Biosciences) and data was analyzed using FlowJo (TreeStar, Inc). DAPI (Invitrogen) was used for exclusion of dead cells. Whole-cell lysates were prepared in NP-40 lysis buffer (20mM Tris, pH 7.5, 5% (vol/vol) glycerol, 150mM NaCl, 5mM β-mercaptoethanol and 0.5% (vol/vol) Nonidet P-40). Primary antibodies used were as follows: anti-Ptbp2 (ab57619; Abcam), anti-AID (22) and anti-Gapdh (loading control) (6C5; Millipore).
ChIP analysis
Knock-down in CH12 cells was described previously (15). ChIP assays were performed as described previously (15, 23). PCR amplification and detection was carried out on a Bio-Rad CFX96 system and threshold cycle (Ct) values were calculated using CFX Manager software by setting the threshold within the linear phase using a log amplification plot. For detection of specific DNA sequences in ChIP samples, iQ SYBR Green Supermix (Bio-Rad) was used for Sμ and p53 amplifications, while FastStart Universal Probe Mix (Roche) was used for VDJ amplification along with FAM-labeled universal probe #45 (Roche). Melt-curve analysis was used for SYBR reactions to verify the presence of a single amplicon of the correct size. ChIP “relative units” were calculated by first normalizing the Ct value of each antibody sample by the Ct value of the input sample (Ctantibody/Ctinput). The inverse of this normalized antibody Ct value was then taken (1/NormCtantibody). The resulting value for the non-specific IgG antibody sample was then subtracted from each of the specific antibody (SA) samples to obtain the final “relative units” ((1/NormCtSA) − (1/NormCtIgG)). Primer sequences are listed in Supplementary Table 2 and primer positions within the Igh locus are depicted in Supplementary Figure 1A.
Mutation analysis
Genomic DNA was isolated from 1.5 – 3 × 106 CH12 cells cultured with or without CIT for 96 hours. Amplification of sequences for mutation analysis was done using Phusion DNA polymerase (New England Biolabs). PCR products were cloned into Zero Blunt TOPO cloning kit (Invitrogen). Individual clones were sequenced with vector-derived M13 primers and mutation frequency was calculated by dividing the total number of unique mutations by the total number of bases sequenced. Mutation data is plotted as a pie chart showing the portion of total sequences containing 0, 1 or 2 mutations. The number of individual clones sequenced is indicated at the center of each chart, and the mutation frequency (unique mutations per bases sequenced) and the number of sequences containing deletions, if applicable, are indicated below. The VDJ sequence expressed by CH12 cells is GenBank M34581.1. Primer sequences are listed in Supplementary Table 2 and primer positions within the Igh locus are depicted in Supplementary Figure 1A.
Statistical analysis
A two-tailed paired Student’s t-Test was used to assess the significance of ChIP data, and a p ≤ 0.05 was considered significant. A z-test for comparing proportions of two independent groups was used to assess the significance of mutation frequencies, and a p ≤ 0.05 was considered significant.
Results
AID binds to S regions but not to V gene segments in CH12 cells
Upon stimulation with a combination of anti-CD40, interleukin-4 (IL-4) and transforming growth factor β (TGF-β) (henceforth referred to as CIT), the mouse B lymphoma line CH12 expresses AID and undergoes robust CSR to IgA (21, 24) (Fig. 1A, B). Given that CSR in CH12 cells is dependent on several of the known factors implicated in CSR of primary B lymphocytes (7, 15, 25), these cells serve as a bona fide model system to elucidate CSR. Despite the high frequency of CSR, CIT-stimulated CH12 cells do not undergo endogenous SHM (26).
FIGURE 1.
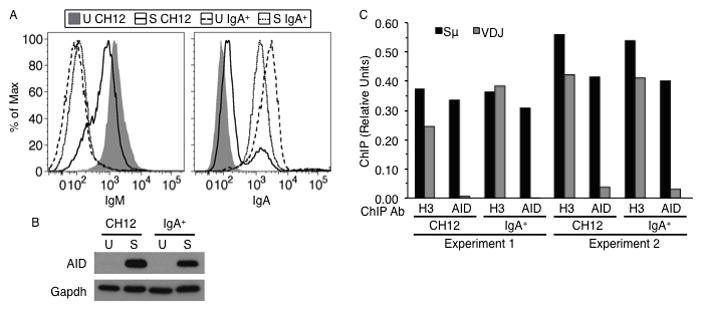
AID expressed in IgA+ cells does not bind the expressed Igh V gene. (A) Flow cytometry analysis of IgM (left) or IgA (right) expression on unstimulated (U) or CIT-stimulated (S) CH12 or IgA+ cells. (B) Western blot analysis of whole cell extracts using AID and Gapdh (loading control) antibodies. (C) ChIP analysis to detect binding of AID or histone H3 to Sμ and Igh V gene (VDJ) in stimulated CH12 or IgA+ cells. Two independent experiments are shown. ‘ChIP (Relative Units)’ is defined as the reciprocal of the quotient of the crossing threshold (Ct) value of specific antibody immunoprecipitation signal and the Ct value of input signal, with non-specific IgG immunoprecipitation signal (background) subtracted from this value.
