
Fig 5. CaMKII and Mechanisms of arrhythmia.
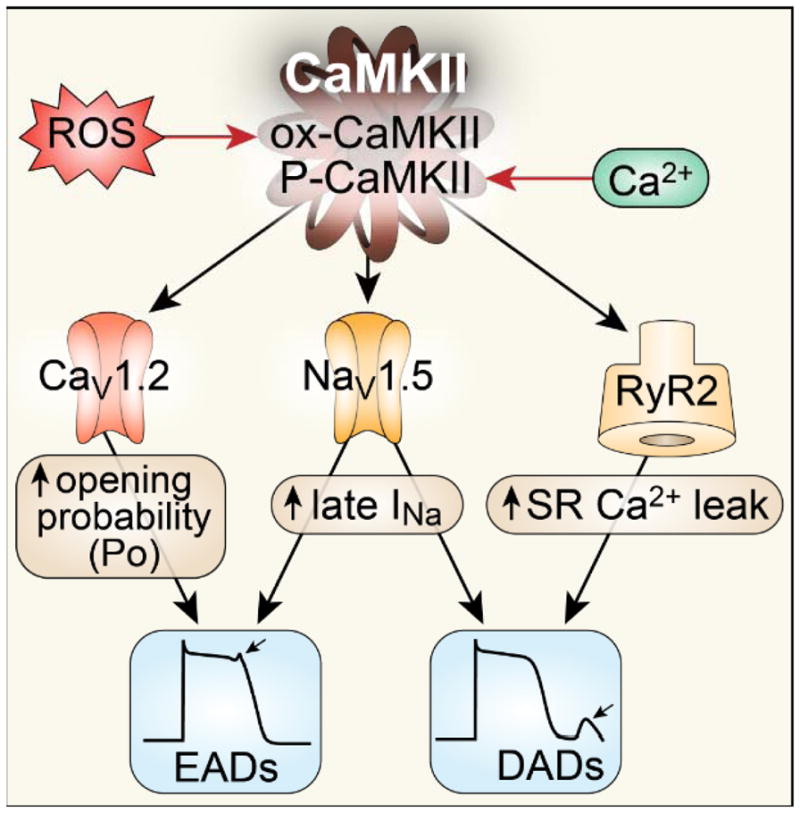
Sustained activation of CaMKII by oxidative stress and elevated [Ca2+]i contributes to arrhythmia in heart failure by several mechanisms: 1) CaMKII phosphorylates L-type Ca channels (CaV1.2) to increase its open probability, causing early afterdepolarizations (EADs). Increased ICa also contributes to action potential prolongation, augmented [Ca2+]i and DADs. 2) CaMKII phosphorylates Na+ channels (NaV1.5) and enhances the long-lasting late INa (gain of function) promoting EADs and increasing subsarcolemmal [Na+]i to favor delayed afterdepolarizations (DADs). 3) CaMKII favors phosphorylation of RyR2 to increase SR Ca2+ leak, which shifts Na+/Ca2+ exchanger (NCX) to a forward mode, causing DADs. CaMKII contributes to arrhythmogenic structural features of injured myocardium by promoting myocyte death and collagen deposition.