


Abstract
Temperature sensitive (Ts) mutants of proteins provide experimentalists with a powerful and reversible way of conditionally expressing genes. The technique has been widely used in determining the role of gene and gene products in several cellular processes. Traditionally, Ts mutants are generated by random mutagenesis and then selected though laborious large-scale screening. Our web server, TSpred (http://mspc.bii.a-star.edu.sg/TSpred/), now enables users to rationally design Ts mutants for their proteins of interest. TSpred uses hydrophobicity and hydrophobic moment, deduced from primary sequence and residue depth, inferred from 3D structures to predict/identify buried hydrophobic residues. Mutating these residues leads to the creation of Ts mutants. Our method has been experimentally validated in 36 positions in six different proteins. It is an attractive proposition for Ts mutant engineering as it proposes a small number of mutations and with high precision. The accompanying web server is simple and intuitive to use and can handle proteins and protein complexes of different sizes.
INTRODUCTION
Temperature sensitive (Ts) mutants of a protein are those whose levels of activity decrease when temperature rises above a certain restrictive temperature. Below the restrictive temperature, in permissive temperatures, the mutants and the wild type protein have similar activity levels. Ts mutants are powerful tools to study protein function in vivo and in cell culture (1,2). They provide a reversible mechanism to lower the level of a protein simply by changing the temperature of growth (3). There are several practical applications of engineering Ts mutants. In developmental biology, such mutants would provide valuable insight into the functioning of essential genes and those used in multiple phases of development. Ts mutants have also been utilized in many to investigate protein folding pathways (4), macro-molecular assembly (5), controlling the genotype of a cell in vivo (6), nervous system defects (7), phenotypic effects (8,9), coordination between different genes (9) pinpointing the phase at which genes are functioning during the cell-division cycle (10), controlled cell-arrest and synchronization of cells or gene functions by means of reversible cell arrest (11,12) and conditional expression of genes in Drosophila (13,14).
Ts mutants possess several advantages over other methods such as CRISPER and RNAi (15,16) in producing conditional expression/induction/repression of genes. The advantages include fast temporal response, high reversibility and the applicability to any tissue type or developmental stage of an organism. Various strategies have been proposed to construct Ts mutants, such as by fusion to a heat-sensitive degron (17–19) or insertion of a Ts intein (20). However, Ts mutants are still most commonly generated by random mutagenesis. The process involves using chemical mutagens, ultraviolet (UV) radiation or error prone Polymerase chain reaction (PCR) techniques to introduce random mutations to protein-coding deoxyribonucleic acid (DNA). A screening procedure is then adopted to select Ts mutants (7,21). This procedure is typically laborious and expensive, as a large number of mutants need to be screened. For instance, identifying Ts mutants in the fruit fly Drosophila melanogaster involves the screening of several hundreds of thousands of progeny (7). This method is not feasible for model organisms with long generation times, and where it is impractical to obtain large numbers of progeny (22).
To overcome the difficulties posed by random mutagenesis, we have previously demonstrated that it is possible to accurately predict, purely from protein sequence, a small subset of candidate positions, that when mutated, are likely to result in a Ts mutant (13,14,22,23). The method is based on the observation that mutations at buried residue positions can cause large changes in protein thermal stability. Further, the likelihood of an amino acid residing at a buried position can be inferred from its hydrophobicity and that of its flanking residues. Two parameters, namely the average hydrophobicity (24,25) and hydrophobic moment (26) were computed to estimate this likelihood. Following prediction of buried positions, substitutions were suggested at such positions to generate a Ts phenotype. The mechanisms responsible for temperature sensitive phenotype for different proteins could be case-specific and is dependent on complex factors, such as the rate of protein synthesis, susceptibility to proteolysis and whether chaperones are involved in the folding or degradation of the protein. Instead of speculating on the exact mechanism, our approach is to suggest mutations with a set of stereochemically diverse amino acids. It is assumed that these mutations will destabilize the protein to different extents, and that at least one mutant is likely to be temperature sensitive. Through large-scale analysis of known Ts mutants, prediction rules were generated based on the two parameters described above (22). This strategy for choosing mutant substitutions has been experimentally tested previously on CcdB, TBP (TATA binding protein), T4 lysozyme and Gal4 (13,23).
In addition to the sequence-based method described above, the present study enhances Ts mutant prediction by incorporating structural information of the protein. This structural information can be inferred from existing protein structures in the Protein Data Bank (PDB) or from homology models. To determine the degree of burial of an amino acid in a protein, the residue depth measure was used. Depth is defined as the distance of any atom/residue to the closest bulk water (27). It has been shown to accurately measure burial and parameterizes the local protein environment (28,29). Importantly, the depth measure correlates well with structural stability (27) and free energy change of cavity-creating mutations in globular proteins (27,29).