To test if the failure to undergo SHM is due to preferred recruitment of AID to the S regions as opposed to the V gene, we carried out chromatin immunoprecipitation (ChIP) experiments in CH12 cells with AID antibody, using histone H3 antibody as a positive control. We analyzed the DNA-protein complexes for the presence of Sμ, Igh V gene (VDJ) and Trp53 (which encodes p53; non-Ig control) genomic sequence using sequence specific primers (Supplementary Fig. 1A and Supplementary Table 2). AID did not associate with Igh regions in unstimulated cells, or with p53 under any condition (Supplementary Fig. 1B, C and F). As expected, AID binding to Sμ was readily observed in stimulated cells; however, no AID was detected at the Igh V gene segment (Fig. 1C, Supplementary Fig. 1F).
One plausible explanation for the observed specificity is that S regions, being rich in RGYW sequence motifs and having the ability to form single-strand (ss) DNA in the context of R-loops, are significantly better than V genes in recruiting AID (18, 19, 27, 28). We therefore hypothesized that in “terminally switched” IgA+ CH12 cells, which cannot undergo further CSR, the Igh V gene would have a better chance of competing favorably against the smaller recombined S region for AID binding. To test this notion, we purified IgA+ cells from CIT-stimulated CH12 cells and subcloned an IgA+ clone by serial dilution (Fig. 1A). When grown in the absence of cytokines, the IgA+ clone expressed no detectable AID, while robust AID expression was induced upon stimulation with CIT (Fig. 1B). As in CH12 cells, AID did not associate with Igh regions in unstimulated IgA+ cells, or with Trp53 (p53; non-Ig control) genomic sequence under any condition (Supplementary Fig. 1B, C and F). However, contrary to our predictions, AID binding to the Igh V gene segment was not detected in CIT-stimulated IgA+ cells (Fig. 1C, Supplementary Fig. 1F). Instead, AID was found associated with the recombined Sμ–Sα DNA (Fig. 1C, Supplementary Fig. 1F). Thus, AID exhibits intra-Igh locus specificity with a bias for binding to S regions, not only in bulk CH12 cells undergoing CSR, but also in IgA+ cells where the Igh V gene might be expected to have a better chance of competing favorably against the remaining S region for AID binding.
Activated CH12 cells do not undergo SHM
To test the possibility that the failure to detect AID at the V gene is not due to the transient nature of the interaction, we isolated genomic DNA from IgA+ cells and sequenced the expressed Igh V gene using sequence specific primers (Supplementary Fig. 1A and Supplementary Table 2). The rate of mutation of the Igh V gene (VDJ) in CIT-stimulated IgA+ cells was similar to that observed for unstimulated IgA+ cells, suggesting that SHM was not induced (Fig. 2A, Supplementary Table 1A). In contrast, the recombined Sμ–Sα sequence in CIT-stimulated IgA+ cells had significant levels of mutations (Fig. 2B, Supplementary Table 1B). Furthermore, the original recombined Sμ–Sα junction present in the unstimulated IgA+ cell line underwent deletions to create novel S junctions (Fig. 2B, 2C). Thus, while AID in IgA+ cells bound to and mutated the remaining S region, it neither associated with nor induced SHM at the Igh V gene.
FIGURE 2.
