

Abstract
Immune adult horses have CD8+ cytotoxic T lymphocytes (CTLs) that recognize and lyse Rhodococcus equi-infected cells in an equine lymphocyte alloantigen (ELA)-A [classical major histocompatibility complex (MHC) class I]-unrestricted fashion. As protein antigens are MHC class I-restricted, the lack of restriction suggests that the bacterial antigens being recognized by the host are not proteins. The goals of this study were to test the hypothesis that these CTLs recognize unique R. equi cell-wall lipids related to mycobacterial lipids. Initial experiments showed that treatment of soluble R. equi antigen with broadly reactive proteases did not significantly diminish the ability of the antigen to stimulate R. equi-specific CTLs. R. equi-specific CTLs were also shown to lyse target cells (equine macrophages) pulsed with an R. equi lipid extract. Analysis of the R. equi lipid by TLC and MS (MALDI-TOF and ES) indicated that the extracted antigen consisted of three primary fractions: trehalose monomycolate (TMM), trehalose dimycolate (TDM) and cardiolipin (CL). ELA-A-mismatched cells pulsed with purified TMM and CL, but not the TDM fraction, were recognized and lysed by R. equi-specific CTLs. Because of their role in immune clearance and pathogenesis, transcription of the cytokines gamma interferon (IFN-γ) and interleukin-4 (IL-4) was also measured in response to R. equi lipids by using real-time PCR; elevated IFN-γ, but not IL-4, was associated with host clearance of the bacteria. The whole-cell R. equi lipid and all three R. equi lipid fractions resulted in marked increases in IFN-γ transcription, but no increase in IL-4 transcription. Together, these data support the hypothesis that immune recognition of unique lipids in the bacterial cell wall is an important component of the protective immune response to R. equi. The results also identify potential lipid antigens not previously shown to be recognized by CTLs in an important, naturally occurring actinomycete bacterial pathogen.
Introduction
Rhodococcus equi is a Gram-positive and weakly acid-fast nocardioform actinomycete bacterium that is related closely to Mycobacterium tuberculosis (Rahman et al., 2003). The organism is found as a soil saprophyte in nearly all environments and can replicate where herbivore manure provides the necessary nutrients (Takai, 1997). R. equi is also a facultative intracellular pathogen that survives within macrophages and replicates within membrane-bound vesicles. The disease manifests primarily as pyogranulomatous pneumonia, producing lesions similar to tuberculosis (Hondalus, 1997; Takai, 1997). In general, only foals younger than about 4–6 months of age, immunocompromised horses and immunosuppressed humans are susceptible (Hondalus, 1997; Perez et al., 2002; Takai, 1997; Torres-Tortosa et al., 2003). As a result of human immunodeficiency virus/AIDS, the use of immunosuppressive drugs in transplantation procedures and the immunosuppressive side effects of cancer chemotherapy, the number of susceptible humans is rising (Linder, 1997; Zinner, 1999).
In young horses, rhodococcal pneumonia represents a common, severely debilitating problem that causes significant morbidity and mortality (Hondalus, 1997; Takai, 1997). Essentially, all foals are exposed to R. equi early in life. Most clear the infection without clinical signs or recover from the disease, and go on to become immune adults. Even on endemic farms, where exposure to virulent strains is presumably high and a significant percentage of foals develop severe pulmonary lesions, adult horses remain unaffected. Immune adult horses represent, therefore, a system by which protective immune mechanisms can be studied (i.e. the correlates of immunity can be defined). An important long-term goal of these studies is a vaccination strategy that would induce protective responses early in life and thereby prevent pneumonia in all foals.