The aim of this study is to predict, a small set of residues that when appropriately mutated, result in temperature sensitive mutants of a given protein. Our server reports the predictions by both the sequence- and structure-based methods. These mutants can be readily tested experimentally. It should be noted that no attempt is made to identify all possible Ts mutants.
In the sections below we first describe the methods used for sequence- and structure-based predictions. This is followed by a description of the benchmark results. The server functionality is then described and illustrated with a case study.
MATERIALS AND METHODS
In designing Ts mutants we have made use of the observation that the Ts phenotype correlates with decreased protein stability (30,31). In general, this reduction in stability, and hence protein activity, can be attributed to the loss of thermal stabilization contributed by hydrophobic residues in the protein core. Accordingly, numerous experimental studies have shown that significant destabilization in protein stability and activity can be achieved by mutating buried residues as compared to mutating residues at the surface (32–34). Our strategy to predict Ts mutant positions is to target buried positions occupied by hydrophobic residues. We identify such positions using both sequence and 3D structural information.
Prediction based on primary sequence
The details of the sequence-based predictions have been described at length in an earlier study (22). Qualitatively, the method was based on the observations that the seven residues, Cys, Phe, Ile, Val, Trp, Met and Leu were observed to have an average side chain solvent accessible area of less than 20%. Cysteines are not considered, as they could also be involved with disulphide bond formation or metal ion coordination. The other six residues are mutation targets if predicted as buried. Burial predictions were made using the local average Rose hydrophobicity (22,35) and the hydrophobic moment of the target residue and its flanking neighbours. A set of rules based on these values, predicted degree of residue burial.
Prediction based on 3D structure
3D structural information would make it easier to identify hydrophobic residue positions that are buried in the core of the protein. We estimate degree of burial by the depth measure (27–29). The depth of a residue (or atom) measures its distance to the closest bulk molecule of bulk solvent (water). In earlier studies we have made a convincing case of how depth gives a more stratified description of residue burial/environment (27) than the widely used solvent accessibility measure.
The average depths of amino acid residues that correspond to an average of 5% side chain accessibility were determined from a non-redundant dataset of 561 proteins extracted from the PDB (36). This dataset was non-redundant to 30% in sequence identity and consisted of single domain (chain length of 120–180 amino acids), high-resolution (resolution ≤ 1.7 Å, R-free ≤ 0.2) protein structures in the PDB (http://mspc.bii.a-star.edu.sg/TSpred/supplementary_data.html). The threshold depth values of Val, Ile, Leu, Met, Phe and Trp were determined to be 6.25, 6.75, 6.75, 7.00, 7.00 and 7.00 Å, respectively. Residues with depths greater than their respective threshold would be predicted as Ts mutant positions. Note, the 5% accessibility cut off value was determined in an earlier study (23) as an optimal value to define buried residues.
Homology modeling protocol
When no experimentally determined structure is available for a protein sequence of interest (query sequence), a homology model is constructed. Residue burial is then inferred from the 3D model. The TSpred server makes use of an automated homology modeling pipeline that includes steps for template selection, target-template alignment, structure modeling and model assessment. Suitable structural templates are searched in the PDB database using three iterations of PSI-BLAST (version 2.2.28) (37) utilizing the BLOSUM62 substitution matrix (38). A stringent e-value cut-off of 0.0001 was used to identify homologs. Among the hits, the one with the best e-value is chosen as template. Next, a sequence alignment between query sequence and the template sequence was constructed using SALIGN (39). The resulting alignment is input to the automodel protocol of MODELLER (40) to construct a 3D model of the protein. The model is assessed for accuracy/reliability with the GA341 (41,42) and Discrete Optimized Protein Energy (DOPE) statistical potentials (43). Residue burial information is only taken from those models that satisfy a stringent GA341 and DOPE cutoff of 0.75 and −1, respectively. When no suitable templates are available or the constructed models do not satisfy the GA341 and DOPE criteria, the burial prediction is made using the sequence-based method alone.
Suggested mutations
At the predicted positions, we suggest that the original residue be mutated to Ala, Trp, Asn, Asp or Pro. While the current program does not rank order amongst these mutations, the suggestion is that one or more of them are likely to result in a Ts phenotype.
Benchmark datasets
We benchmarked our method by examining the agreement between our predictions with experimentally validated Ts mutants. Thirty six mutants from a set of six proteins, for which extensive mutagenesis data exists, constituted our benchmark. The proteins were gene V (PDB: 1YHA), lambda repressor (PDB: 1LMB), T4 Lysozyme (PDB: 2LZM), CcdB (PDB: 3VUB), Gal4 (PDB: 3CQQ) and Ura3 (PDB: 1DQW)) (13,23,44–49). The performance of our predictions was assessed by coverage (the number of predictions) and precision (number of true positives / number of predictions). Results from both sequence-based and structure-based methods are reported.