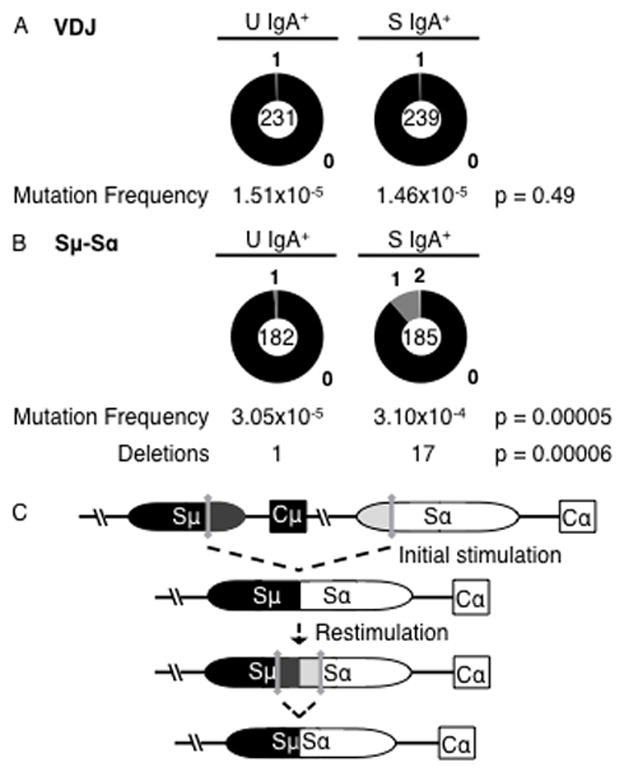
AID does not induce SHM in CH12 cells. (A) Mutation frequency of the Igh V region (VDJ). Unstimulated (U) IgA+ cells accumulated 1 mutation in 66,297 bases, and stimulated (S) IgA+ cells had 1 mutation in 68,593 bases. (B) Mutation frequency of Sμ–Sα junction. Unstimulated (U) IgA+ cells accumulated 2 mutations in 65,515 bases and 1 sequence with a deletion, and CIT-stimulated (S) IgA+ cells had 20 unique mutations in 64,568 bases and 17 sequences with a deletion. Data were obtained from two independent samples. p-values were calculated using z-test; p ≤ 0.05 was considered significant. (C) Diagram depicting the generation of deletions at the Sμ–Sα junction in CIT-stimulated IgA+ cells. DSBs generated in Sμ and Sα upon initial stimulation of an IgM+ CH12 cell are synapsed to form an initial Sμ–Sα junction. Stimulation of an IgA+ cell generates a new set of DSBs and results in the formation of a new Sμ–Sα junction, deleting the intervening switch sequence.
Localization of AID at the Igh locus is altered upon Ptbp2 depletion
The RNA binding protein Ptbp2 interacts with AID and facilitates the recruitment of AID to S regions (15). We investigated the possibility that in Ptbp2-depleted cells, AID that fails to bind to S regions can be “retargeted” to the V gene segment. Using an shRNA directed against the 3′ untranslated region of Ptbp2 mRNA, we knocked-down Ptbp2 expression in CH12 cells, with “scrambled” shRNA (Scr) serving as a control (Fig. 3A). As expected (15), AID binding to S regions was significantly reduced (~2-fold, p = 0.003) and CSR was impaired (Fig. 3B, 3C, Supplementary Fig. 1G). Strikingly, the reduction in AID at S regions was accompanied by a significant increase (>6-fold, p = 0.01) in AID binding specifically to the Igh V gene of stimulated Ptbp2-depleted cells (Fig. 3C, Supplementary Fig. 1D, 1G). AID was not substantially enriched at other regions of the Igh locus, including 1kb upstream and downstream of the rearranged VDJ (5′Vh1-53 and 3′Jh2, respectively), Cμ and the Iμ promoter (Fig. 3D). Surprisingly, AID was not detected at the expressed VJκ light chain locus (Fig. 3D). This could be due to occlusion of the antibody binding epitope while AID is bound to the light chain locus, or, more interestingly, a suggestion that the level of Ptbp2 preferentially effects AID relocalization within the Igh locus during SHM; thus, only the binding of AID to the S region and Igh V gene exons is altered upon Ptbp2 depletion. Finally, AID was not found to be associated with control genomic sequences, such as Trp53 (p53) or other non-Ig genes that have been shown to be upregulated in B cells undergoing CSR (23) (Fig. 3E). Thus, when the association of AID with S regions is impaired, its interaction with the expressed Igh V gene segment is specifically and significantly promoted.
FIGURE 3.