Although immune control of R. equi infection is complex, with both antibody and T lymphocytes playing roles, the intracellular location of the bacteria within infected macrophages dictates that a cell-mediated response is most significant. As with M. tuberculosis, the protective cell-mediated immune response encompasses both CD4+ and CD8+ T lymphocytes and the secretion of gamma interferon (IFN-γ) (Grotzke & Lewinsohn, 2005; Hines et al., 2001; Kanaly et al., 1993, 1995, 1996; Patton et al., 2004; Stenger & Modlin, 1999). IFN-γ, which activates macrophages and upregulates phagolysosomal fusion, appears critical. Antigen-specific T helper 1-like (TH1) CD4+ T lymphocytes are the predominant producers of IFN-γ and appear to be absolutely required for immune clearance (Hines et al., 2003; Kanaly et al., 1993, 1995, 1996). CD8+ T lymphocytes can also produce IFN-γ, but probably have a more substantial role as cytotoxic T lymphocytes (CTLs) (Hines et al., 2003; Patton et al., 2004).
Recently, our laboratory demonstrated that all immune adult horses tested have R. equi-specific cytotoxic effector cells. These studies showed that the effector cells are primarily CD8+CD4− T lymphocytes, require antigen stimulation and lyse R. equi-infected cells in a classical major histocompatibility complex (MHC) class I [equine lymphocyte alloantigen (ELA)-A]-unrestricted manner (Patton et al., 2004). Moreover, depletion studies using anti-CD3 and anti-CD2 antibodies showed that the effector cells are CTLs bearing T-cell receptors (TCRs), and not natural killer (NK) cells. Importantly, the R. equi-specific, ELA-A-unrestricted CTLs were shown not to be present when foals are most susceptible to rhodococcal pneumonia (i.e. the first few weeks of life) and then appeared as foals matured – presumably reflecting a developing adaptive immune response (Patton et al., 2005). Whilst this work defined the lymphocyte population involved in a potentially protective immune response in horses and showed a correlation between the presence of CTLs and age-associated susceptibility, it did not define the bacterial antigen(s) involved.
One possible explanation for the MHC class I-unrestricted killing is that the antigens presented by infected cells and recognized by CTLs are not peptides. Specifically, based on the similarity to M. tuberculosis, an important consideration is presentation of bacterial lipid antigen by CD1 molecules (Karakousis et al., 2004; Moody & Porcelli, 2003; Ulrichs et al., 2003). Members of the CD1 receptor family are MHC-like molecules that have a specialized polar binding groove that binds lipid (Brigl & Brenner, 2004; Hava et al., 2005; Roura-mir & Moody, 2003). In humans, CD1-restricted T lymphocytes recognize lipid antigens of M. tuberculosis via their TCRs and are postulated to play an important role in immunity (Boom et al., 2003; Li et al., 1996; Szereday et al., 2003).
In this report, we tested the hypothesis that the R. equi antigens recognized by CTLs from immune adult horses are not peptides, but bacterial lipids. The antigen recognized by equine CTLs was further fractionated by TLC and characterized by MALDI-TOF MS and ES-MS analyses. We also investigated the ability of TLC-purified R. equi lipids to stimulate production of IFN-γ and interleukin-4 (IL-4). This work addresses the role of immune recognition of bacterial lipid antigens in protecting against an important, naturally occurring disease of domestic animals.
Methods
Horses.
Adult horses of various breeds, currently maintained in the Washington State University Veterinary Teaching Hospital herd, were selected based on MHC haplotype and used in accordance with the Washington State University institutional animal care and use committee. Using previously described serological reagents, equine ELA-A MHC class I haplotypes were determined by the microcytotoxicity assay (Zhang et al., 1998). Venous blood was collected from the jugular vein of each horse using 500 ml evacuated containers (Baxter) containing 80 ml Anticoagulant Citrate Dextrose A (Baxter) per 420 ml blood. Peripheral blood mononuclear cells (PBMCs) were isolated from venous blood using the Ficoll–Hypaque technique. PBMCs were then used either as effector cells or for isolation of peripheral blood adherent cells (PBACs). PBACs are synonymously referred to as monocyte-derived macrophage target cells.
Bacteria.
The plasmid-bearing virulent R. equi strain ATCC 33701 expressing the protein VapA was utilized in the experiments and grown using previously published methods (Patton et al., 2004). The number of bacteria ml−1 was estimated with an A600 reading of 0.050 (Beckman DU-64), equivalent to 1.5×108 R. equi cells ml−1. The bacterial concentration was confirmed by plating serial dilutions on brain–heart infusion (BHI; Becton Dickinson) plates and calculating the c.f.u. ml−1.
