

Background: Oligomeric complexes of APP, APLP1, and APLP2 contribute to synapse formation and structure.
Results: Zinc binding to the E2 domain of APP and APLPs promotes their oligomerization in the cell, most notably with APLP1.
Conclusion: Extracellular zinc is a regulator for structure and function of APP and APLPs.
Significance: Novel insight into how APP and APLP function is regulated at the molecular level.
Keywords: Amyloid Precursor Protein (APP), Fluorescence Resonance Energy Transfer (FRET), Mass Spectrometry (MS), Metalloprotein, Zinc
Abstract
The amyloid precursor protein (APP) and the APP-like proteins 1 and 2 (APLP1 and APLP2) are a family of multidomain transmembrane proteins possessing homo- and heterotypic contact sites in their ectodomains. We previously reported that divalent metal ions dictate the conformation of the extracellular APP E2 domain (Dahms, S. O., Könnig, I., Roeser, D., Gührs, K.-H., Mayer, M. C., Kaden, D., Multhaup, G., and Than, M. E. (2012) J. Mol. Biol. 416, 438–452), but unresolved is the nature and functional importance of metal ion binding to APLP1 and APLP2. We found here that zinc ions bound to APP and APLP1 E2 domains and mediated their oligomerization, whereas the APLP2 E2 domain interacted more weakly with zinc possessing a less surface-exposed zinc-binding site, and stayed monomeric. Copper ions bound to E2 domains of all three proteins. Fluorescence resonance energy transfer (FRET) analyses examined the effect of metal ion binding to APP and APLPs in the cellular context in real time. Zinc ions specifically induced APP and APLP1 oligomerization and forced APLP1 into multimeric clusters at the plasma membrane consistent with zinc concentrations in the blood and brain. The observed effects were mediated by a novel zinc-binding site within the APLP1 E2 domain as APLP1 deletion mutants revealed. Based upon its cellular localization and its dominant response to zinc ions, APLP1 is mainly affected by extracellular zinc among the APP family proteins. We conclude that zinc binding and APP/APLP oligomerization are intimately linked, and we propose that this represents a novel mechanism for regulating APP/APLP protein function at the molecular level.
Introduction
The amyloid precursor protein (APP)3 and the APP-like proteins 1 and 2 (APLP1 and APLP2) are the mammalian representatives of an evolutionarily conserved protein superfamily (1). Harboring the potentially neurotoxic amyloid β (Aβ) peptide, APP is hypothesized to be the causative molecular factor in the etiology of Alzheimer disease (2). Although APLP1 and APLP2 lack the Aβ region, all three proteins are similarly processed by secretases (3–5), and all three paralogs are capable of forming homo- and heterodimers in living cells (6–8). Previously, we demonstrated that APP dimerization plays a major role in Aβ peptide generation (9). Although it is well established that APP and its processing products are critical in the context of Alzheimer disease, the physiological role of APP, APLP1, and APLP2 still remains unclear.
Genetic studies with single and combined knockouts of APP, APLP1, and APLP2 have demonstrated that the APP family is essential to central nervous system development. Notably, mice lacking all three APP/APLP family members survive post-embryonically but exhibit severe impairments in cortical development (10). The apparent redundancy between APP, APLP1, and APLP2 in synapse formation, neuronal differentiation, and synaptic plasticity (11–16) has yet to be fully characterized at the molecular level. For example, APLP2 is required for priming cortical progenitors for neuronal differentiation, a function that is likely shared by APP (17). APP and APLP2 are more similar to each other in terms of localization and dimer formation than APLP1 (7, 18, 19). APLP1 mainly localizes to the cell surface, whereas APP and APLP2 are primarily found in intracellular compartments. APLP1 is exclusively found in the nervous system (18, 20), and APLP1-heparin complexes are only formed by the E2 domain (21, 22). In contrast, APP and APLP2 are ubiquitously expressed and can bind heparin through their E1 and E2 domains (23–25). The ectodomains of APP and APLPs are not only mediators of cell-extracellular matrix interactions (26). All APP family members have been shown to bind metal ions (copper, zinc) through their extracellular E1 domain (27–29). Moreover, we have previously shown that metal ions can also bind to the extracellular E2 domain of APP, thereby affecting the conformation and flexibility of the domain (30).
Because little is known about how extracellular ligand binding to APP, APLP1, and APLP2 affects these proteins in the cell, we have now examined zinc and copper binding to isolated domains of APP and APLPs, as well as to the full-length proteins at the cellular level. Our detailed biophysical findings have determined that zinc and copper can bind to the E2 domain of all APP family members, but zinc binding is sterically restricted in the case of APLP2. Live cell FRET analyses and confocal microscopy revealed that zinc ions mediate the oligomerization of APP and APLPs, but APLP1 is predominantly regulated by zinc among the three family members. Notably, we have determined that APLP1 oligomerization is mediated by a novel zinc-binding site in its E2 domain. We also present detailed biochemical evidence that zinc binding and oligomerization are intimately linked, impacting the structural and functional properties of APLP1 as well as of APP and APLP2.
EXPERIMENTAL PROCEDURES
Plasmids
APP, APLP1, APLP1-ΔE1-AcD, and APLP2 were cloned in pcDNA3-CFP, pcDNA3-YFP, and pcDNA3-FLAG custom-made vectors as described previously (7). Point mutations in the APLP1 sequence were introduced by PCR-based site-directed mutagenesis using partially overlapping primers (31). APLP1 deletion constructs were generated from the full-length sequence by PCR-mediated deletion (32). Amino acids 290–494, 290–580, or 614–650 were deleted to generate APLP1 ΔE2, APLP1 ΔE2-JMR, or APLP1 ΔCT, respectively. Recombinant E2 and E2-JMR proteins of APP, APLP1, and APLP2 were expressed in Pichia pastoris with the pPICZα vector system (Invitrogen). Sequences encoding amino acids 365–566 of APP, 290–495 or 290–566 of APLP1, and 370–565 of APLP2 were introduced into the pPICZα vector via the EcoRI restriction site at the 5′ end and with an additional stop codon at the 3′ end. For APP E2, an additional ATT codon was introduced between the EcoRI restriction site and the APP sequence. Sequences of all vectors were verified by DNA sequencing (GATC).
Recombinant Proteins
P. pastoris cultures were precultured in BMGY, pelleted, and resuspended in expression medium BMMY (pH 6 or 7) with 0.5% methanol at A600 of 1.0. Supernatants containing APP E2, APLP1 E2, APLP1 E2-JMR or APLP2 E2 were collected 24 h after methanol induction and purified in a two-step chromatography procedure using HiTrap Blue HP (GE Healthcare) columns and a HiLoad 16/60 Superdex 200 column (GE Healthcare).
Immobilized Metal Ion Affinity Chromatography (IMAC)
Purified recombinant APP E2, APLP1 E2, APLP2 E2, and APLP1 E2-JMR were preincubated with EDTA, extensively dialyzed against 50 mm sodium acetate, pH 7.0, containing 100 mm NaCl, and then applied to a zinc- or copper-loaded chelating Sepharose 6B (Pharmacia) column. For batch binding analyses, three wash steps were performed (50 mm sodium acetate, pH 7.0, containing 1 m NaCl) prior to elution (50 mm sodium acetate, pH 7.0, containing 100 mm NaCl and 50 mm EDTA). For molecular dissection of the APLP1 zinc-binding site, column-bound APLP1 E2-JMR was incubated with 10 μg/ml trypsin (Roche Applied Science) at 37 °C or 10 μg/ml GluC (Roche Applied Science) in digestion buffer (100 mm NH4HCO3, pH 8.0) at 25 °C overnight. Nonspecifically bound material was removed using 50 mm sodium acetate, pH 7.0, with increasing salt concentrations as follows: 100 mm (Wash 1), 500 mm (Wash 2), 1 m NaCl (Wash 3). Specifically bound peptides were eluted from the column using 50 mm sodium acetate, pH 7.0, containing 10 mm NaCl and 50 mm EDTA. Fractions were washed and concentrated with C18 reversed-phase tips (Varian) in 50–80% acetonitrile containing 0.1% TCA. MALDI-MS analysis was performed using an Ultraflex-II TOF/TOF instrument (Bruker Daltonics) with α-cyano-4-hydroxycinnamic acid as the matrix. Fragment-ion spectra for peptide sequencing were recorded with the LIFT method (33).