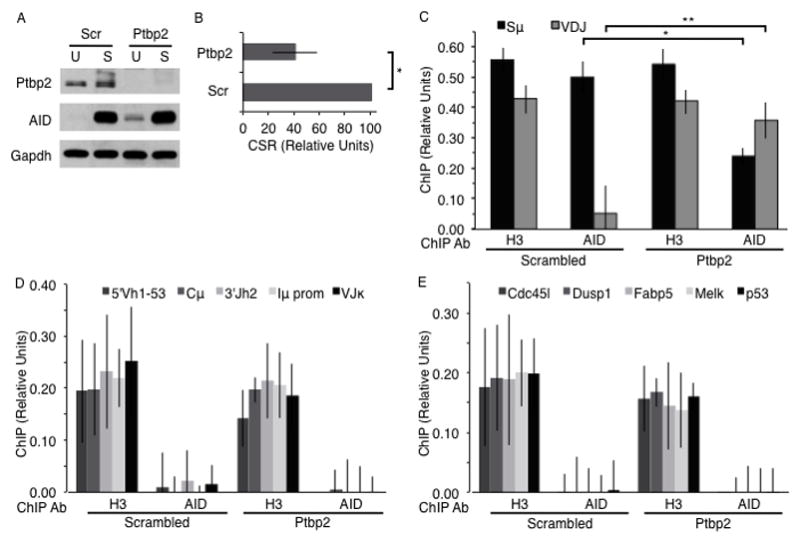
AID is retargeted from Sμ to the Igh V gene in Ptbp2-depleted CH12 cells. (A) Western blot of Ptbp2, AID and Gapdh in whole cell extracts of unstimulated (U) or CIT-stimulated (S) CH12 cells expressing scrambled (Scr) or Ptbp2 shRNA constructs. Data is representative of 6 independent experiments. (B) Quantification of CSR in stimulated cells. CSR frequency in Ptbp2-depleted cells was expressed as a percent of CSR in Scr cells, which was arbitrarily set to 100; n = 6. *p = 0.0004 (paired, two-tailed Student’s t-Test). (C) ChIP analyses of histone H3 (positive control) and AID bound to Sμ and Igh V gene (VDJ) DNA sequence in stimulated cells expressing either scrambled or Ptbp2 shRNA; n = 4. *p = 0.003, **p = 0.01 (paired, two-tailed Student’s t-Test). (D) AID or H3 binding to region 5′ of VDJ (5′Vh1-53), Cμ, region 3′ of VDJ (3′Jh2), Iμ promoter (Iμ prom), or Ig light chain V gene (VJκ) in CIT-stimulated (S) scrambled or Ptbp2-depleted (Ptbp2) CH12 cells; n = 4. (E) AID or H3 binding to Cdc45l, Dusp1, Fabp5, Melk, and p53 gene regions in CIT-stimulated scrambled and Ptbp2-depleted (Ptbp2) CH12 cells; n = 4. Data represent the mean and error bars show standard deviation from the mean.
AID bound to the Igh V gene in PTBP2-depleted cells is phosphorylated
AID is phosphorylated at serine-38 (S38) and mutation of S38 to alanine impairs the ability of AID to mediate SHM and CSR (23, 29–33). To determine if AID bound to the V gene was phosphorylated at S38 (pS38-AID), we carried out ChIP experiments using pS38-AID-specific antibody (23). In accordance with AID bound to Sμ, the amount of pS38-AID localized to Sμ was reduced in Ptbp2-depleted CH12 cells (Fig. 4A). This reduction was accompanied by a significant increase (~7-fold, p = 0.05) in pS38-AID levels specifically associated with the Igh V gene (Fig. 4A). Localization of pS38-AID to Trp53 (p53; non-Ig control) genomic sequence was not amplified from pS38-AID samples (Supplementary Fig. 1E). Thus, AID is not only specifically targeted to the V gene segment in Ptbp2-depleted CH12 cells; it is also phosphorylated at S38.
FIGURE 4.
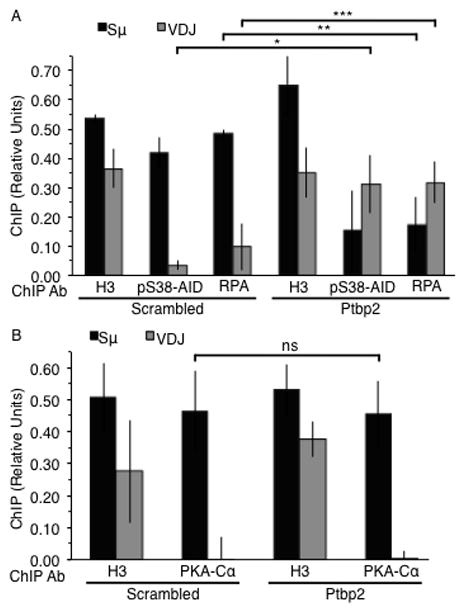
pS38-AID and RPA, but not PKA-Cα, are retargeted from Sμ to the Igh V gene in Ptbp2-depleted CH12 cells. ChIP analyses of histone H3 (positive control) and (A) pS38-AID and RPA or (B) PKA-Cα bound to Sμ and Igh V gene (VDJ) DNA sequence in stimulated cells expressing either scrambled or Ptbp2 shRNA constructs; n = 3. *p = 0.05, **p = 0.03, ***p = 0.01 (paired, two-tailed Student’s t-Test). Data represents mean and error bars show standard deviation from mean.