For a query sequence without a PDB entry, structural information was inferred from a homology model. The accuracy of the model is largely determined by sequence identity to the structural template used. To gauge the effect of template sequence identity on Ts mutant prediction performance, we built models of T4 lysozyme using 15 templates of varying sequence similarity. The templates were identified by searching DBAli (http://www.salilab.org/DBAli/) (50) for structures similar to T4 lysozyme (PDB 2LZM). We defined similarity as a minimum MAMMOTH P-value of 10 and at least 100 equivalent positions (61% structure overlap with T4 lysozyme). The selected templates were between 22 and 91% identical in sequence to T4 lysozyme and all identifiable by PSI-BLAST search with an e-value cutoff of 0.0001.
RESULTS
Performance on experimentally validated benchmark
Our methods successfully predicted all 36 experimentally validated Ts mutant positions in the six proteins, CcdB, Lysozyme, gene V, gal4, Ura3 and Lamda repressor (Table 1). The sequence-based method correctly predicted 22 and the structure-based method 28 of the 36 cases. The two methods were correct together in 14 of the cases. In addition to the correct predictions, the sequence-based method made one confirmed false positive identification, Val53 in CcdB. Our methods also made 71 other predictions in these six proteins that are yet to be experimentally validated (http://mspc.bii.a-star.edu.sg/TSpred/supplementary_data.html).
Table 1.
Ts mutant position as predicted by sequence-based, structured-based or both methods
| Protein | PDB ID | Chain length | Residue position | Residue type | Prediction method |
|---|---|---|---|---|---|
| gene V | 1YHA | 87 | 35 | VAL | Both |
| 45 | VAL | Both | |||
| 47 | ILE | Structure | |||
| 63 | VAL | Structure | |||
| 81 | LEU | Structure | |||
| 78 | ILE | Sequence | |||
| lambda repressor | 1LMB | 92 | 51 | PHE | Both |
| 65 | LEU | Both | |||
| 76 | PHE | Both | |||
| 84 | ILE | Both | |||
| 18 | LEU | Structure | |||
| 36 | VAL | Structure | |||
| 47 | VAL | Structure | |||
| T4 lysozyme | 2LZM | 164 | 6 | MET | Both |
| 102 | MET | Both | |||
| 149 | VAL | Structure | |||
| 153 | PHE | Structure | |||
| 103 | VAL | Sequence | |||
| CcdB | 3VUB | 101 | 17 | PHE | Both |
| 18 | VAL | Both | |||
| 33 | VAL | Both | |||
| 34 | ILE | Both | |||
| 54 | VAL | Both | |||
| 5 | VAL | Structure | |||
| 36 | LEU | Structure | |||
| 63 | MET | Structure | |||
| 50 | LEU | Sequence | |||
| 53* | VAL | Sequence | |||
| 96 | LEU | Sequence | |||
| 97 | MET | Sequence | |||
| 98 | PHE | Sequence | |||
| Gal4 | 3COQ | 88 | 68 | PHE | Both |
| 69 | LEU | Sequence | |||
| 70 | LEU | Sequence | |||
| Ura3 | 1DQW | 267 | 25 | MET | Structure |
| 32 | LEU | Structure | |||
| 118 | ILE | Structure |
The wild-type residue (three-letter amino acid code) at the position is listed under residue type. * The prediction of VAL53 in CcdB as a Ts mutant position is a false positive identification.
The amino acids that we target for mutation—Val, Ile, Leu, Met, Phe and Trp constitute 29.2% (544 residue positions) of the amino acids in these six proteins. Only 19.8% (108 residue positions) of these are identified by our predictions as potential Ts mutant positions. The sequence based method accurately identifies 53% (57 positions) and the structure-based method identifies 79% (85 positions), while 32% (34 positions) is common to both the methods.
Effect of model accuracy on prediction
In the case of T4 lysozyme, 15 homology models were built using templates with sequence identity ranging from 22 to 91%. The aim was to determine the efficacy of structure-based prediction with homology models of varying accuracy. Out of five experimentally validated Ts mutant positions, high accuracy models (template sequence identity >40%) correctly identified four mutant positions (Table 2). For models built with low target-template sequence identity (<25%) templates, their DOPE and GA341 scores failed to cross the acceptance threshold. These models only predict one or two of the validated positions. The sequence-based method, being independent of the model accuracy, predicted three positions correctly in each of the cases. Note, for both the structure- and sequence-based methods many other positions are predicted by our program. As these positions have not been validated or invalidated as Ts mutation sites, these are not considered as false positives of our method.
Table 2.