Preparation of protease-treated soluble R. equi antigen (SRA).
SRA was prepared using previously published methods (Patton et al., 2004) and the protein concentration was identified using a bicinchoninic acid (BCA) protein assay kit (Pierce). Briefly, a set of dilutions using 2.0 mg BSA ml−1 (Pierce) and a series of SRA dilutions were incubated with the BCA reagents and absorbance was measured at 550 nm (Thermo Electron Multiscan MCC). A standard curve was generated by plotting the known albumin protein concentrations against the measured light absorbances, and the protein content of the SRA was identified by comparison against the standard curve. The SRA was then digested by proteinase K (Fermentas) using a 1 : 5 ratio of protease : SRA protein, measured in mg, based on the method of Beckman et al. (1994). The digestion was conducted in a shaking water bath for 14 h at 37 °C and 200 r.p.m., followed by heat inactivation of the enzyme in a water bath for 15 min at 65 °C. Subsequently, the sample was digested with subtilisin (Sigma-Aldrich) in an identical manner. Remnant peptides were removed by dialysing the sample in PBS against a 10 000 Da molecular mass cut-off membrane (Pierce). The sample was then run on a polyacrylamide gel and evaluation for protein was done as per the manufacturer’s directions using a SilverSNAP Stain kit II (Pierce). SRA and the aforementioned proteases were used as positive and negative controls, respectively.
Extraction and purification of R. equi lipid.
One colony of R. equi ATCC 33701 was inoculated into 10 ml BHI broth and incubated at 37 °C with shaking for 8 h. The inoculum was transferred into 1 l BHI broth and incubated with shaking for an additional 3–5 days. The bacteria were collected by centrifugation at 15 000 g</italic> for 15 min at 4 °C. The pellets were then washed twice in PBS. The process was repeated until a 20 ml bacterial pellet was obtained. The cell-wall lipids were then extracted using previously published methods (Beckman et al., 1994; Folch et al., 1957). Briefly, the pellet was resuspended in 20 vols chloroform/methanol (2 : 1, v/v) and incubated overnight at 37 °C with shaking. A 20 % volume of sterile 0.9 % NaCl (Abbott Laboratories) was added to the suspension and incubated for 2 h at 37 °C with shaking. The sample was then allowed to settle into a top aqueous layer, middle insoluble layer and bottom organic layer. The organic layer was collected and dried on a rotary evaporator. The sample was weighed and then resuspended in PBS, with sonication, at a concentration of 10 µg ml−1. Individual 2 ml aliquots were stored at −20 °C. The fractions, consisting of trehalose 6-monomycolate (TMM), trehalose 6,6′-dimycolate (TDM) and cardiolipin (diphosphatidylglycerol; CL), were identified as follows.
Following extraction of R. equi lipids, the glycolipid and phospholipid components were purified as described previously (Rao et al., 2005; Ueda et al., 2001). Briefly, TMM, TDM and CL were purified completely by preparative TLC on silica gel G (Uniplate; 20×20 cm, 250 µm; Analtech) until a single spot was obtained. The TLC plate was developed repeatedly with chloroform/methanol/acetone/acetic acid (90 : 10 : 6 : 1 and 80 : 20 : 6 : 1, v/v) and chloroform/methanol/water (90 : 10 : 1, v/v). After exposure of the TLC plate to iodine vapour, the lipid bands were marked and then the silica gel was scraped off. TMM, TDM and CL were eluted with chloroform/methanol (2 : 1, v/v). The final products were weighed and the purity and quantity were examined by TLC. Products were visualized by spraying with 20 % H2SO4 in ethanol and charring for 3 min at 180 °C, and CL was detected by Dittmer’s reagent (Dittmer & Lester, 1964).