Dot-blot and Immunodetection
Fractions were transferred to nitrocellulose membranes (Macherey-Nagel) with a 96-well dot-blot device (Schleicher & Schuell). For detection of APLP1 E2 and APLP2 E2, we generated the polyclonal antibodies anti-APLP1 E2 and anti-APLP2 E2. Rabbits were immunized with synthetic peptides (purchased from PSL) containing sequences of APLP1 E2 (349ALNEHFQSILQTLEE363) or APLP2 E2 (485RPHRILQALRR495), both with a C-terminal cysteine added for C-terminal conjugation to keyhole limpet hemocyanin (Thermo Fisher Scientific). APLP1 E2 antiserum was purified on a HiTrap Protein A HP column (GE Healthcare). The antisera displayed no cross-reactivity with other APP family proteins. Immunodetection was performed with anti-APLP1 E2 (1:2000) or anti-APLP2 E2 (1:500), followed by incubation with HRP-coupled anti-rabbit IgG antibody (1:10,000; Promega), Detection of APP E2 was performed with 5 μg/ml heparin-biotin in PBS-T with 100 μm ZnCl2 followed by incubation with polystreptavidin-HRP (1:30,000; Thermo Fisher Scientific). Protein signals were detected by ECL.
Surface Plasmon Resonance (SPR)
SPR analyses were performed using a BIAcore 3000 system (GE Healthcare). Recombinant E2 domains (diluted in 10 mm sodium acetate, pH 5.0) were amine-coupled to SCB CMD 500L sensor chips (XanTec Bioanalytics) according to the manufacturer's instructions as follows: immobilized densities of 3600 RU (APP E2), 3400 RU (APLP1 E2), and 2900 RU (APLP2 E2). Metal binding analyses were performed as described previously (30).
Intrinsic Fluorescence Spectroscopy (IFS)
Prior to analysis, protein samples were preincubated with EDTA and extensively dialyzed against 50 mm sodium acetate, pH 6.8, containing 150 mm NaCl. Dialyzed proteins were diluted to 0.5 μm and recorded on a Jasco FP-6500 spectrofluorometer (temperature = 25 °C, path length = 1 cm, constant stirring = 450 rpm). In the presence of buffer (“native” protein scan), ZnCl2, and/or EDTA (inhibit metal-binding), fluorescence spectra were recorded using an excitation wavelength of 295 nm (slit width = 5 nm), and emission was recorded from 305 to 400 nm (slit width = 5 nm; scanning speed = 50 nm/min). The ActA peptide (Ac-SFEWPPPPT-NH2) was kindly provided by Dr. Robert Opitz (FMP Berlin). For each set of experimental conditions, the acquired spectra were corrected for dilution effects and for background emission of the buffer.
Size-exclusion Chromatography (SEC) and Static Light Scattering (SLS)
For gel filtration-coupled SEC-SLS analyses, 100 μl of 50 μm APLP1 E2 in HBS (10 mm HEPES, pH 7.1, containing 135 mm NaCl, 6 mm KCl, and 5.5 mm glucose) was applied to a calibrated Superdex 200 10/300 GL column (GE Healthcare). SLS (refractive index and right angle light scattering) data were recorded using VE 3580 RI and 270 dual detectors (Viscotek) and evaluated using the OmniSEC software (Viscotek). Values are expressed as means ± S.D. of three independent runs.
In additional experiments, APP E2, APLP1 E2, or APLP2 E2 (400 μl at 4 μm each) were loaded on a calibrated Superose 12 10/300 GL column (GE Healthcare) equilibrated in 50 mm sodium acetate, pH 6.8, containing 150 mm NaCl, with or without 10 μm ZnCl2. Prior to application, protein samples were preincubated with EDTA, extensively dialyzed against the running buffer, incubated for 10 min, and centrifuged. For better comparison, spectra were normalized to their highest absorption signal.
HEK293 and Neuronal Cell Culture
Cells were grown in an incubator at 37 °C and 5% CO2 on glass coverslips. HEK293 cells were grown in DMEM (PAA) supplemented with 10% FBS (PAA), 2 mm glutamine, 1 mm pyruvate and were transiently transfected using polyethyleneimine or FuGENE 6 transfection reagent (Roche Applied Science) with the indicated plasmids. Primary hippocampal cultures were isolated from Wistar rat pups (E19) for nucleofection with pcDNA3-APLP1-YFP using the rat neuron Nucleofector kit according to the manufacturer's instructions (Lonza). Neuron growth medium consists of a 1:1 mixture of DMEM/F-12 medium (Invitrogen) with 0.3% glucose, 20% FBS, 50 μg/ml gentamicin, 15 mm HEPES/NaOH, pH 7.3, and neurobasal medium (Invitrogen) with 2% B27 supplement (Invitrogen), 2 mm GlutaMAX (Invitrogen).
Live Cell Imaging and Fluorescence Resonance Energy Transfer (FRET)
HEK293 cells and hippocampal neurons were imaged 1 day after transfection using a confocal microscope (LSM 510 Meta, Carl Zeiss, with an α-Plan-Fluar 100×/1.45 objective or TCS SP8, Leica Microsystems, with a 63×/1.4 objective) at room temperature in HBS. HBS containing ZnCl2, CuCl2, CoCl2, MgCl2, CaCl2, or EDTA was added to the indicated final concentration. FRET measurements were performed with 4 × 4 binning using an inverted epifluorescence microscope (DMI6000B, Leica Microsystems) with sensitized emission (SE) recordings and the manufacturer's FRET SE wizard within the LAS AF software suite. FRET efficiency was calculated by correction for donor cross-talk and acceptor cross-excitation (34). For this, transfected HEK293 cells grown on a coverslip were placed in a perfusion chamber and rinsed with HBS. Cells were then perfused with HBS containing ZnCl2, CuCl2, CoCl2, MgCl2, CaCl2, (NH4)2Fe(SO4)2, (NH4)Fe(SO4)2, or EDTA by a multivalve perfusion system (Warner Instruments). For concentration-response analyses (5 min perfusion at each concentration), HBS was supplemented with 0.3 μm to 3 mm ZnCl2 (APP wild type (WT), APLP1 WT, APLP1ΔE1-AcD) or 1 μm to 10 mm ZnCl2 (all other constructs). Zinc incubations were followed by washing solutions I (HBS containing 1 mm EDTA) and II (HBS only). EC50 values were calculated by fitting the curves with a Levenberg-Marquardt algorithm to a three-parameter Hill equation.
Statistical Analysis
Data are expressed as means ± S.E. All statistical analyses were performed by Welch's t test.
RESULTS
Zinc and Copper Binding to E2 Domains of APP, APLP1, and APLP2
Previously, we identified two metal-binding sites in the APP E2 domain that can regulate the conformation of the domain (Fig. 1, A and B) (30). This finding prompted us to study zinc and copper binding to the E2 domains of the other two mammalian APP family members, APLP1 and APLP2. Initially, we performed IMAC to examine metal binding to recombinant E2 domains of APP, APLP1, and APLP2. Although APP E2 and APLP1 E2 bound to the zinc-charged IMAC columns and were specifically eluted, the E2 domain of APLP2 was not retained by immobilized zinc and was detected in the flow-through and the wash fractions (Fig. 1C). However, all three E2 domains bound to copper-charged IMAC columns (Fig. 1C). To cross-validate these findings, we then performed SPR analyses in the reverse orientation; the E2 domains of APP, APLP1, and APLP2 were amine-coupled to SPR sensor chips, and zinc or copper chloride were titrated over all immobilized surfaces (Fig. 1D). The strong signal responses (i.e. RU-bound) with all three E2 domains indicated that zinc was interacting with all three proteins in a similar manner. Likewise, titration with copper yielded a strong increase of the RU. Although no obvious differences in association with copper ions to the proteins on the sensor chip were observed, APLP1 E2 displayed the slowest dissociation and APP E2 the fastest.
FIGURE 1.