Ts mutant prediction in T4 lysozyme when homology models (identified by their templates) of different accuracies are used
| Template quality | Number of predictions | Experimentally validated mutant positions | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB:chain | Sequence ID (%) | DOPE | GA341 | Sequence | Structure | Both | M6 | M102 | V103 | V149 | F153 |
| 1pqj:A | 90.8 | −2.08 | 1.00 | 3 (8) | 4 (11) | 2 (3) | Both | Both | Sequence | Structure | Structure |
| 1d3n:A | 86.1 | −1.96 | 1.00 | 3 (8) | 4 (11) | 2 (3) | Both | Both | Sequence | Structure | Structure |
| 1t8a:A | 81.6 | −1.65 | 1.00 | 3 (8) | 4 (13) | 2 (3) | Both | Both | Sequence | Structure | Structure |
| 1cx6:A | 79.9 | −2.03 | 1.00 | 3 (8) | 4 (11) | 2 (3) | Both | Both | Sequence | Structure | Structure |
| 1lpy:A | 78.8 | −1.95 | 1.00 | 3 (8) | 4 (10) | 2 (3) | Both | Both | Sequence | Structure | Structure |
| 1swz:A | 77.5 | −2.21 | 1.00 | 3 (8) | 4 (13) | 2 (3) | Both | Both | Sequence | Structure | Structure |
| 1lwk:A | 77.0 | −1.74 | 1.00 | 3 (8) | 4 (12) | 2 (3) | Both | Both | Sequence | Structure | Structure |
| 1swy:A | 74.5 | −2.22 | 1.00 | 3 (8) | 4 (12) | 2 (3) | Both | Both | Sequence | Structure | Structure |
| 1sx2:A | 72.4 | −2.28 | 1.00 | 3 (8) | 4 (13) | 2 (4) | Both | Both | Sequence | Structure | Structure |
| 1wth:A | 43.2 | −1.49 | 1.00 | 3 (8) | 3 (12) | 1 (2) | Sequence | Both | Sequence | Structure | Structure |
| 1k28:A | 43.2 | −1.43 | 1.00 | 3 (8) | 5 (15) | 3 (5) | Both | Both | Both | Structure | Structure |
| 2anv:A | 24.2 | 0.54 | 0.12 | 3 (8) | 1 (11) | 1 (2) | Sequence | Both | Sequence | ||
| 2anx:B | 23.9 | 0.60 | 0.08 | 3 (8) | 2 (11) | 2 (3) | Sequence | Both | Both | ||
| 2anv:B | 23.5 | 0.49 | 0.13 | 3 (8) | 2 (11) | 2 (3) | Sequence | Both | Both | ||
| 2anx:A | 22.1 | 0.64 | 0.13 | 3 (8) | 1 (10) | 1 (2) | Sequence | Both | Sequence | ||
The number of predictions made by the sequence-based, structure-based or both methods are listed for each of the models with the number of experimentally validated predictions within brackets. The performance of the different models on the experimentally validated mutant positions are additionally shown in separate columns.
Server description
Our server supports both sequence and structure inputs, and several options are provided for each. For input sequences, users could either specify a database, GenBank (51) or UniProt (52,53), identification number or upload the protein amino acid sequence in FASTA format. If the input consists of multiple sequences, Ts mutant predictions are made for each of the sequences separately. For input structures, the users could either specify the four-letter PDB code with optional additional letters to select specific chains of the protein (the biological unit is used for predictions) or upload a file in PDB format. User uploaded structures are used for prediction without any model assessment.
Our server uses sequence and structure information to make Ts mutant position predictions. The web server outputs the residue positions that have been predicted as Ts mutation targets and prediction method (structure-based, sequence-based or both). In the case where a homology model was used, the server only displays the results of the structure-based method if the models satisfy the assessment criteria.
The user has the option to review details of the hydrophobic moment, average hydrophobicity and residues flanking of predicted mutant positions. In cases where 3D structural data have been used in making the Ts mutant predictions, the PDB structure or the homology model is displayed using a Jmol plugin (http://www.jmol.org/). The backbone chain trace of the proteins is coloured according to the residue depth and the mutant positions are shown as spheres centered on the Cα atoms. The spheres are also coloured according to residue depth. In the case of homology models, the user is notified about the target-template sequence identity and the alignment used in model construction is provided.
Server availability
The server is freely accessible at http://mspc.bii.a-star.edu.sg/TSpred and has no login requirements.
Case study
We demonstrate the functioning of our server using Escherichia coli CcdB protein as an example. CcdB is a poison of DNA gyrase and is a potent cytotoxin (10,11). CcdB is 101 amino acid residues long and its functional form (biological unit) is a homodimer. CcdB was chosen because saturation mutagenesis experiments have been performed (23) on this protein and can be used for validation.
The sequence-based method identified 10 target positions (F17, V18, V33, L34, L50, V53, V54, L96, M97, F98) and the structure-based method identified 14 target positions (V5, F17, V18, V20, M32, V33, L34, L36, M63, M68, I90, I94, M97, F98) (Figure 1). Eight Ts mutant targets (V5, V20, M32, L36, M63, M68, I90, I94) were exclusively predicted by the structure-based method. All the predictions were experimentally verified (100% accuracy). Four target positions were exclusively predicted by the sequence-based method, viz. L50, V53, V54 and L96. Of these, V53 is a false positive. V54 (depth 6.18 Å) only marginally missed the depth threshold (6.25 Å) for structure prediction; L50 and L96 were not buried but their side chains were within 4 Å of active site residues. It was proposed that these residues were important in maintaining the conformation of active site residues, and were hence potential Ts mutant positions. The sequence- and structure-based methods when applied independently, predict ∼10–15% of all residues to be potential Ts mutant positions, corresponding to 50–75 mutation suggestions (five mutations at each position). There are six predictions common to both methods—F17, V18, V33, L34, M97 and F98. All these residues were experimentally verified Ts mutants (23). The structure-based method helps reduce the false positives from sequence-based method and reduces the number of predictions to 6% of all residues or equivalently ∼30 mutant suggestions in CcdB.