AID phosphorylated at S38 interacts with the ssDNA binding protein replication protein A (RPA) (29, 30). To test if RPA localization is also altered in Ptbp2-depleted cells, we carried out ChIP analyses using an antibody specific for the 32 kDa subunit of RPA. RPA levels at Sμ were significantly reduced (~2.5-fold, p = 0.03) in Ptbp2-depleted CH12 cells (Fig. 4A). In contrast, there was a significant increase (~3-fold, p = 0.01) in RPA localization specifically to the Igh V gene segment (Fig. 4A, Supplementary Fig. 1E).
Phosphorylation of AID at S38 is mediated by protein kinase A (PKA) (23, 30, 31, 34), and it is believed that AID is phosphorylated by S region-bound PKA to activate the CSR cascade (23, 35). In keeping with AID-independent recruitment of PKA to S regions (23), the catalytic subunit of PKA (PKA-Cα) was detected at Sμ in CIT-stimulated Ptbp2-depleted cells (Fig. 4B). However, PKA-Cα was not detected at the Igh V gene segment (Fig. 4B), even though phosphorylated AID was readily detectable (Fig. 4A). Thus, not all proteins known to bind S regions are retargeted to the V gene in the absence of Ptbp2, thereby ruling out the possibility that the binding of AID to the Igh V gene is due to general deregulation of protein-DNA associations in Ptbp2-depleted CH12 cells.
Our findings that PKA-Cα is associated with S regions but not with the Igh V gene, regardless of Ptbp2 expression, lends credence to the proposal (23) that AID phosphorylation has a different role in SHM than in CSR. It is generally believed that transcribed S regions form R-loops, allowing AID to access S regions independent of its phosphorylation status (36). Phosphorylation of AID at S38 is still required to both promote formation of DSBs at S regions through interaction with APE1 (35), and for the repair of DSBs through recruitment of RPA to S regions (8, 23). In contrast, transcribed V genes do not readily reveal ssDNA in the context of R-loops, and it is likely that AID only binds V genes in the context of a pS38-AID•RPA complex, due to the ability of RPA to bind and stabilize ssDNA within transcription bubbles (29). Our findings that pS38-AID and RPA, but not PKA-Cα, are detected at the Igh V gene support the notion that AID binding to V genes requires prior phosphorylation. However, we cannot exclude the possibilities that PKA-Cα was not detected at the V gene segment due to the transient nature of the interaction, or that other proteins bound to the V gene segment mask the antibody-binding site of V gene-bound PKA-Cα. Additionally, we cannot rule out the possibility that AID bound to the variable region exons is not phosphorylated by PKA, but is instead modified by an as yet unidentified kinase.
AID localization to the V gene does not induce SHM
To determine if AID binding induced SHM, we sequenced the Igh V gene (VDJ). Surprisingly, the mutation frequency in Ptbp2-depleted cells stimulated for 96 hours was similar to that in scrambled cells (Fig. 5A and Supplementary Table 1C). Moreover, Ptpb2-depleted cells stimulated continuously for 3 weeks did not accumulate additional mutations (Supplementary Table 1C). While the mutation frequency was higher than in unstimulated cells (Supplementary Table 1C), it was still considerably lower than typical SHM (32, 33, 37). The absence of SHM activity in Ptbp2-depleted cells cannot be attributed to a deficiency in Igh V gene (VDJ) transcription, as transcription through this region was not markedly altered (<2-fold change) upon Ptbp2-depletion (Supplementary Fig. 2A). Most importantly, there was no correlation between the levels of AID bound to the V gene and SHM frequency (Fig. 3C, 5A). In contrast, the region 5′ of the Sμ repetitive region (34) was highly mutated (Fig. 5B and Supplementary Table 1D). A majority of the mutations in this region were transition mutations and all mutations occurred at C:G base pairs (Fig. 5C). This spectrum suggests that mutations in CH12 cells arise mainly from replication across deaminated residues, with a small portion repaired via BER engagement. The lack of mutation at A:T bases could indicate that the MMR pathway is either not active, or is not recruited to the Igh locus in CH12 cells (38). Additionally, there was a marked lack of G to T mutations in scrambled cells while Ptbp2-depleted cells showed both G to A and G to T. The reason behind this intriguing observation is not clear at present, but it is possible that alteration in AID and/or Ptbp2 at S regions could modulate the engagement of error-prone polymerases during CSR.
FIGURE 5.