Zinc and copper binding properties of APP, APLP1, and APLP2 E2 domains. A, schematic representation of the amyloid precursor protein family: SP, signal peptide; E1 and E2, two conserved regions of the ectodomain; AcD, acidic stretch linker region between E1 and E2; JMR, juxtamembrane region; Aβ, amyloid β sequence; PM, plasma membrane. B, metal-binding residues of metal-binding sites 1 and 2 (M1 and M2) observed in crystals of the APP E2 domain (30). C, recombinant E2 domains were loaded onto an IMAC column charged with immobilized zinc or copper ions; flow-through (FT), washes (W1–3), and elution volumes were analyzed by dot-blotting. D, overlays of reference-subtracted SPR titrations for ZnCl2 or CuCl2 binding (1–100 μm ZnCl2 or 2.5–50 μm CuCl2 at 30 μl/min) to amine-coupled E2 domains (∼3300 RU each). E, corrected intrinsic fluorescence titrations for ZnCl2 binding (0–25 μm) to E2 domains in solution (0.5 μm) in the absence or presence of 250 μm EDTA. F, intrinsic fluorescence repeated (0–100 μm ZnCl2 binding) with ActA control peptide. C–F, blots and spectra shown are representatives of at least three independent experiments.
The apparent discord between the zinc-immobilized IMAC and E2-immobilized SPR outcomes (i.e. contradictory zinc-binding ability of APLP2 E2 domain) was further resolved using solution-phase IFS. Conserved tryptophan (Trp) residues within the E2 domains of APP (residue 413), APLP1 (residue 332), and APLP2 (residue 412) were specifically excited to monitor for zinc-induced conformational changes. The emission spectra revealed that the fluorescence maxima of the native proteins were around 350 nm (Fig. 1E), indicating highly solvent-accessible Trp residues (35), which agrees with the known x-ray crystal structures for APP E2 and APLP1 E2 (30, 36, 37). The presence of increasing zinc concentration induced small fluorescence decreases in APP E2 with a concomitant blue shift of 3–4 nm (Fig. 1E1). This was anticipated because Trp-413 is only minimally influenced by global structural changes in APP E2 when co-crystallized with metal ions (30). Consistent with major perturbations in the local Trp environments, zinc induced large, dose-dependent fluorescence decreases in APLP1 E2 (with concomitant blue shift of 5 nm; Fig. 1E2) and APLP2 E2 (no emission wavelength shift; Fig. 1E3). Notably, the significant fluorescence decreases at 25 μm ZnCl2 were reversed by addition of a 10-fold excess of EDTA. To exclude a generic effect of zinc ions on the Trp residue in the E2 domains in APP, APLP1, and APLP2, we repeated our fluorescence assay using a derivative of the ActA class of EVH1 ligands (38). As a negative control, this peptide contained a single Trp residue but no histidines or cysteines, which are the major zinc-binding residues of proteins (39). Titrating zinc to even higher concentrations (100 μm) did not significantly alter the emission spectrum of native ActA (Fig. 1F; only 2–3% decrease due to minor photobleaching effect encountered in all fluorescence assays), thus validating the specific binding effect of zinc on the E2 domains tested.
Overall, we have utilized three complementary technologies (IMAC, SPR, and IFS) to demonstrate that zinc and copper can specifically bind to the E2 domains of APP, APLP1, and APLP2 in a reversible, dose-dependent manner. Although the IFS experiments indicate that zinc binding can induce conformational rearrangements in all E2 domains, comparison between the IMAC (i.e. only 2 or 3 of 4/5 zinc coordination sites available when chelated to column) and SPR (i.e. zinc ions in solution and all 4/5 zinc coordination sites available) experiments suggests that the zinc-binding sites in APP E2 and APLP1 E2 are more surface-exposed compared with APLP2 E2 (i.e. similar zinc binding properties to APP E2 and APLP1 E2 in SPR, but poorly retained by Zn-IMAC column).
Zinc Induces Oligomerization of E2 Domains in APP and APLP1 but Not APLP2
Previous IFS analyses with APLP1 E2 showed that a fluorescence decrease can be a result of dimerization (37). Thus, we employed SEC to monitor APP, APLP1, and APLP2 oligomerization in solution. In the absence of zinc, recombinant APLP1 E2 yielded a single peak with an apparent massrh of 41.8 ± 0.2 kDa (Fig. 2A), which falls between the predictions for monomeric (theoretical mass of 24.4 kDa) or dimeric APLP1 E2. Subsequent shape- and conformation-independent SLS analyses confirmed an absolute massabs of 26.6 ± 0.1 kDa (Fig. 2A), which is in good agreement with the theoretical mass of monomeric APLP1 E2. In the absence of zinc, we conclude that recombinantly purified APLP1 E2 is monomeric at the employed experimental conditions. Similarly to APLP1 E2, the E2 domain of APP was also shown to exist in a monomeric form in solution (40). In the presence of 10 μm ZnCl2, a significant peak broadening was observed with APP E2 and APLP1 E2, which corresponded to the theoretical mass of dimers (Fig. 2, B and C). In contrast, there was no change in the elution profile of APLP2 E2 in the presence of 10 μm ZnCl2 (Fig. 2D). A further increase of the zinc concentration beyond 10 μm ZnCl2 rather led to a precipitation of recombinant APLP2 E2 than changing the elution profile. In solution, we conclude that the binding of zinc ions can induce a structural change that promotes the self-interaction of E2 domains in APP and APLP1 but not APLP2.
FIGURE 2.
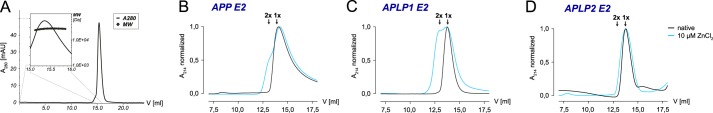
Oligomerization of recombinant APP, APLP1, and APLP2 E2 domains. A, gel filtration chromatography coupled to static light scattering (SEC-SLS) to analyze APLP1 E2 (line, absorbance at 280 nm; symbols, SLS-based molecular weight) as a function of retention volume. B–D, SECs for E2 domains in the absence (black lines) and presence (cyan lines) of 10 μm zinc; retention volumes at which monomers (1×) and dimers (2×) elute are indicated. A–D, chromatograms shown are representative of at least three independent experiments.
Zinc Induces Oligomerization of APP Family Proteins in Cells
To test if zinc-induced E2 interaction influences oligomerization of the full-length proteins in living cells, we measured fluorescence resonance energy transfer (FRET) of CFP- and YFP-tagged APP, APLP1, and APLP2 in HEK293 cells. We utilized the sensitized emission-FRET (SE-FRET) method (34), which is optimal for detecting relative changes in FRET efficiency with high temporal resolution rather than the exact absolute FRET values. A multivalve perfusion system (Warner Instruments) with a high buffer exchange rate also enabled us to monitor responses in real time.
APLP1-CFP- and APLP1-YFP-overexpressing cells exhibited basal levels of FRET indicating the presence of APLP1 dimers, whereupon ZnCl2 perfusion strongly increased FRET efficiency within seconds (Fig. 3, A and B). Subsequent washing with EDTA quickly reduced the FRET signals back to basal levels, thus indicating a shift in the APLP1 monomer/dimer equilibrium or the formation of higher APLP1 oligomers by multimerization of preformed APLP1 dimers. Strong conformational rearrangements that decrease the distance from CFP to YFP (thereby increasing FRET efficiency) could also not be ruled out, but they appear to be unlikely due to our C-terminal fluorophore fusions that permit little conformational flexibility. To determine whether APLP1 oligomerization is specifically induced by zinc ions, we studied if other divalent metal ions affect the FRET efficiency. Oligomerization was increased in the presence of 50 μm Zn2+ and 50 μm Cu2+, whereas 1 mm Ca2+, 1 mm Mg2+, 50 μm Fe2+, or 50 μm Fe3+ ions had no influence on FRET efficiency (Fig. 3, C and D). 150 μm CoCl2, a concentration far exceeding the physiological range (41) yielded only minimal increases in APLP1 FRET efficiency, thus validating the specific increases in APLP1 oligomerization that were observed with zinc and copper ions. This is in line with our previous observation that copper and zinc ions bind to the same site in the APP E2 domain (30). Physiological concentrations of free zinc ions generally exceed copper ion concentrations by magnitudes (42, 43) due to the high number of copper chelation sites provided by proteins (44, 45).
FIGURE 3.