Figure 1.
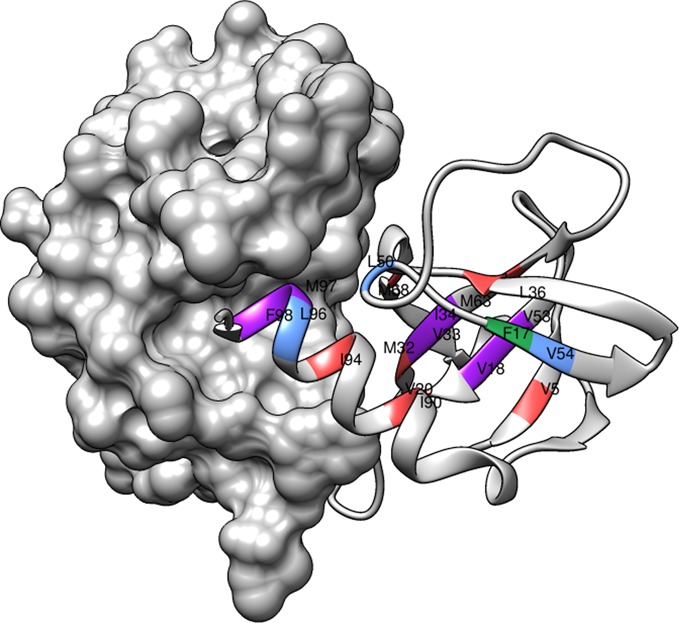
A functionally active CcdB dimer with one monomer in surface representation and the other in ribbon representation. The labeled location of the predictions made by the sequence-based, structure-based and both predictions are coloured blue, red and purple, respectively. The false positive identification is shown in green. The figure was rendered using Chimera (54).
We also compared the coverage between sequence- and structure-based predictions. The saturation mutagenesis data shows that CcdB consists of 55 possible Ts positions (23). As our algorithm aims primarily to predict buried hydrophobic residues as Ts mutants, active site (I24, I25, D26, E87, N88, K91, N92, N95, W99, G100, I101), dimerization site (Q2, V20, Q21, S22, I25, T27, M32, T66, M68, A69, I94, N95, M97, F98, W99, G100) and non-hydrophobic residues were excluded from the analysis. Only 18 residues (F3, V5, L16, F17, V18, V33, I34, L36, L41, L50, V54, I56, M63, M64, V80, L83, I90, L96) remain as potential prediction targets. The sequence-based method correctly predicted seven (39%), and structure-based method predicted eight (44%) of all targets. Only four (22%) residues were common between the two methods. Eleven (61%) of all targets can be identified by either one of the methods.
As saturated mutagenesis experiments have been performed for CcdB, it enabled us to quantify the number of substitutions at a predicted position that would lead to a temperature sensitive mutant. On average 6.5 substitutions led to Ts mutants at the positions predicted by the structure-based method, as compared to 3.3 substitutions predicted by sequence-based method (Table 3).
Table 3.
Number of substitutions that led to a Ts mutant at positions identified by different prediction modes in Escherichia coli protein CcdB
| Prediction method | Number of predictions | Average number of Ts mutants |
|---|---|---|
| Exclusively sequence-based | 3 | 3.3 |
| Exclusively structure-based | 4 | 6.5 |
| Sequence-based | 7 | 5.1 |
| Structure-based | 8 | 6.5 |
| Sequence- and structure-based | 4 | 6.5 |
| Sequence- or structure-based | 11 | 5.6 |
DISCUSSION and CONCLUSION
Our predictions are based on the assumption that mutating buried hydrophobic residues would destabilize a protein, hence sometimes rendering it temperature sensitive. We have employed two methods, sequence-based and structure-based to make these predictions. The sequence-based method estimates the degree of burial of a residue by the hydrophobicity and hydrophobic moment of it and its neighbouring residues. The structure-based method computes residue depth (extent of burial), a 3D structure or accurate homology model. The aim is to identify a small number of residue positions in proteins and suggests substitutions that are likely to produce Ts mutants. These potential Ts mutants can then be constructed and experimentally screened. Our methods do not attempt to predict either all buried residues or all possible Ts mutants.