Rhodococcus terrae OCU 70014 cell-wall glycolipids, consisting of mannose monomycolate (MMM), glucose monomycolate (GMM), TDM, fructose monomycolate (FMM) and TMM, were extracted and purified similarly using previously established methods (Natsuhara et al., 1990; Ueda et al., 2001).
MALDI-TOF MS analysis.
The molecular species of the intact TMM and TDM were detected by MALDI-TOF MS with an Ultraflex II (Bruker Daltonics). The samples were dissolved in chloroform/methanol (2 : 1, v/v) at a concentration of 1 mg ml−1, and 1 µl was applied directly to the sample plate, followed by addition of 1 µl 2,5-dihydroxybenzoic acid [10 mg ml−1 in chloroform/methanol (1 : 1, v/v)] as a matrix. It was analysed in the reflectron mode with an accelerating voltage operating in positive mode of 20 kV (Bhatt et al., 2007).
ES-MS analysis.
For negative-ion MS, CL was converted to the ammonium salt form as described by Catucci et al. (2004). The molecular species of CL was detected by ES-MS with an LCQ (Thermo Fisher Scientific). ES-MS was carried out by infusion mode, and the conditions were as follows: flow rate, 10 µl min−1; collision energy, 35 %. The CL was dissolved in methanol at a concentration of 0.2 mg ml−1 and was analysed in the negative mode.
Effector-cell preparation and stimulation.
In total, 108 PBMCs in antibiotic-free complete medium (Hyclone) were plated in 75 cm2 flasks at 4×106 cells ml−1 in 25 ml medium. The cells were stimulated with one of the following: (i) 6×106 R. equi ATCC 33701 cells (approx. 0.3 m.o.i. in monocytes, approximately 20 % monocytes in PBMCs), (ii) 2 µg SRA ml−1, or (iii) 2 µg protease-digested SRA ml−1. In the experiments using whole R. equi, following 1 h incubation at 37 °C with 5 % CO2, 0.05 mg gentamicin ml−1 (Sigma-Aldrich) was added to kill extracellular bacteria. Culture flasks were then incubated for 5 days at 37 °C with 5 % CO2, followed by resting for 2 days without antigenic stimulation. On day 5 of the incubation, the culture flasks were examined with an inverting microscope to confirm lymphocyte proliferation around PBACs and to assess for loss of macrophages. At the low m.o.i. and with the continued presence of extracellular antibiotics, the loss of macrophages due to necrosis was minimal. Resting consisted of collecting the non-adherent cells from the flasks and washing them in antibiotic complete medium by centrifugation (600 g at 4 °C), followed by resuspending the cells in 25 ml fresh medium and incubating at 37 °C with 5 % CO2. Immediately prior to use in CTL assays, the effector cells were washed once in antibiotic-free complete medium and counted.
Target-cell isolation.
ELA-A-mismatched PBACs were harvested from PBMCs for use as target cells as described previously (Patton et al., 2004). Briefly, 150 cm2 Petri dishes were coated with 2 % sterile gelatin (J. T. Baker) by incubating the dishes for 2 h at 37 °C with 5 % CO2. The gelatin was then removed and the dishes were air-dried for 30 min. The dishes were next coated with 6 ml heat-inactivated horse serum (Invitrogen) by incubation for 30 min at 37 °C with 5 % CO2. Serum was removed and PBMCs were plated on dishes coated with gelatin and serum at a density of 1.5×108 cells per dish in 20 ml antibiotic-free complete medium. Cells were incubated for 15–19 h at 37 °C with 5 % CO2. Following incubation, non-adherent cells were resuspended in the culture medium by repeated pipetting (10 times) and then removed by washing each plate with 10 ml Hanks’ salt solution (Hyclone) and repeated pipetting (10 times). Adhered cells were eluted with 20 ml antibiotic-free complete medium containing 20 % heat-inactivated horse serum mixed 1 : 1 (v/v) with 10 mM EDTA (J. T. Baker) for 25 min at 37 °C with 5 % CO2. The cells were then collected by vigorous pipetting of the eluting fluid 10 times, and subsequently each dish was washed twice with 10 ml Hanks’ salt solution per wash. Eluted cells were combined, washed once with Hanks’ salt solution and suspended in 5 ml antibiotic-free complete medium. The eluted cells were counted and added to 96-well plates at 105 cells per well in 100 µl antibiotic-free complete medium. Thirty minutes prior to adding the cells, 20 µl commercial heat-inactivated horse serum was added to each well.