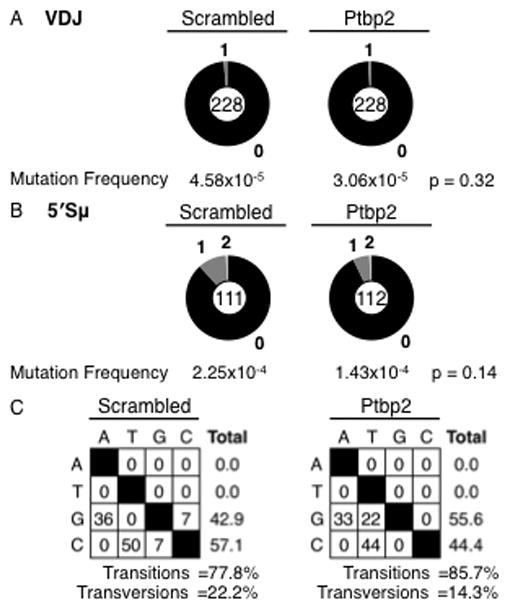
AID binding to the Igh V gene does not induce SHM. (A) Mutation analysis of Igh V gene (VDJ) segment in CIT-stimulated CH12 cells expressing either scrambled or Ptbp2 shRNA constructs. Scrambled cells accumulated 3 mutations in 65,436 bases and Ptbp2 cells had 2 mutations in 65,436 bases sequenced. (B) Mutation analysis of 5′ Sμ region. Scrambled cells accumulated 14 mutations in 62,160 bases and Ptbp2 cells had 9 mutations in 62,720 bases sequenced. Data represent four independent samples. p-values were calculated using z-test; p ≤ 0.05 was considered significant. (C) Characterization of mutations found in 5′ Sμ region. Percent nucleotide substitutions from residue “x” (down left side) to residue “y” (along top), with total percent substitution for each residue listed on the right. Percentage transitions or transversions are indicated below.
Many of the mutations identified in the region 5′ of the Sμ repetitive region and V gene of stimulated scrambled and Ptbp2-depleted cells were in RGYW hot-spot motifs (Supplementary Fig. 2B and Supplementary Table 1E). Of the total bases sequenced for mutation analysis, the amount of available RGYW motifs in the Igh V gene (VDJ) is approximately 61% more than the amount available in the region 5′ of the Sμ repetitive region (based on numbers listed in Supplementary Table 1C, 1D and 1E); however, the percentage of RGYW motifs mutated in the region 5′ of the Sμ repetitive region is much higher than in the Igh V gene (VDJ) (Supplementary Fig. 2B and Supplementary Table 1E). Consequently, if the RGYW motifs found in the Igh V gene were being mutated at the same frequency as the RGYW motifs in the region 5′ of the Sμ repetitive region, the number of RGYW motifs expected to be mutated at the Igh V gene would be significantly higher (Supplementary Fig. 2C and Supplementary Table 1E). Thus, the lack of detectable SHM in Ptbp2-depleted CH12 cells is not due to a deficiency in the amount of available hot-spot motifs in this region, rather it is likely due to the absence in expression of required repair mediators or uncharacterized SHM-specific factors in CH12 cells. In conclusion, even though known mediators of SHM, namely AID and RPA, were abundantly present at the Igh V gene and the bound AID was phosphorylated at S38, SHM was not induced.
Discussion
Our results clearly demonstrate that S regions are preferred targets for AID binding. Even in cells that have undergone CSR to the last CH gene, AID is efficiently recruited to the remaining S region in lieu of the transcribed Igh V gene. This strong preference for S regions might represent a physiological mechanism for AID regulation, wherein S regions act as a “sink” to prevent AID from interacting with non-Ig genomic sequences. Being non-coding, S regions could sustain mutations and deletions without having any deleterious effect on cell viability. This is evident from the abundant mutations and deletions observed at Sμ–Sα junctions upon re-expression of AID in IgA+ cells. Thus, active recruitment of AID to S regions probably functions as a default pathway to sequester AID activity away from other genomic targets.
If AID expressed in a B cell is indeed actively recruited to the S regions, the question that follows is how AID is targeted to the V genes to initiate SHM. Our studies clearly demonstrate that reduction in Ptbp2 levels promotes binding of AID to Igh V gene segments. Thus, in a B cell undergoing SHM, modulating Ptbp2 expression or perturbing the interaction between AID and Ptbp2 through post-translational modifications of AID and/or Ptbp2 could increase the amount of non-S region bound AID (Figure 6). The relative expression of Ptbp2 in germinal center B cells undergoing SHM versus CSR will shed further light into the differential role of this protein during an immune response.
FIGURE 6.