Live cell FRET analysis of APP and APLP oligomerization in the presence of metal ions. HEK293 cells were transiently transfected with APLP1-CFP and APLP1-YFP (A–D) or APLP1-CFP and GypA-YFP (E). A, representative CFP, YFP, and overlay images of cells used for FRET measurements, monitored without binning. Scale bar, 10 μm. B, real time analysis of FRET signal changes upon perfusion with 50 μm ZnCl2 (90–330 s) followed by 500 μm EDTA; displayed are single-cell curves (gray) and averaged FRET signals (black) of SE measurements of five cells. C and D, single-cell curves of SE. FRET signals reveal a specific increase only for 50 μm Zn2+ and 50 μm Cu2+ but not for 50 μm Fe2+, 50 μm Fe3+ (C), 1 mm Ca2+, 1 mm Mg2+, or 50 μm Co2+ (D); a minimal increase of less than 1% was observed for 150 μm Co2+ (D); cells were incubated with metal ions and washed in continuous cycles using a perfusion device. E, GypA-YFP/APLP1-CFP pair shows a slight decrease of the baseline FRET signal in the presence of zinc between APLP1 and GypA most likely due to increased homophilic oligomerization of APLP1; displayed are single-cell curves (gray) and averaged FRET signals (black) of SE measurements of five cells. A–E, measurements presented are representative of at least three independent transfections.
To validate the specificity of APLP1 oligomerization upon zinc exposure, we also tested APLP1-CFP in combination with glycophorin A-YFP (GypA), another single-pass transmembrane protein localized to the plasma membrane (46). Whereas a low basal level of FRET was observed due to random plasma membrane encounters between APLP1-CFP and GypA-YFP (Fig. 3E), the presence of zinc prompted a slight decrease in the nonspecific FRET signal indicative of fewer encounters of APLP1 and GypA (likely due to the increased APLP1-YFP oligomerization that lowered the number of freely diffusing APLP1 entities).
FRET efficiencies between APP-CFP and APP-YFP or APLP2-CFP and APLP2-YFP were also increased by zinc (Fig. 4A) but were lower compared with APLP1 because of their different subcellular localization. Compared with APLP1, which is mainly found at the cell surface, APP and APLP2 are more strongly retained in intracellular compartments (7), which means that a higher proportion of these molecules are not accessible to exogenously applied zinc ions. To highlight the physiological relevance of our findings, we further assessed the concentration-dependent oligomerization of APP, APLP1, and APLP2 by zinc. By plotting the relative FRET signal increases as a function of ZnCl2 concentration, the sigmoidal curves are indicative of concentration-dependent zinc reactions. Accordingly, zinc mediated APLP1 oligomerization at low micromolar concentrations with an EC50 value (13.9 ± 2.1 μm) that matches the physiological zinc concentrations in the blood and in the brain at active zinc vesicle-containing synapses (47, 48). Zinc-induced oligomerization of APP (EC50 = 9.2 ± 3.0 μm) was comparable with APLP1, whereas significantly higher, nonphysiological zinc concentrations (EC50 = 325 ± 64 μm) were required for APLP2 oligomerization (Fig. 4B).
FIGURE 4.
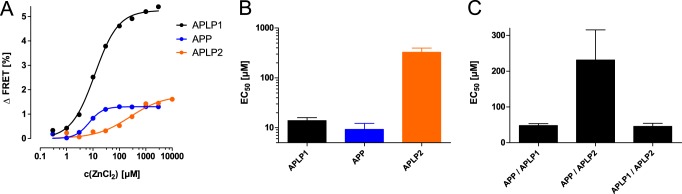
Concentration-dependent FRET analysis of zinc-mediated oligomerization of APP, APLP1, and APLP2. A, increased FRET efficiencies as a function of increasing zinc concentrations reveal sigmoidal concentration responses for homophilic APP, APLP1, and APLP2 FRET pairs, each fitted with a logistic function. B and C, EC50 values for FRET increases after zinc treatment (n = 4–6 experiments with 6–12 cells per experiment) with FRET homoligomer (B) or hetero-oligomer pairs (C; APP-CFP/APLP1-YFP, APP-CFP/APLP2-YFP, or APLP2-CFP/APLP1-YFP).
Similar to our homo-oligomerization outcomes, additional FRET analyses showed that hetero-oligomerization between APP/APLP1 and APLP1/APLP2 (EC50 values ∼50 μm in both cases) was increased in the presence of zinc (Fig. 4C). In contrast, APP/APLP2 hetero-oligomerization was affected at significantly higher zinc concentrations (EC50 = 231 ± 76 μm). This finding suggests that APLP2 preferentially forms hetero-oligomers with APLP1 (rather than homo-oligomers) at low zinc concentrations.
Overall, we have demonstrated that zinc increases the oligomerization of APP family members in the plasma membrane of living cells. Moreover, we have shown that the zinc-induced oligomerization of these full-length proteins in vivo resembles oligomerization of the isolated E2 domains of APP, APLP1, and APLP2 in vitro at low micromolar zinc concentrations. Based upon its subcellular localization and maximal sensitivity, oligomerization of APLP1 appears to be predominantly affected by extracellular zinc ions among the APP family members.
Zinc Induces Cluster Formation of APLP1 and APLP2 at the Plasma Membrane
To analyze whether zinc binding and oligomerization of APP family proteins in the plasma membrane affect their localization, we examined YFP-tagged APP, APLP1, and APLP2 in living HEK293 cells with confocal laser scanning microscopy (cLSM). At basal conditions, APLP1 was primarily localized to the cell surface, whereas APLP2 was strongly retained in intracellular compartments (Fig. 5A). APP levels at the plasma membrane were below the limit of detection, preventing an analysis of membrane-localized APP by cLSM. The addition of zinc ions to the incubation buffer resulted in the formation of large, distinguishable clusters of APLP1 or APLP2 at the plasma membrane (Fig. 5A). The effect occurred within 30 s after the addition of zinc and was stronger for APLP1 than for APLP2, consistent with a larger impact of zinc on APLP1 oligomerization compared with APLP2. Chelation of the metal ions by EDTA reversed the Zn2+ effect, and the cluster-like structures disappeared (Fig. 5A). As a negative control, GypA was used. GypA distribution at the plasma membrane was not altered by zinc addition (Fig. 5A). Thus, zinc ions specifically triggered the redistribution of APLP1 at the plasma membrane, with an attenuated effect on APLP2.
FIGURE 5.

Plasma membrane distribution of APLPs in the presence of metal ions. Plasmids encoding for APLP1, APLP2, and GypA with C-terminal YFP tags were transiently transfected in HEK293 cells (A and B) or rat hippocampal neurons (C) and imaged 1 day after transfection by cLSM. Representative images from at least three independent transfections were taken before and 2 min after addition of divalent metal ions to the medium (50 μm ZnCl2, 50 μm CuCl2, 1 mm MgCl2, 1 mm CaCl2, or 50 μm CoCl2) and 2 min after a further addition of EDTA. C, inset, upper left, is a magnification of the boxed neurite illustrating the punctate appearance of APLP1 clusters in the presence of zinc ions. Scale bar, 10 μm.
To further assess the metal ion specificity of APLP1 or APLP2 clustering, we tested other divalent metal ions. Neither 50 μm Co2+ nor concentrations as high as 1 mm Ca2+ or 1 mm Mg2+ ions altered the cellular localization of APLP1 or APLP2 (Fig. 5B). The only other metal ion tested that induced APLP1 and APLP2 clustering was copper (Fig. 5B), which was consistent with copper effecting APLP1 oligomerization as recorded by SE-FRET measurements.
Because APLP1 expression in contrast to APP and APLP2 is restricted to the nervous system (18), we tested whether zinc could induce clustering of APLP1 in the plasma membrane of rat primary hippocampal neurons. Within seconds, zinc ions induced a rapid re-arrangement of APLP1 molecules into clusters along the soma and dendritic plasma membranes, as monitored by cLSM (Fig. 5C). This clustering was reversible by treatment with EDTA, as we had observed using HEK293 cells. Of note, a clear redistribution of APLP1-YFP was not observed in cells with low APLP1-YFP expression levels, which is likely due to interference of endogenous nonfluorescent APLP1.
These observations indicate that APLP1 and APLP2 respond to elevated extracellular zinc concentrations by rapidly forming protein clusters at the plasma membrane. Also, the formation of large distinguishable clusters, which are visible at cLSM resolution, demonstrates that the zinc-increased FRET signal is caused by high APLP multimers instead of a shift in the APLP monomer/dimer equilibrium.