We have shown that, both sequence-based and structure-based methods are capable of accurately identifying Ts mutants. The overlap between the two methods is however low. Specifically, the Jaccard index (ratio of the intersection set to the union set) of the sequence-based and the structure-based predictions is only 0.32. This motivates us to suggest that the experimentalists first mutate positions that are common to the two prediction methods. This strategy substantially reduces (halves) the number of predictions and hence increases precision (90–100% in the case study).
In our benchmark set, of 36 experimentally verified Ts mutants, the sequence-based method identified 22 (59%), and the other 15 (41%) were exclusively identified by the structure-based method. Furthermore, none of the structure-based predictions have been invalidated by experiments as yet. However, hydrophobic environments can also be found on protein surface forming active sites and protein-protein interaction interfaces (55). Mutating these non-buried residues could also lead to decreased functional activity of the protein and a Ts mutant. This is shown in the case of E. coli CcdB. Both a high-resolution structure and an exhaustive set of Ts mutants is available for the protein. We found that eight Ts mutants correctly predicted with the sequence-based method did not satisfy structure-based burial criteria, and most of these residues are part of the protein active site or are part of a protein-protein interaction interface (23). This suggested that the sequence-based method can be utilized when no structural prediction are forthcoming.
The dependency of structure-based prediction on homology model quality was also investigated. Our results show that structure-based prediction performance was not critically dependent on modeling template as long as they passed the assessment criteria.
As mentioned earlier, the sequence-based method also predicts mutations in positions spatially close to active site and protein-protein interfaces to be temperature sensitive (23). However, our analysis with CcdB reveals that these positions are more selective to mutations as compared to buried positions. It is likely that mutations at positions that are not as deep as core residues are less thermally destabilizing.
Without the availability of large scale and exhaustive studies of Ts mutants, it is not possible to accurately gauge the false positive rate of the method. Hence, a rigorous estimation of sensitivity and specificity of the method is not currently possible. However, for practical purpose, usually one or a few Ts mutants are sufficient to facilitate further investigation of a biological system. Using our testing set, we showed that at least a few high confidence predictions could be made for each case.
Engineering temperature sensitive mutant is a versatile technique that can be used for a variety of purposes as it enables an easy and reversible manipulation of protein function. This server has been set up to provide a simple and intuitive tool to rationally design temperature sensitive mutants. We hope that it will be useful in investigating various biological systems.
Acknowledgments
M.S.M. would like to acknowledge the DBT-Wellcome alliance for a senior fellowship. We express our gratitude to Yong Tai Pang for his support in the setting up and maintenance of the web server.
FUNDING
Funding for open access charge: DBT-Wellcome senior fellowship (to M.S.M.).
Conflict of interest. None declared.
REFERENCES
- 1.Horowitz N.H. Biochemical genetics of neurospora. Adv. Genet. Inc. Mol. Genet. Med. 1950;3:33–71. doi: 10.1016/s0065-2660(08)60082-6. [DOI] [PubMed] [Google Scholar]
- 2.Fried M. Cell-transforming ability of a temperature-sensitive mutant of polyoma virus. Proc. Natl. Acad. Sci. U.S.A. 1965;53:486–491. doi: 10.1073/pnas.53.3.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Couso J., Bate M., Martinez-Arias A. A wingless-dependent polar coordinate system in Drosophila imaginal discs. Science. 1993;259:484–489. doi: 10.1126/science.8424170. [DOI] [PubMed] [Google Scholar]
- 4.Goldenberg D.P., Smith D.H., King J. Genetic analysis of the folding pathway for the tail spike protein of phage P22. Proc. Natl. Acad. Sci. U.S.A. 1983;80:7060–7064. doi: 10.1073/pnas.80.23.7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warner J.R., Udem S.A. Temperature sensitive mutations affecting ribosome synthesis in Saccharomyces cerevisiae. J. Mol. Biol. 1972;65:243–257. doi: 10.1016/0022-2836(72)90280-x. [DOI] [PubMed] [Google Scholar]