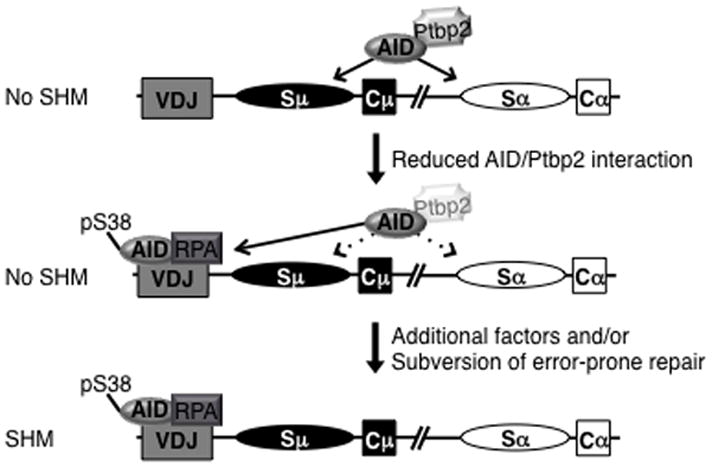
Model for AID binding to Igh V gene upon Ptbp2 depletion. Ptbp2 interacts with AID and actively recruits it to S regions. When the interaction is dampened through an unknown mechanism, AID can then bind to the variable region exons. AID bound to the Igh V gene is phosphorylated and is bound to RPA; however, SHM is not induced. Thus, additional factors/ processes absent or inactive in CH12 cells are required for SHM.
The most striking finding is that despite binding to the Igh V gene, AID cannot induce SHM. Over the past few years, several genome-wide association analyses of AID have reported that AID has the potential to bind to other genomic regions (7, 11, 39). Our findings clearly enforce the notion that binding of AID is not synonymous with mutations. For SHM to occur, at least in the context of CIT-stimulated CH12 cells, additional steps need to occur beyond AID binding. It is feasible that CH12 cells lack a factor(s) required for SHM and/or that CIT-induction is not sufficient to induce the expression of all required proteins. Another likely possibility is that AID targeted to the Igh V gene in Ptbp2-depleted cells is indeed actively deaminating, but high-fidelity repair (6), as opposed to error-prone repair required for SHM, “fixes” the lesions in a non-mutagenic fashion. Under physiological conditions, SHM is associated with germinal center structures found in secondary lymphoid organs, where the interaction of B cells with CD4+ helper T cells (40) could induce SHM promoting factors or subversion of high-fidelity repair pathways that are absent from the in vitro culture system described here.
In conclusion, the data presented here intriguingly suggest that targeting of AID to S regions is the default mechanism in a CSR sufficient B cell line, and may indicate a role for S regions as an AID “sink”, sequestering AID away from the rest of the genome. Our findings identify Ptbp2 as a crucial mediator between S region and Igh V gene AID targeting, and show that AID localization does not determine AID deamination activity or the induction of mutation. Additionally, we describe a cell line-based model system that will be invaluable in further elucidating factors/conditions required for SHM versus CSR.
Supplementary Material
Acknowledgments
This work is supported by National Institutes of Health Grant 1RO1AI072194-01 (to JC) and T32AI007621 and T32CA009149 (to AJM).
We thank Tasaku Honjo, Kyoto University, Japan, for providing us the CH12 cell line. We thank David Schatz, Yale University, and members of the Chaudhuri laboratory for helpful discussions and suggestions.
Abbreviations
- CSR
Class Switch Recombination
- SHM
Somatic Hypermutation
- AID
Activation Induced Cytidine Deaminase
- BER
Base-Excision Repair
- MMR
Mismatch Repair
- RPA
Replication Protein A
- Ptbp2
Polypyrimidine tract binding protein-2
- ChIP
Chromatin Immunoprecipitation
- PKA
Protein Kinase A
- DSBs
Double Strand Breaks
References
- 1.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 2.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 3.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 4.Papavasiliou FN, Schatz DG. Somatic hypermutation of immunoglobulin genes: merging mechanisms for genetic diversity. Cell. 2002;109:S35–44. doi: 10.1016/s0092-8674(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 5.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 7.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, Nieland TJ, Root DE, Casellas R, Nussenzweig MC. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nature immunology. 2011;12:62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR, Ried T, Nussenzweig A, Nussenzweig MC. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Molecular cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, Neuberg D, Monti S, Giallourakis CC, Gostissa M, Alt FW. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig Loci in activated B cells. Molecular cell. 2011;41:232–242. doi: 10.1016/j.molcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HC, 3rd, Nussenzweig MC, Dalla-Favera R. AID is required for germinal center-derived lymphomagenesis. Nature genetics. 2008;40:108–112. doi: 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- 13.Manser T. Mitogen-driven B cell proliferation and differentiation are not accompanied by hypermutation of immunoglobulin variable region genes. J Immunol. 1987;139:234–238. [PubMed] [Google Scholar]