Zinc-induced APLP1 Oligomerization Is Mediated by Histidine Residues 430, 433, 450, and 452 in the APLP1 E2 Domain
According to our novel findings above and based upon partial homology to the two zinc-binding sites in APP (21), we predicted that zinc-binding sites within the E1 and E2 domains of APLP1 may exist. To determine which site is functionally important for zinc-induced APLP1 oligomerization at the cellular level, we generated deletion mutants of APLP1 (Fig. 6A) with deletion of the E1 domain and the adjacent stretch of acidic amino acid residues (ΔE1-AcD), deletion of the E2 domain alone (ΔE2) or in combination with the JMR (ΔE2-JMR), as well as a C-terminal deletion mutant (ΔCT) for additional FRET analyses in HEK293 cells.
FIGURE 6.

FRET analysis of zinc-mediated oligomerization of APLP1 mutants. A, schematic representation of APLP1 deletion mutants: SP, signal peptide; E1 and E2, two conserved regions of the ectodomain; AcD, acidic stretch linker region between E1 and E2; JMR, juxtamembrane region; PM, plasma membrane. B, HEK293 cells were transfected with APLP1 deletion mutants (A) or full-length site-directed APLP1 mutants (4× H/A represents the quadruple point mutation H430A/H433A/H450A/H452A); FRET concentration-response curves for zinc treatment were recorded, and EC50 values were determined (n = 4–6 experiments with 5–12 cells per experiment). Asterisks indicate significant differences to APLP1 WT as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, plasmids encoding for wild-type APLP1 fused to YFP and APLP1-CFP mutant constructs were transiently transfected in HEK293 cells; representative of at least three independent transfections, the cells were imaged by cLSM 1 day post-transfection. Scale bar, 10 μm.
The E2 deletion decreased APLP1 zinc sensitivity ∼10-fold, whereas deletion of the E1 domain or the C-terminal intracellular domain had no significant effect (Fig. 6B). Although this indicated that zinc-induced oligomerization is primarily mediated by the E2 domain, it was not completely abolished as residual oligomer formation was observed (potentially low affinity zinc-binding sites in other regions of APLP1 that compensate for the loss of the E2 domain). As shown previously for the ΔE1-AcD mutant (7), APLP1 localization was not affected by the ΔE2-(JMR) or ΔCT mutations as observed by cLSM (Fig. 6C).
Because zinc had its greatest effects on the APLP1 E2 domain, we used an APLP1 construct containing the E2 domain and additional residues of the adjacent juxtamembrane region (i.e. APLP1 E2-JMR; Fig. 7C) to identify the amino acid residues responsible for zinc binding in the E2 domain of APLP1. APLP1 E2-JMR was specifically bound to a zinc-charged IMAC column and then partially digested using trypsin and GluC (V8) proteases. Non-zinc binding regions of the recombinant protein were washed away, whereas zinc-bound peptides were eluted with EDTA. As analyzed by MALDI-mass spectrometry (MALDI-MS), the trypsin-digested fractions revealed six candidate APLP1 peptides (encompassing amino acids 430–459) as zinc binders (Fig. 7A). The GluC-digested fractions revealed two candidate APLP1 peptides (encompassing amino acids 424–458) as zinc binders (Fig. 7B). Thus, overlapping residues between the two protease digestions indicate that amino acids 430–458 mediate zinc binding to the E2 domain of APLP1 (Fig. 7C).
FIGURE 7.

Identification of the zinc-binding site in APLP1 E2. Recombinant APLP1 E2-JMR was digested with trypsin (A) or GluC (B) while bound to zinc-chelated Sepharose. Load, flow-through (FT), wash (W1–3), and elution volumes were analyzed by MALDI-MS. For trypsin analysis, EDTA elutions were performed using two different acetonitrile concentrations to cover a broad molecular mass range for peptide recovery. Mass spectrum for W3 digested with GluC is not shown because no APLP1 peptides were found in this fraction. Peaks are labeled with the respective peptide mass. Numbers are referring to the amino acid residues in the APLP1 sequence. A, trypsin digestion revealed six zinc-binding peptides harboring amino acids 430–459 of full-length APLP1. The nontryptic peptide with m/z = 987.6 was sequenced and found to result from an internal cleavage between Asp-438 and Pro-439. This internal cleavage is frequently observed in MALDI-MS due to the exceptional susceptibility of the amide linkage C-terminal to an Asp and N-terminal to a Pro residue (67, 68). The peptide with m/z = 3042.5 was sequenced and found to contain an additional EA sequence at its N terminus before the EF linker. This was previously observed in heterologous protein production and described as a result of incomplete processing by P. pastoris signal peptidases (57). B, GluC digestion yielded two zinc-binding peptides comprising amino acids 424–458. C, schematic representation of APLP1 with the E2 sequence from residues 427 to 461; histidines are shown in bold, and the identified zinc-binding region is highlighted; SP, signal peptide; E1 and E2, two conserved regions of the ectodomain; AcD, acidic stretch linker region between E1 and E2; JMR, juxtamembrane region; PM, plasma membrane. D, representative cLSM images of APLP1-YFP 4×H/A (H430A/H433A/H450A/H452A)-transfected HEK293 cells from three independent transfections taken before and after addition of zinc solution to final concentrations of 50 μm ZnCl2 or 200 μm ZnCl2. Scale bar, 10 μm.
Because we observed elution of the APLP1 E2-JMR protein from Zn-IMAC columns at pH 6.0 or lower, we suspected that histidine residues at positions 430, 433, 450, and 452 were the most likely candidates to coordinate zinc ions even though aspartate and glutamate residues are also potentially zinc-chelating residues (39, 49). To test this hypothesis, we mutated the candidate histidines to alanines and then re-evaluated zinc-induced oligomerization of the full-length protein. Similar to the ΔE2 and the ΔE2-JMR deletion constructs, the ability of the 4×H/A point mutant (APLP1 H430A/H433A/H450A/H452A) to respond to zinc was significantly impaired compared with wild-type APLP1 (Fig. 6B). Furthermore, APLP1 clusters at ZnCl2 concentrations of 50 μm were barely detectable for the 4×H/A mutant (Fig. 7D). Only at concentrations of 200 μm zinc or higher, a clustering of the APLP1 mutant was observed although to a much weaker extent than for the wild-type protein (Figs. 7D and 5A).
Overall, we have identified a novel zinc-binding site in the C-terminal part of the APLP1 E2 domain that can mediate its oligomerization at physiological concentrations of extracellular zinc. Histidine residues 433, 450, and 452 are essential for the activity of the novel binding site, whereas histidine 430 is rather dispensable (Fig. 6B).
DISCUSSION
To advance our understanding of protein-metal ion interactions involving the amyloid precursor protein family, the goal of this study was to investigate the binding of zinc and copper to APP and its paralogs APLP1 and APLP2. Through a series of complementary biochemical and cellular studies, we have now shown the following: (i) the E2 domains of APP, APLP1, and APLP2 can all bind zinc and copper, but the zinc ion-binding site of APLP2 E2 is less surface-exposed; (ii) zinc binding can mediate oligomerization of the E2 domains in APP and APLP1 but not APLP2; (iii) zinc ions can rapidly induce homo- and hetero-oligomerization of APP, APLP1, and APLP2 on the cell surface with a concomitant redistribution of APLPs into protein clusters observed in two different cell types, HEK293 cells and primary hippocampal neurons; and (iv) a novel zinc-binding site in the E2 domain of APLP1 promotes its oligomerization.
Based upon APP homology (21), it was proposed that APLP1 contains a zinc-binding site in the E1 domain. Interestingly, our analysis of deletion mutants revealed that APLP1 oligomerization is mediated by a zinc-binding site in the E2 domain. Site-directed mutagenesis further revealed that histidine residues 430, 433, 450, and 452 (in the C-terminal part of the E2 domain) are essential for zinc-mediated oligomerization. Significantly elevated nonphysiological concentrations of zinc could compensate for reduced oligomerization/clustering with the deletion and/or site-directed mutants of APLP1. However, the weaker and/or nonspecific zinc-binding sites likely do not play a major role in APLP1 oligomerization.
Through co-crystallization of recombinant APP with cadmium followed by competitive displacement with copper and zinc, we recently identified two metal-binding sites in the E2 domain (30). The displaced cadmium-binding sites in APP E2 overlap marginally with the zinc-binding histidine residues (430, 433, 450, and 452) identified in APLP1 E2 in this study as follows: metal-binding site 1 (M1) contains His-436 in APP, which is conserved to His-430 in APLP1; metal-binding site 2 (M2) contains His-458, which is conserved to His-452 in APLP1. Of special note, the primary sequence of APP lacks His-450, which we have shown here to be important for zinc-induced APLP1 oligomerization. Consequently, the composition of the zinc-binding site that we have just identified is novel compared with the intramolecular M1 and M2 sites in the E2 domain of APP.