- 6.Balakrishnan R., Backman K. Controllable alteration of cell genotype in bacterial cultures using an excision vector. Gene. 1988;67:97–103. doi: 10.1016/0378-1119(88)90012-1. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki D.T., Grigliatti T., Williamson R. Temperature-sensitive mutations in Drosophila melanogaster, VII. A mutation (parats) causing reversible adult paralysis. Proc. Nat. Acad. Sci. U.S.A. 1971;68:890–893. doi: 10.1073/pnas.68.5.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novick P., Botstein D. Phenotypic analysis of temperature-sensitive yeast actin mutants. Cell. 1985;40:405–416. doi: 10.1016/0092-8674(85)90154-0. [DOI] [PubMed] [Google Scholar]
- 9.Hereford L.M., Hartwell L.H. Sequential gene function in the initiation of Saccharomyces cerevisiae DNA synthesis. J. Mol. Biol. 1974;84:445–461. doi: 10.1016/0022-2836(74)90451-3. [DOI] [PubMed] [Google Scholar]
- 10.Hartwell L.H., Culotti J., Reid B. Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc. Natl. Acad. Sci. U.S.A. 1970;66:352–359. doi: 10.1073/pnas.66.2.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bucking-Throm E., Duntze W., Hartwell L.H., Manney T.R. Reversible arrest of haploid yeast cells in the initiation of DNA synthesis by a diffusible sex factor. Exp. Cell Res. 1973;76:99–110. doi: 10.1016/0014-4827(73)90424-2. [DOI] [PubMed] [Google Scholar]
- 12.Wilkinson L.E., Pringle J.R. Transient G1 arrest of S. cerevisiae cells of mating type alpha by a factor produced by cells of mating type a. Exp. Cell Res. 1974;89:175–187. doi: 10.1016/0014-4827(74)90200-6. [DOI] [PubMed] [Google Scholar]
- 13.Mondal K., Dastidar A.G., Singh G., Madhusudhanan S., Gande S.L., VijayRaghavan K., Varadarajan R. Design and isolation of temperature-sensitive mutants of Gal4 in yeast and Drosophila. J. Mol. Biol. 2007;370:939–950. doi: 10.1016/j.jmb.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 14.Mondal K., VijayRaghavan K., Varadarajan R. Design and utility of temperature-sensitive Gal4 mutants for conditional gene expression in Drosophila. Fly. 2007;1:282–286. doi: 10.4161/fly.5251. [DOI] [PubMed] [Google Scholar]
- 15.Dorfman M., Gomes J.E., O’Rourke S., Bowerman B. Using RNA interference to identify specific modifiers of a temperature-sensitive, embryonic-lethal mutation in the Caenorhabditis elegans ubiquitin-like Nedd8 protein modification pathway E1-activating gene rfl-1. Genetics. 2009;182:1035–1049. doi: 10.1534/genetics.109.104885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajagopalan S., Liling Z., Liu J., Balasubramanian M. The N-degron approach to create temperature-sensitive mutants in Schizosaccharomyces pombe. Methods. 2004;33:206–212. doi: 10.1016/j.ymeth.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 18.Kanemaki M., Sanchez-Diaz A., Gambus A., Labib K. Functional proteomic identification of DNA replication proteins by induced proteolysis in vivo. Nature. 2003;423:720–725. doi: 10.1038/nature01692. [DOI] [PubMed] [Google Scholar]
- 19.Dohmen R., Wu P., Varshavsky A. Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science. 1994;263:1273–1276. doi: 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- 20.Zeidler M.P., Tan C., Bellaiche Y., Cherry S., Hader S., Gayko U., Perrimon N. Temperature-sensitive control of protein activity by conditionally splicing inteins. Nat. Biotechnol. 2004;22:871–876. doi: 10.1038/nbt979. [DOI] [PubMed] [Google Scholar]
- 21.Patrick M., Duncan R., Coombs K.M. Generation and genetic characterization of avian reovirus temperature-sensitive mutants. Virology. 2001;284:113–122. doi: 10.1006/viro.2001.0915. [DOI] [PubMed] [Google Scholar]
- 22.Varadarajan R., Nagarajaram H.A., Ramakrishnan C. A procedure for the prediction of temperature-sensitive mutants of a globular protein based solely on the amino acid sequence. Proc. Natl. Acad. Sci. U.S.A. 1996;93:13908–13913. doi: 10.1073/pnas.93.24.13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bajaj K., Dewan P.C., Chakrabarti P., Goswami D., Barua B., Baliga C., Varadarajan R. Structural correlates of the temperature sensitive phenotype derived from saturation mutagenesis studies of CcdB†. Biochemistry. 2008;47:12964–12973. doi: 10.1021/bi8014345. [DOI] [PubMed] [Google Scholar]
- 24.Hopp T.P., Woods K.R. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl. Acad. Sci. U.S.A. 1981;78:3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rose G.D., Roy S. Hydrophobic basis of packing in globular proteins. Proc. Natl. Acad. Sci. U.S.A. 1980;77:4643–4647. doi: 10.1073/pnas.77.8.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisenberg D. Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem. 1984;53:595–623. doi: 10.1146/annurev.bi.53.070184.003115. [DOI] [PubMed] [Google Scholar]
- 27.Chakravarty S., Varadarajan R. Residue depth: a novel parameter for the analysis of protein structure and stability. Structure. 1999;7:723–732. doi: 10.1016/s0969-2126(99)80097-5. [DOI] [PubMed] [Google Scholar]