- 14.Nagaoka H, Muramatsu M, Yamamura N, Kinoshita K, Honjo T. Activation-induced deaminase (AID)-directed hypermutation in the immunoglobulin Smu region: implication of AID involvement in a common step of class switch recombination and somatic hypermutation. The Journal of experimental medicine. 2002;195:529–534. doi: 10.1084/jem.20012144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nature immunology. 2011;12:160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, Januszyk K, Gregory RI, Deng H, Lima CD, Alt FW. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, Thomas LM, Al-Qahtani A, White CA, Park SR, Steinacker P, Li Z, Yates J, 3rd, Herron B, Otto M, Zan H, Fu H, Casali P. 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nature structural & molecular biology. 2010;17:1124–1135. doi: 10.1038/nsmb.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian M, Alt FW. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. The Journal of biological chemistry. 2000;275:24163–24172. doi: 10.1074/jbc.M003343200. [DOI] [PubMed] [Google Scholar]
- 19.Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature immunology. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- 20.Michael N, Shen HM, Longerich S, Kim N, Longacre A, Storb U. The E box motif CAGGTG enhances somatic hypermutation without enhancing transcription. Immunity. 2003;19:235–242. doi: 10.1016/s1074-7613(03)00204-8. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura M, Kondo S, Sugai M, Nazarea M, Imamura S, Honjo T. High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int Immunol. 1996;8:193–201. doi: 10.1093/intimm/8.2.193. [DOI] [PubMed] [Google Scholar]
- 22.Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- 23.Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, Chaudhuri J. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nature immunology. 2009;10:420–426. doi: 10.1038/ni.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muramatsu M, V, Sankaranand S, Anant S, Sugai M, Kinoshita K, Davidson NO, Honjo T. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. The Journal of biological chemistry. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 25.Han L, Yu K. Altered kinetics of nonhomologous end joining and class switch recombination in ligase IV--deficient B cells. The Journal of experimental medicine. 2008;205:2745–2753. doi: 10.1084/jem.20081623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eto T, Kinoshita K, Yoshikawa K, Muramatsu M, Honjo T. RNA-editing cytidine deaminase Apobec-1 is unable to induce somatic hypermutation in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12895–12898. doi: 10.1073/pnas.2135587100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Misaghi S, Garris CS, Sun Y, Nguyen A, Zhang J, Sebrell A, Senger K, Yan D, Lorenzo MN, Heldens S, Lee WP, Xu M, Wu J, DeForge L, Sai T, Dixit VM, Zarrin AA. Increased targeting of donor switch region and IgE in Sgamma1-deficient B cells. J Immunol. 2010;185:166–173. doi: 10.4049/jimmunol.1000515. [DOI] [PubMed] [Google Scholar]
- 28.Zhang T, Franklin A, Boboila C, McQuay A, Gallagher MP, Manis JP, Khamlichi AA, Alt FW. Downstream class switching leads to IgE antibody production by B lymphocytes lacking IgM switch regions. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3040–3045. doi: 10.1073/pnas.0915072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 30.Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP, Alt FW. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438:508–511. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 31.Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:395–400. doi: 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. The Journal of experimental medicine. 2008;205:2585–2594. doi: 10.1084/jem.20081319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng HL, Vuong BQ, Basu U, Franklin A, Schwer B, Astarita J, Phan RT, Datta A, Manis J, Alt FW, Chaudhuri J. Integrity of the AID serine-38 phosphorylation site is critical for class switch recombination and somatic hypermutation in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2717–2722. doi: 10.1073/pnas.0812304106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, Nussenzweig MC. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8798–8803. doi: 10.1073/pnas.0603272103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, Pucella JN, Ucher AJ, Donghia NM, Gu X, Nicolas L, Nowak U, Rahman N, Strout MP, Mills KD, Stavnezer J, Chaudhuri J. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nature immunology. 2013;14:1183–1189. doi: 10.1038/ni.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matthews AJ, Zheng S, Dimenna LJ, Chaudhuri J. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Advances in immunology. 2014;122:1–57. doi: 10.1016/B978-0-12-800267-4.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKean D, Huppi K, Bell M, Staudt L, Gerhard W, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chahwan R, Edelmann W, Scharff MD, Roa S. AIDing antibody diversity by error-prone mismatch repair. Seminars in immunology. 2012;24:293–300. doi: 10.1016/j.smim.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, Casellas R, Nussenzweig MC. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamel KM, V, Liarski M, Clark MR. Germinal center B-cells. Autoimmunity. 2012;45:333–347. doi: 10.3109/08916934.2012.665524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.