Two of the histidines (430 and 433) in this study were previously identified as part of a heparin-binding site when the crystal structure of the APLP1 E2 domain was determined (22). This is particularly interesting given that zinc can strengthen the binding of polyanion heparin to APP and APLPs (21), an effect originally attributed to the E1 domain although never experimentally proven. Consistent with our findings, the cross-interaction of zinc and heparin could implicate the C-terminal part of the E2 domain as a general regulatory site for APLP1 oligomerization, as well as for binding interactions with components of the extracellular matrix. Notably, several extracellular matrix protein interactions in non-neuronal tissues have been shown to be regulated by zinc (50, 51). It is possible that locally elevated concentrations of zinc could also facilitate such interactions in the brain by the ability of zinc to modulate extracellular matrix interactions with APP/APLPs.
Although the majority of zinc ions remains tightly bound as protein cofactors in the brain, low (∼20 nm) concentrations of “free” zinc in the extracellular fluid (52) can reach up to 100 μm after exocytosis of zinc-filled vesicles at the synapse (47). Similarly, high local concentrations of copper have been recorded at the synapse (53), although their significance is far less understood compared with zinc (54). Because this study demonstrates that APP and APLP1 oligomerization was induced at low micromolar concentrations of zinc whereas APLP2 oligomerization required significantly higher concentrations, this suggests that APP and APLP1 oligomerization could be linked to the active state of zincergic synapses. These synapses are predominantly found in cortical and limbic structures of the brain with a particularly high abundance of zincergic terminals in the hippocampus (55); interestingly, this coincides with high APLP1 expression in the hippocampal region (56, 58). On the subcellular level, APLP1 is mainly expressed at the cell surface, whereas APP is more strongly retained in intracellular compartments of the cell (7, 59). This indicates that among the APP family members APLP1 is predominantly regulated by zinc ions, which is consistent with our present hetero-oligomerization analyses, i.e. in the presence of zinc, APLP1 mediated the formation of hetero-oligomer pairs with APP and APLP2. Notably, APLP1 may regulate the number of zinc-induced APP oligomers because APP localization to the plasma membrane is linked to APLP1 expression (7).
Consistent with the overall theme of this study, the effect of zinc ions on protein oligomerization has previously been reported for the ProSAP/Shank family (60, 61). ProSAP1/Shank2 and ProSAP2/Shank3 are zinc-dependent postsynaptic protein scaffolds that are involved in synaptogenesis and the maintenance of immature synapse stability (61). Notably, APP and APLPs are also present in the synaptic cleft both at presynaptic and postsynaptic compartments (14, 62), where APP and APLP homo- and hetero-oligomers function as structural components of the synapse (63–66); APP has also been shown to promote synaptogenesis (14). If zinc is a key regulator of APP/APLP oligomerization at the synapse, this adds another layer of complexity to the structural role that APP/APLPs play in synaptogenesis. Taken together, the formation of oligomeric APP/APLP clusters (predominantly by APLP1) may serve as zinc-dependent interaction hubs for the structural network of APP protein family members.
Acknowledgments
We thank Dr. Robert Opitz for the ActA derivative peptide and for assistance with fluorescence spectroscopy, Christoph Weise for advice with MALDI-TOF MS, and Lisa Henning for help with confocal microscopy.
This work was supported by Deutsche Forschungsgemeinschaft Grants MU901, SFB740, GRK1123, and Exc 257 NeuroCure, German Federal Ministry of Education and Research through the Kompetenznetz Degenerative Demenzen Förderkennzeichen 01 GI 1004G, and Canadian Institute of Health Research Grant MOP-133411 (to G. M.).
- APP
- amyloid precursor protein
- APLP
- amyloid precursor-like protein
- Aβ
- amyloid-β
- CFP
- cyan fluorescent protein
- JMR
- juxtamembrane region
- SPR
- surface plasmon resonance
- IFS
- intrinsic fluorescence spectroscopy
- SEC
- size-exclusion chromatography
- SLS
- static light scattering
- SE
- sensitized emission
- IMAC
- immobilized metal ion affinity chromatography
- GypA
- glycophorin A
- RU
- response unit
- cLSM
- confocal laser scanning microscopy.
REFERENCES
- 1. Coulson E. J., Paliga K., Beyreuther K., Masters C. L. (2000) What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem. Int. 36, 175–184 [DOI] [PubMed] [Google Scholar]
- 2. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 3. Eggert S., Paliga K., Soba P., Evin G., Masters C. L., Weidemann A., Beyreuther K. (2004) The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves α-, β-, γ-, and ϵ-like cleavages: modulation of APLP-1 processing by N-glycosylation. J. Biol. Chem. 279, 18146–18156 [DOI] [PubMed] [Google Scholar]
- 4. Li Q., Südhof T. C. (2004) Cleavage of amyloid-β precursor protein and amyloid-β precursor-like protein by BACE 1. J. Biol. Chem. 279, 10542–10550 [DOI] [PubMed] [Google Scholar]
- 5. Kuhn P.-H., Koroniak K., Hogl S., Colombo A., Zeitschel U., Willem M., Volbracht C., Schepers U., Imhof A., Hoffmeister A., Haass C., Rossner S., Bräse S., Lichtenthaler S. F. (2012) Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 31, 3157–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Soba P., Eggert S., Wagner K., Zentgraf H., Siehl K., Kreger S., Löwer A., Langer A., Merdes G., Paro R., Masters C. L., Müller U., Kins S., Beyreuther K. (2005) Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 24, 3624–3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kaden D., Voigt P., Munter L.-M., Bobowski K. D., Schaefer M., Multhaup G. (2009) Subcellular localization and dimerization of APLP1 are strikingly different from APP and APLP2. J. Cell Sci. 122, 368–377 [DOI] [PubMed] [Google Scholar]
- 8. Isbert S., Wagner K., Eggert S., Schweitzer A., Multhaup G., Weggen S., Kins S., Pietrzik C. U. (2012) APP dimer formation is initiated in the endoplasmic reticulum and differs between APP isoforms. Cell. Mol. Life Sci. 69, 1353–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Munter L.-M., Voigt P., Harmeier A., Kaden D., Gottschalk K. E., Weise C., Pipkorn R., Schaefer M., Langosch D., Multhaup G. (2007) GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Aβ42. EMBO J. 26, 1702–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Herms J., Anliker B., Heber S., Ring S., Fuhrmann M., Kretzschmar H., Sisodia S., Müller U. (2004) Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 23, 4106–4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng H., Jiang M., Trumbauer M. E., Sirinathsinghji D. J., Hopkins R., Smith D. W., Heavens R. P., Dawson G. R., Boyce S., Conner M. W., Stevens K. A., Slunt H. H., Sisoda S. S., Chen H. Y., Van der Ploeg L. H. (1995) β-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81, 525–531 [DOI] [PubMed] [Google Scholar]
- 12. von Koch C. S., Zheng H., Chen H., Trumbauer M., Thinakaran G., van der Ploeg L. H., Price D. L., Sisodia S. S. (1997) Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol. Aging 18, 661–669 [DOI] [PubMed] [Google Scholar]
