
Fig. 5.
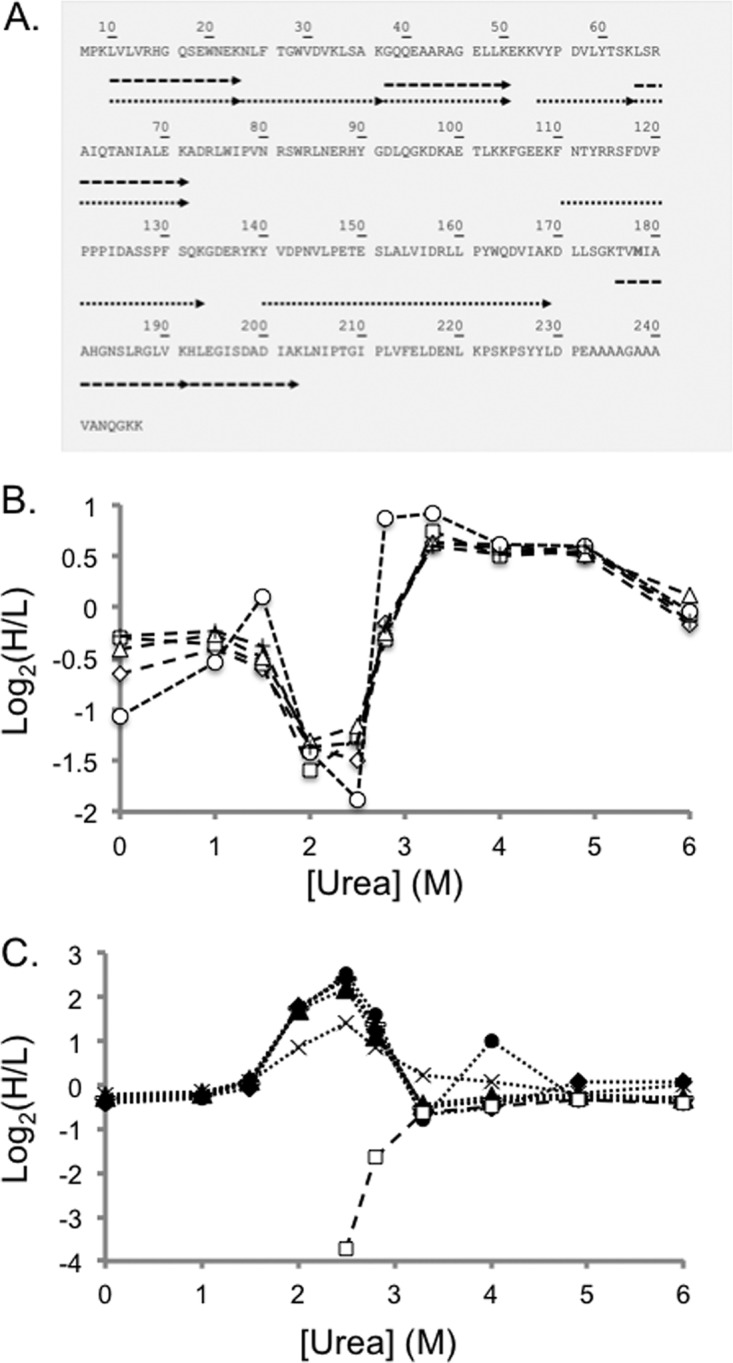
Representative SILAC-SPROX data obtained on a protein hit, PGM-1, identified in the ATP-binding experiments using the gel-based approach. Shown in A, is a schematic representation of the sequence coverage obtained for PGM-1 in Gel Experiment 2. Each arrow represents a peptide identified in the bottom-up proteomics analysis. The dashed and dotted arrows represent peptides detected from the full-length protein and CnBr fragments, respectively. Shown in B, are the SILAC-SPROX data obtained in Gel Experiment 2 on the five nonmethionine containing peptides identified from gel bands in the 25–30 kDa size range that includes intact PGM-1. These peptides included: GQQEAARAGELLK (open diamonds), LSRAIQTANIALEK (open triangles), VYPDVLYTSK (crosses), HLEGISDADIAK (open squares), and LVLVRHGQSEWNEK (open circles). Shown in C, are the SILAC-SPROX data obtained in Gel Experiment 2 on the seven the nonmethionine containing peptides identified from gel bands in the 20–25 kDa size range that included the 21 kDa CnBr fragment of PGM-1. These peptides included all the peptides in B, (same symbols but filled) and two additional peptides, NLFTGWVDVK (bars) and YVDPNVLPETESLALVIDRLLPYWQDVIAK (x's). The HLEGISDADIAK peptide, which is unique to the full-length 27 kDa protein was identified in both A, and B, but in both cases displayed the behavior expected of being derived from the intact protein. This suggests that the gel electrophoresis and cutting strategy may not have completely separated all the intact protein from the CnBr-fragment. Nevertheless, the ability of the nonmethionine containing peptide to report on the ATP binding properties of the protein was not compromised.