Cytotoxicity assay.
Each well of target cells was labelled with 50 µl antibiotic-free complete medium containing 100 µCi (3.7×109 Bq) 51Cr ml−1 (PerkinElmer). Following 15 h incubation, the targets were: (i) infected with live R. equi by adding 50 µl antibiotic-free complete medium containing 107 live R. equi cells ml−1, (ii) pulsed with R. equi lipid extract by adding 100 µl antibiotic-free complete medium containing 250 µg sonicated lipid ml−1, or (iii) pulsed with purified lipid components by adding 100 µl antibiotic-free complete medium containing 250 µg sonicated TDM, CL or TMM ml−1. One hour post-R. equi infection, 20 µl complete medium containing 0.05 mg gentamicin sulfate ml−1 was added to each culture well. Following an additional 7 h incubation, the cells were washed four times with complete medium containing 0.05 mg gentamicin sulfate ml−1 to remove extracellular 51Cr. Effector cells were then added to target cells and incubated for an additional 4 h at 37 °C with 5 % CO2. Following incubation, 50 µl supernatant was removed and the amount of 51Cr was measured by using a MicroBeta plate reader (PerkinElmer). The following formula for percentage specific lysis was used: [(E−S)/(T−S)]×100, where E is the mean of three test wells, S is the mean spontaneous release from three target cell wells without effector cells, and T is the mean total release from three target cell wells with 2 % Triton X-100 (Sigma-Aldrich). As reported previously, significant lysis was defined as 3 sem above the negative-control value (McGuire et al., 1994; Patton et al., 2004, 2005; Siliciano et al., 1985).
Cytokine PCR.
In total, 3×107 PBMCs in antibiotic-free complete medium were plated in 25 cm2 flasks at 4×106 cells ml−1. The cells were stimulated with one of the following: (i) 2×106 live R. equi ATCC 33701 cells, (ii) 800 µg R. equi lipid extract, or (iii) 800 µg purified lipid components TDM, CL or TMM. In the experiments using whole, live R. equi, following a 1 h incubation at 37 °C with 5 % CO2, 0.05 mg gentamicin ml−1 was added to kill extracellular bacteria. Culture flasks were then incubated for 18 h at 37 °C with 5 % CO2. The PBMCs were centrifuged and RNA was extracted from the pellets by using an RNeasy Plus mini kit (Qiagen).
To eliminate contaminating DNA, RNA was treated with Turbo DNase (Ambion Applied Biosystems) for 30 min at 37 °C following the manufacturer’s instructions. The concentration of RNA in samples was estimated by using A260 and A280 (Beckman DU-64). RNA was converted to cDNA in a two-step process. For the first step, 1 µg RNA, 3.3 µM random hexamers (Applied Biosystems) and 0.67 mM dNTPs (Applied Biosystems) were mixed with water to a total volume of 15 µl, and the sample was incubated for 5 min at 70 °C. For the second step, the entire sample was mixed with 5 µl buffer, 4 mM DTT (Invitrogen), 2 mM MgCl2, 20 U RNase inhibitor (Applied Biosystems) and 200 U reverse transcriptase (Promega), and incubated at 37 °C for 60 min. To confirm that no genomic DNA was present, portions of the DNase-treated RNA samples were used in cDNA reaction mixtures without reverse transcriptase.