- 28.Tan K.P., Varadarajan R., Madhusudhan M.S. DEPTH: a web server to compute depth and predict small-molecule binding cavities in proteins. Nucleic. Acids Res. 2011;39:W242–W248. doi: 10.1093/nar/gkr356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan K.P., Nguyen T.B., Patel S., Varadarajan R., Madhusudhan M.S. Depth: a web server to compute depth, cavity sizes, detect potential small-molecule ligand-binding cavities and predict the pKa of ionizable residues in proteins. Nucleic Acids Res. 2013;41:W314–W321. doi: 10.1093/nar/gkt503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parsell D.A., Sauer R.T. The structural stability of a protein is an important determinant of its proteolytic susceptibility in Escherichia coli. J. Biol. Chem. 1989;264:7590–7595. [PubMed] [Google Scholar]
- 31.Pakula A.A., Sauer R.T. Genetic analysis of protein stability and function. Annu. Rev. Genet. 1989;23:289–310. doi: 10.1146/annurev.ge.23.120189.001445. [DOI] [PubMed] [Google Scholar]
- 32.Pakula A.A., Young V.B., Sauer R.T. Bacteriophage lambda cro mutations: effects on activity and intracellular degradation. Proc. Natl. Acad. Sci. U.S.A. 1986;83:8829–8833. doi: 10.1073/pnas.83.23.8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kellis J.T., Nyberg K., Fersht A.R. Energetics of complementary side chain packing in a protein hydrophobic core. Biochemistry. 1989;28:4914–4922. doi: 10.1021/bi00437a058. [DOI] [PubMed] [Google Scholar]
- 34.Shortle D., Stites W.E., Meeker A.K. Contributions of the large hydrophobic amino acids to the stability of staphylococcal nuclease. Biochemistry. 1990;29:8033–8041. doi: 10.1021/bi00487a007. [DOI] [PubMed] [Google Scholar]
- 35.Rose G.D., Geselowitz A.R., Lesser G.J., Lee R.H., Zehfus M.H. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 36.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 38.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braberg H., Webb B.M., Tjioe E., Pieper U., Sali A., Madhusudhan M.S. SALIGN: a web server for alignment of multiple protein sequences and structures. Bioinformatics. 2012;28:2072–2073. doi: 10.1093/bioinformatics/bts302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Šali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 41.Melo F., Sanchez R., Sali A. Statistical potentials for fold assessment. Protein Sci. 2002;11:430–448. doi: 10.1002/pro.110430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.John B., Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen M.Y., Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sandberg W.S., Schlunk P.M., Zabin H.B., Terwilliger T.C. Relationship between in vivo activity and in vitro measures of function and stability of a protein. Biochemistry. 1995;34:11970–11978. doi: 10.1021/bi00037a039. [DOI] [PubMed] [Google Scholar]
- 45.Alber T., Sun D.P., Nye J.A., Muchmore D.C., Matthews B.W. Temperature-sensitive mutations of bacteriophage T4 lysozyme occur at sites with low mobility and low solvent accessibility in the folded protein. Biochemistry. 1987;26:3754–3758. doi: 10.1021/bi00387a002. [DOI] [PubMed] [Google Scholar]
- 46.Hecht M.H., Nelson H.C., Sauer R.T. Mutations in lambda repressor's amino-terminal domain: implications for protein stability and DNA binding. Proc. Natl. Acad. Sci. U.S.A. 1983;80:2676–2680. doi: 10.1073/pnas.80.9.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim W.A., Sauer R.T. Alternative packing arrangements in the hydrophobic core of lambda repressor. Nature. 1989;339:31–36. doi: 10.1038/339031a0. [DOI] [PubMed] [Google Scholar]
- 48.Jakubowska A., Korona R. Lack of evolutionary conservation at positions important for thermal stability in the yeast ODCase protein. Mol. Biol. Evol. 2009;26:1431–1434. doi: 10.1093/molbev/msp066. [DOI] [PubMed] [Google Scholar]
- 49.Chakshusmathi G., Mondal K., Lakshmi G.S., Singh G., Roy A., Ch R.B., Madhusudhanan S., Varadarajan R. Design of temperature-sensitive mutants solely from amino acid sequence. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7925–7930. doi: 10.1073/pnas.0402222101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marti-Renom M.A., Pieper U., Madhusudhan M.S., Rossi A., Eswar N., Davis F.P., Al-Shahrour F., Dopazo J., Sali A. DBAli tools: mining the protein structure space. Nucleic Acids Res. 2007;35:W393–W397. doi: 10.1093/nar/gkm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2009;37:D26–D31. doi: 10.1093/nar/gkn723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leinonen R., Diez F.G., Binns D., Fleischmann W., Lopez R., Apweiler R. UniProt archive. Bioinformatics. 2004;20:3236–3237. doi: 10.1093/bioinformatics/bth191. [DOI] [PubMed] [Google Scholar]
- 53.Consortium T.U. Ongoing and future developments at the universal protein resource. Nucleic Acids Res. 2011;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 55.Tsai C.J., Lin S.L., Meng E.C., Nussinov R. Studies of protein-protein interfaces: a statistical analysis ofthe hydrophobic effect. ProteinSci. 1997;6:53–64. doi: 10.1002/pro.5560060106. [DOI] [PMC free article] [PubMed] [Google Scholar]