- 13. Heber S., Herms J., Gajic V., Hainfellner J., Aguzzi A., Rülicke T., von Kretzschmar H., von Koch C., Sisodia S., Tremml P., Lipp H. P., Wolfer D. P., Müller U. (2000) Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J. Neurosci. 20, 7951–7963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Z., Wang B., Yang L., Guo Q., Aithmitti N., Songyang Z., Zheng H. (2009) Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J. Neurosci. 29, 10788–10801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Weyer S. W., Klevanski M., Delekate A., Voikar V., Aydin D., Hick M., Filippov M., Drost N., Schaller K. L., Saar M., Vogt M. A., Gass P., Samanta A., Jäschke A., Korte M., Wolfer D. P., Caldwell J. H., Müller U. C. (2011) APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J. 30, 2266–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tyan S.-H., Shih A. Y.-J., Walsh J. J., Maruyama H., Sarsoza F., Ku L., Eggert S., Hof P. R., Koo E. H., Dickstein D. L. (2012) Amyloid precursor protein (APP) regulates synaptic structure and function. Mol. Cell. Neurosci. 51, 43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shariati S. A., Lau P., Hassan B. A., Müller U., Dotti C. G., De Strooper B., Gärtner A. (2013) APLP2 regulates neuronal stem cell differentiation during cortical development. J. Cell Sci. 126, 1268–1277 [DOI] [PubMed] [Google Scholar]
- 18. Lorent K., Overbergh L., Moechars D., De Strooper B., Van Leuven F., Van den Berghe H. (1995) Expression in mouse embryos and in adult mouse brain of three members of the amyloid precursor protein family, of the α-2-macroglobulin receptor/low density lipoprotein receptor-related protein and of its ligands apolipoprotein E, lipoprotein lipase, α-2-macroglobulin and the 40,000 molecular weight receptor-associated protein. Neuroscience 65, 1009–1025 [DOI] [PubMed] [Google Scholar]
- 19. Thinakaran G., Kitt C. A., Roskams A. J., Slunt H. H., Masliah E., von Koch C., Ginsberg S. D., Ronnett G. V., Reed R. R., Price D. L. (1995) Distribution of an APP homolog, APLP2, in the mouse olfactory system: a potential role for APLP2 in axogenesis. J. Neurosci. 15, 6314–6326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Su A. I., Wiltshire T., Batalov S., Lapp H., Ching K. A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G., Cooke M. P., Walker J. R., Hogenesch J. B. (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U.S.A. 101, 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bush A. I., Pettingell W. H., Jr., de Paradis M., Tanzi R. E., Wasco W. (1994) The amyloid β-protein precursor and its mammalian homologues. Evidence for a zinc-modulated heparin-binding superfamily. J. Biol. Chem. 269, 26618–26621 [PubMed] [Google Scholar]
- 22. Xue Y., Lee S., Ha Y. (2011) Crystal structure of amyloid precursor-like protein 1 and heparin complex suggests a dual role of heparin in E2 dimerization. Proc. Natl. Acad. Sci. U.S.A. 108, 16229–16234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Multhaup G. (1994) Identification and regulation of the high affinity binding site of the Alzheimer's disease amyloid protein precursor (APP) to glycosaminoglycans. Biochimie 76, 304–311 [DOI] [PubMed] [Google Scholar]
- 24. Small D. H., Nurcombe V., Reed G., Clarris H., Moir R., Beyreuther K., Masters C. L. (1994) A heparin-binding domain in the amyloid protein precursor of Alzheimer's disease is involved in the regulation of neurite outgrowth. J. Neurosci. 14, 2117–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Multhaup G., Mechler H., Masters C. L. (1995) Characterization of the high affinity heparin binding site of the Alzheimer's disease β-amyloid precursor protein (APP) and its enhancement by zinc (II). J. Mol. Recognit. 8, 247–257 [DOI] [PubMed] [Google Scholar]
- 26. Kaden D., Munter L. M., Reif B., Multhaup G. (2012) The amyloid precursor protein and its homologues: structural and functional aspects of native and pathogenic oligomerization. Eur. J. Cell Biol. 91, 234–239 [DOI] [PubMed] [Google Scholar]
- 27. Bush A. I., Multhaup G., Moir R. D., Williamson T. G., Small D. H., Rumble B., Pollwein P., Beyreuther K., Masters C. L. (1993) A novel zinc(II)-binding site modulates the function of the β A4 amyloid protein precursor of Alzheimer's disease. J. Biol. Chem. 268, 16109–16112 [PubMed] [Google Scholar]
- 28. Hesse L., Beher D., Masters C. L., Multhaup G. (1994) The β A4 amyloid precursor protein binding to copper. FEBS Lett. 349, 109–116 [DOI] [PubMed] [Google Scholar]
- 29. Duce J. A., Tsatsanis A., Cater M. A., James S. A., Robb E., Wikhe K., Leong S. L., Perez K., Johanssen T., Greenough M. A., Cho H.-H., Galatis D., Moir R. D., Masters C. L., McLean C., Tanzi R. E., Cappai R., Barnham K. J., Ciccotosto G. D., Rogers J. T., Bush A. I. (2010) Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell 142, 857–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dahms S. O., Könnig I., Roeser D., Gührs K.-H., Mayer M. C., Kaden D., Multhaup G., Than M. E. (2012) Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain. J. Mol. Biol. 416, 438–452 [DOI] [PubMed] [Google Scholar]
- 31. Zheng L., Baumann U., Reymond J.-L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hansson M. D., Rzeznicka K., Rosenbäck M., Hansson M., Sirijovski N. (2008) PCR-mediated deletion of plasmid DNA. Anal. Biochem. 375, 373–375 [DOI] [PubMed] [Google Scholar]
- 33. Suckau D., Resemann A., Schuerenberg M., Hufnagel P., Franzen J., Holle A. (2003) A novel MALDI LIFT-TOF/TOF mass spectrometer for proteomics. Anal. Bioanal. Chem. 376, 952–965 [DOI] [PubMed] [Google Scholar]
- 34. Wouters F. S., Verveer P. J., Bastiaens P. I. (2001) Imaging biochemistry inside cells. Trends Cell Biol. 11, 203–211 [DOI] [PubMed] [Google Scholar]
- 35. Shen C., Menon R., Das D., Bansal N., Nahar N., Guduru N., Jaegle S., Peckham J., Reshetnyak Y. K. (2008) The protein fluorescence and structural toolkit: Database and programs for the analysis of protein fluorescence and structural data. Proteins 71, 1744–1754 [DOI] [PubMed] [Google Scholar]
- 36. Wang Y., Ha Y. (2004) The x-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol. Cell 15, 343–353 [DOI] [PubMed] [Google Scholar]
- 37. Lee S., Xue Y., Hu J., Wang Y., Liu X., Demeler B., Ha Y. (2011) The E2 domains of APP and APLP1 share a conserved mode of dimerization. Biochemistry 50, 5453–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ball L. J., Jarchau T., Oschkinat H., Walter U. (2002) EVH1 domains: structure, function and interactions. FEBS Lett. 513, 45–52 [DOI] [PubMed] [Google Scholar]
- 39. Vallee B. L., Falchuk K. H. (1993) The biochemical basis of zinc physiology. Physiol. Rev. 73, 79–118 [DOI] [PubMed] [Google Scholar]
- 40. Coburger I., Dahms S. O., Roeser D., Gührs K.-H., Hortschansky P., Than M. E. (2013) Analysis of the overall structure of the multidomain amyloid precursor protein (APP). PLoS ONE 8, e81926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Parr R. M., Taylor D. M. (1964) The concentrations of cobalt, copper, iron and zinc in some normal human tissues as determined by neutron-activation analysis. Biochem. J. 91, 424–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Que E. L., Domaille D. W., Chang C. J. (2008) Metals in neurobiology: probing their chemistry and biology with molecular imaging. Chem. Rev. 108, 1517–1549 [DOI] [PubMed] [Google Scholar]
- 43. Akatsu H., Hori A., Yamamoto T., Yoshida M., Mimuro M., Hashizume Y., Tooyama I., Yezdimer E. M. (2012) Transition metal abnormalities in progressive dementias. Biometals 25, 337–350 [DOI] [PubMed] [Google Scholar]
- 44. Hemdan E. S., Zhao Y. J., Sulkowski E., Porath J. (1989) Surface topography of histidine residues: a facile probe by immobilized metal ion affinity chromatography. Proc. Natl. Acad. Sci. U.S.A. 86, 1811–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Halloran T. V. (2000) Metallochaperones, an intracellular shuttle service for metal ions. J. Biol. Chem. 275, 25057–25060 [DOI] [PubMed] [Google Scholar]