The cDNA was then used to detect expression of IFN-γ and IL-4. Expression of equine IFN-γ, IL-4 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in cells was determined by performing real-time PCR with previously described primers (Lopez et al., 2002). Briefly, the fluorescent dye SYBR Green (Bio-Rad Laboratories) was used to measure amplified products in a thermocycler (iCycler; Bio-Rad Laboratories). Standard reactions were performed in 25 µl volumes using cDNA diluted 1 : 5 in a master mix containing 2× SYBR Green, 4 µM primer and 1 mM MgCl2. Standard curves were generated by using a plasmid diluted 1 : 5 with the master mix. All standard curves were run in duplicate and all unknowns in triplicate, and amplification was performed using the following procedure: 2 min at 50 °C and 10 min at 95 °C, followed by 45 cycles of 95 °C for 30 s and 55 °C for 60 s, and then one cycle of 72 °C for 7 min. Transcript levels were determined for each sample by comparing the threshold cycle values of IFN-γ, IL-4 and GAPDH with the corresponding standard curves. The transcript levels were normalized by dividing the copy numbers of IFN-γ and IL-4 by that of GAPDH. In each real-time PCR, reaction mixtures containing no cDNA template were included to control for extraneous DNA within the reagents. To determine statistical significance, the normalized copy numbers of IFN-γ and IL-4 were transformed logarithmically and analysed in a one-way ANOVA, using the mean value of the three horses. In instances where P<0.05, t-tests were run to determine whether the R. equi lipid and the lipid components differed from the control or live R. equi values.
Results
Protease sensitivity of R. equi antigen that stimulates CTLs
T lymphocytes from immune adult horses have previously been shown to effectively lyse R. equi-infected macrophages in an MHC class I-unrestricted manner (Patton et al., 2004). Effector cells were CD8+CD3+ CTLs and required antigen stimulation (typically a 3–5 day stimulation with live R. equi or SRA) to produce significant killing of infected target cells in the standard assay. To test whether the antigen(s) that stimulated R. equi-specific CTLs was protein or non-protein, SRA was digested sequentially with two broadly reactive proteases, proteinase K and subtilisin. The total protein in the protease-digested SRA was reduced to near that of a protease-treated PBS negative control. To confirm that the residual protein in the digested SRA was only heat-inactivated protease, the digested antigen was characterized via silver-stained SDS/PAGE gels and immunoblot for the presence of protein bands and virulence-associated plasmid A (VapA), respectively. When compared against control SRA and sonicated R. equi by silver staining, the protease-treated SRA lacked comparable banding, indicating complete digestion. Rare low-mass residual bands that remained matched the protease-alone controls (see Supplementary Fig. S1, available with the online version of this paper). Immunoblots of the protease-treated SRA demonstrated complete to near-complete loss of antigen, including a faint to absent 17–20 kDa band corresponding to the abundant, immunodominant VapA lipoprotein (data not shown).
When used to stimulate equine PBMCs, the protease-treated SRA induced CTLs that effectively lysed R. equi-infected, MHC class I-mismatched macrophages (Fig. 1a). The lysis was statistically significant compared with uninfected cells, but not significantly different from effector cells stimulated with the untreated SRA control (Fig. 1b). Increasing the ratio of effector cells to target cells resulted in increased target-cell lysis, as determined by release of chromium. These data suggested that the R. equi antigen(s) recognized by equine CTLs were not primarily proteins.
Fig. 1.
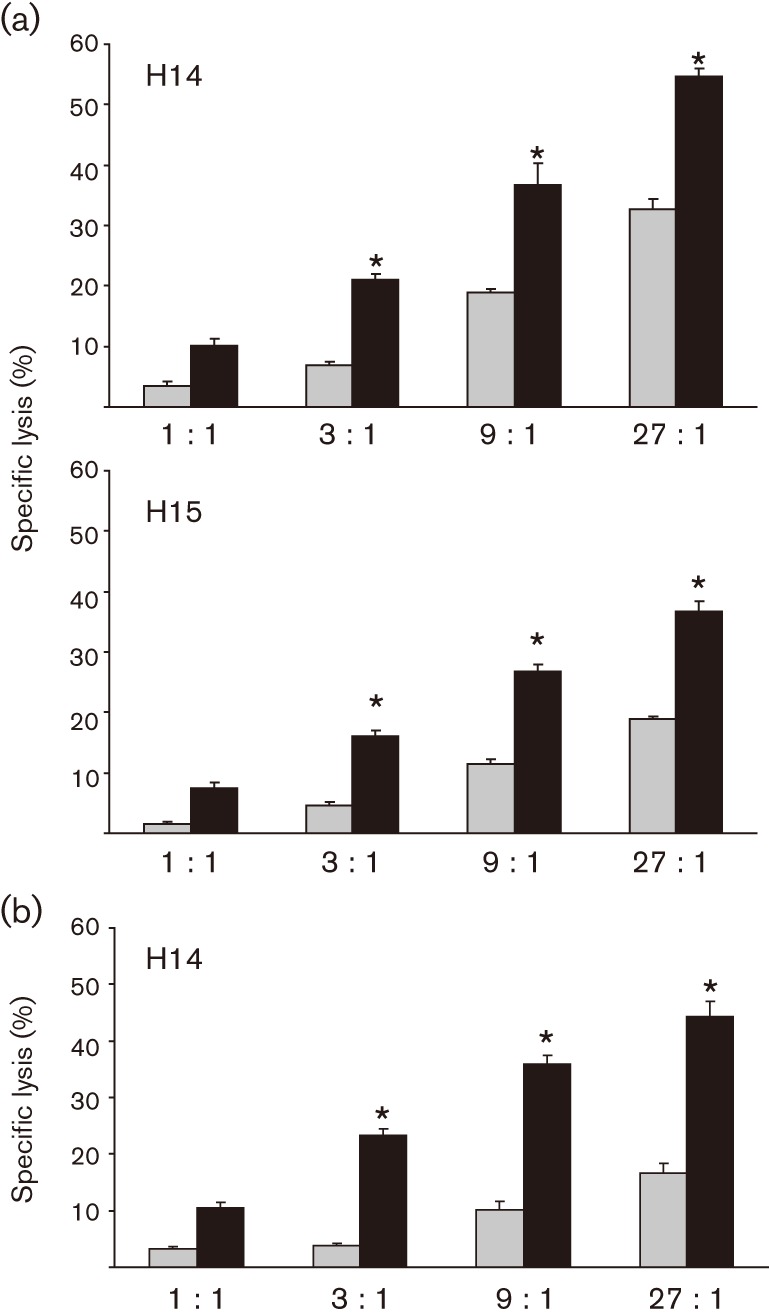
Antigen that stimulates R. equi-specific, MHC class I-unrestricted CTLs is not protease-sensitive. PBMCs from immune adult horses were stimulated for 5 days with 2 µg protease-treated SRA ml−1 (a) or untreated SRA (b), rested for 2 days and then tested for CTL activity. Target cells were infected (black bars) with 5 m.o.i. R. equi ATCC 33701 for 9 h prior to the addition of effector cells at effector : target (E : T) ratios of 1 : 1, 3 : 1, 9 : 1 and 27 : 1. An asterisk indicates statistically significant lysis of target cells [>3 sem compared with the corresponding uninfected control (grey bars)]. Animal identification numbers are H14 and H15. These results were repeated in a total of four (a) and two (b) independent experiments.