- 46. Segrest J. P., Kahane I., Jackson R. L., Marchesi V. T. (1973) Major glycoprotein of the human erythrocyte membrane: evidence for an amphipathic molecular structure. Arch. Biochem. Biophys. 155, 167–183 [DOI] [PubMed] [Google Scholar]
- 47. Sensi S. L., Paoletti P., Koh J.-Y., Aizenman E., Bush A. I., Hershfinkel M. (2011) The neurophysiology and pathology of brain zinc. J. Neurosci. 31, 16076–16085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jansen J., Rosenkranz E., Overbeck S., Warmuth S., Mocchegiani E., Giacconi R., Weiskirchen R., Karges W., Rink L. (2012) Disturbed zinc homeostasis in diabetic patients by in vitro and in vivo analysis of insulinomimetic activity of zinc. J. Nutr. Biochem. 23, 1458–1466 [DOI] [PubMed] [Google Scholar]
- 49. Auld D. S. (2001) Zinc coordination sphere in biochemical zinc sites. Biometals 14, 271–313 [DOI] [PubMed] [Google Scholar]
- 50. Dugan T. A., Yang V. W., McQuillan D. J., Höök M. (2003) Decorin binds fibrinogen in a Zn2+-dependent interaction. J. Biol. Chem. 278, 13655–13662 [DOI] [PubMed] [Google Scholar]
- 51. Graille M., Pagano M., Rose T., Ravaux M. R., van Tilbeurgh H. (2010) Zinc induces structural reorganization of gelatin binding domain from human fibronectin and affects collagen binding. Structure 18, 710–718 [DOI] [PubMed] [Google Scholar]
- 52. Frederickson C. J., Giblin L. J., Krezel A., McAdoo D. J., Mueller R. N., Muelle R. N., Zeng Y., Balaji R. V., Masalha R., Thompson R. B., Fierke C. A., Sarvey J. M., de Valdenebro M., Prough D. S., Zornow M. H. (2006) Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp. Neurol. 198, 285–293 [DOI] [PubMed] [Google Scholar]
- 53. Gaier E. D., Eipper B. A., Mains R. E. (2013) Copper signaling in the mammalian nervous system: synaptic effects. J. Neurosci. Res. 91, 2–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tamano H., Takeda A. (2011) Dynamic action of neurometals at the synapse. Metallomics 3, 656–661 [DOI] [PubMed] [Google Scholar]
- 55. Sindreu C., Storm D. R. (2011) Modulation of neuronal signal transduction and memory formation by synaptic zinc. Front. Behav. Neurosci. 5, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lein E. S., Hawrylycz M. J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A. F., Boguski M. S., Brockway K. S., Byrnes E. J., Chen L., Chen L., Chen T.-M., Chin M. C., Chong J., Crook B. E., Czaplinska A., Dang C. N., Datta S., Dee N. R., Desaki A. L., Desta T., Diep E., Dolbeare T. A., Donelan M. J., Dong H.-W., Dougherty J. G., Duncan B. J., Ebbert A. J., Eichele G., Estin L. K., Faber C., Facer B. A., Fields R., Fischer S. R., Fliss T. P., Frensley C., Gates S. N., Glattfelder K. J., Halverson K. R., Hart M. R., Hohmann J. G., Howell M. P., Jeung D. P., Johnson R. A., Karr P. T., Kawal R., Kidney J. M., Knapik R. H., Kuan C. L., Lake J. H., Laramee A. R., Larsen K. D., Lau C., Lemon T. A., Liang A. J., Liu Y., Luong L. T., Michaels J., Morgan J. J., Morgan R. J., Mortrud M. T., Mosqueda N. F., Ng L. L., Ng R., Orta G. J., Overly C. C., Pak T. H., Parry S. E., Pathak S. D., Pearson O. C., Puchalski R. B., Riley Z. L., Rockett H. R., Rowland S. A., Royall J. J., Ruiz M. J., Sarno N. R., Schaffnit K., Shapovalova N. V., Sivisay T., Slaughterbeck C. R., Smith S. C., Smith K. A., Smith B. I., Sodt A. J., Stewart N. N., Stumpf K.-R., Sunkin S. M., Sutram M., Tam A., Teemer C. D., Thaller C., Thompson C. L., Varnam L. R., Visel A., Whitlock R. M., Wohnoutka P. E., Wolkey C. K., Wong V. Y., Wood M., Yaylaoglu M. B., Young R. C., Youngstrom B. L., Yuan X. F., Zhang B., Zwingman T. A., Jones A. R. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 [DOI] [PubMed] [Google Scholar]
- 57. Brocca S., Schmidt-Dannert C., Lotti M., Alberghina L., Schmid R. D. (1998) Design, total synthesis, and functional overexpression of the Candida rugosa lipl gene coding for a major industrial lipase. Protein Sci. 7, 1415–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Diez-Roux G., Banfi S., Sultan M., Geffers L., Anand S., Rozado D., Magen A., Canidio E., Pagani M., Peluso I., Lin-Marq N., Koch M., Bilio M., Cantiello I., Verde R., De Masi C., Bianchi S. A., Cicchini J., Perroud E., Mehmeti S., Dagand E., Schrinner S., Nürnberger A., Schmidt K., Metz K., Zwingmann C., Brieske N., Springer C., Hernandez A. M., Herzog S., Grabbe F., Sieverding C., Fischer B., Schrader K., Brockmeyer M., Dettmer S., Helbig C., Alunni V., Battaini M.-A., Mura C., Henrichsen C. N., Garcia-Lopez R., Echevarria D., Puelles E., Garcia-Calero E., Kruse S., Uhr M., Kauck C., Feng G., Milyaev N., Ong C. K., Kumar L., Lam M., Semple C. A., Gyenesei A., Mundlos S., Radelof U., Lehrach H., Sarmientos P., Reymond A., Davidson D. R., Dollé P., Antonarakis S. E., Yaspo M.-L., Martinez S., Baldock R. A., Eichele G., Ballabio A. (2011) A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 9, e1000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Guo Q., Li H., Gaddam S. S., Justice N. J., Robertson C. S., Zheng H. (2012) Amyloid precursor protein revisited: neuron-specific expression and highly stable nature of soluble derivatives. J. Biol. Chem. 287, 2437–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Baron M. K., Boeckers T. M., Vaida B., Faham S., Gingery M., Sawaya M. R., Salyer D., Gundelfinger E. D., Bowie J. U. (2006) An architectural framework that may lie at the core of the postsynaptic density. Science 311, 531–535 [DOI] [PubMed] [Google Scholar]
- 61. Grabrucker A. M., Knight M. J., Proepper C., Bockmann J., Joubert M., Rowan M., Nienhaus G. U., Garner C. C., Bowie J. U., Kreutz M. R., Gundelfinger E. D., Boeckers T. M. (2011) Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 30, 569–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Baumkötter F., Wagner K., Eggert S., Wild K., Kins S. (2012) Structural aspects and physiological consequences of APP/APLP trans-dimerization. Exp. Brain Res. 217, 389–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim T. W., Wu K., Xu J. L., McAuliffe G., Tanzi R. E., Wasco W., Black I. B. (1995) Selective localization of amyloid precursor-like protein 1 in the cerebral cortex postsynaptic density. Mol. Brain Res. 32, 36–44 [DOI] [PubMed] [Google Scholar]
- 64. Bayer T. A., Paliga K., Weggen S., Wiestler O. D., Beyreuther K., Multhaup G. (1997) Amyloid precursor-like protein 1 accumulates in neuritic plaques in Alzheimer's disease. Acta Neuropathol. 94, 519–524 [DOI] [PubMed] [Google Scholar]
- 65. Lyckman A. W., Confaloni A. M., Thinakaran G., Sisodia S. S., Moya K. L. (1998) Post-translational processing and turnover kinetics of presynaptically targeted amyloid precursor superfamily proteins in the central nervous system. J. Biol. Chem. 273, 11100–11106 [DOI] [PubMed] [Google Scholar]
- 66. Hoe H.-S., Fu Z., Makarova A., Lee J.-Y., Lu C., Feng L., Pajoohesh-Ganji A., Matsuoka Y., Hyman B. T., Ehlers M. D., Vicini S., Pak D. T., Rebeck G. W. (2009) The effects of amyloid precursor protein on postsynaptic composition and activity. J. Biol. Chem. 284, 8495–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gu C., Somogyi Á., Wysocki V. H., Medzihradszky K. F. (1999) Fragmentation of protonated oligopeptides XLDVLQ (X=L, H, K or R) by surface induced dissociation: additional evidence for the “mobile proton” model. Anal. Chim. Acta 397, 247–256 [Google Scholar]
- 68. Wattenberg A., Organ A. J., Schneider K., Tyldesley R., Bordoli R., Bateman R. H. (2002) Sequence dependent fragmentation of peptides generated by MALDI quadrupole time-of-flight (MALDI Q-TOF) mass spectrometry and its implications for protein identification. J. Am. Soc. Mass Spectrom. 13, 772–783 [DOI] [PubMed] [Google Scholar]