R. equi-stimulated effector cells recognize and lyse lipid-pulsed target cells
R. equi ATCC 33701 lipid antigen was prepared by using a chloroform/methanol extraction method. The antigen was characterized by silver stain and immunoblot, and was indistinguishable from protease-digested SRA sample run as a control. In initial experiments, R. equi lipid was used to stimulate equine PBMCs and the resulting CTLs were tested for the ability to recognize and kill infected cells (as in previous experiments). Initial stimulation by bacterial lipids rather than live R. equi was selected to allow expansion of a population of CTLs that would be specific for lipid. Whilst this demonstrated significant killing of R. equi-infected target cells, background killing was also high, due to apparent carryover of lipid antigen to uninfected cells in the CTL assay (data not shown). To address this problem, effector cells were stimulated instead for 5 days with R. equi ATCC 33701, rested for 2 days, and then tested for the ability to recognize and lyse target cells that had been pulsed with lipid antigen. Controls included PBMCs that had been cultured under identical conditions, but not stimulated with R. equi. As in previous studies, lysis of lipid-pulsed targets was compared with that of uninfected cells not exposed to R. equi antigen. Preliminary serial dilution experiments showed that an R. equi lipid concentration of 250 µg ml−1 was optimal for pulsing macrophages in the CTL assay, and is similar to the 200 µg ml−1 lipid concentration utilized with similar experiments in Mycobacterium (Beckman et al., 1994). As demonstrated in Fig. 2, R. equi-specific CTLs produced significant lysis of lipid-pulsed, MHC class I-mismatched target cells. As in previous experiments, PBMCs not stimulated with R. equi did not produce significant lysis. These data suggest strongly that the antigen recognized by CTLs is lipid in nature. Because this antigen is a mixture of bacterial lipids and as a number of mycobacterial lipids and glycolipids have been shown to be presented via the CD1 antigen-presenting system (Bricard & Porcelli, 2007; Willcox et al., 2007), we sought to fractionate and characterize the recognized lipids further.
Fig. 2.
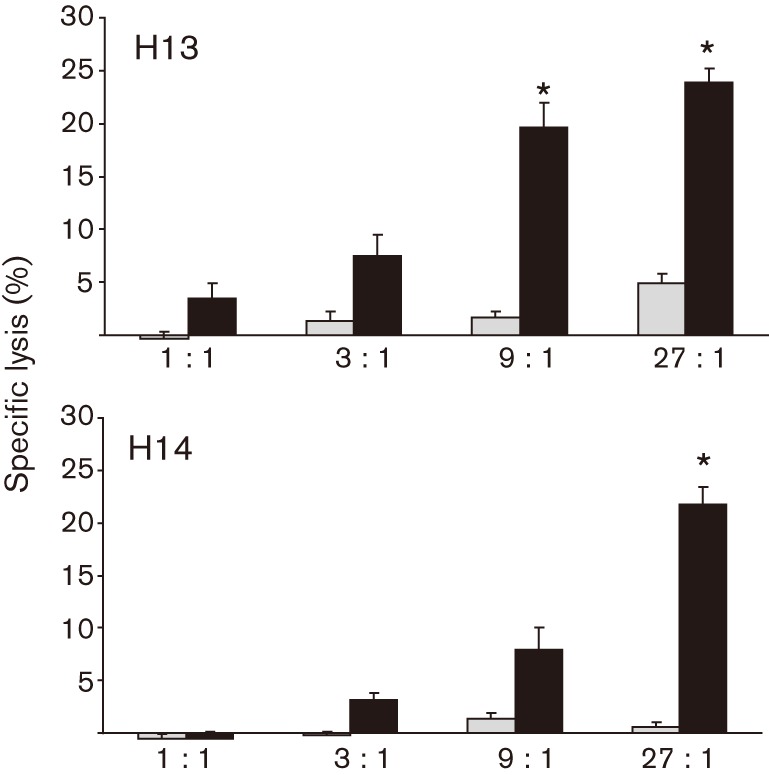
R. equi-specific CTLs recognize and lyse MHC class I-mismatched target cells pulsed with R. equi cell-wall lipid. PBMCs were stimulated for 5 days with live, virulent R. equi ATCC 33701, then rested for 2 days before testing in the CTL assay. Target cells (equine macrophages) were pulsed (black bars) with R. equi lipid for 9 h prior to the addition of effector cells at the indicated E : T ratios of 1 : 1, 3 : 1, 9 : 1 and 27 : 1. An asterisk indicates statistically significant lysis of target cells [>3 sem compared with the corresponding non-pulsed target-cell control (grey bars)]. Two representative animals (H13 and H14) are shown. These results have been confirmed in two additional experiments.
Purification and structural definition of R. equi cell-wall lipids
Two mycoloyl glycolipids, TMM and TDM, and the phospholipid CL were detected as the major lipid components on the developing TLC, and were purified by preparative TLC. All spots were visualized by spraying with 20 % H2SO4 and charring, and only CL containing a phospho group was detected as a spot of blue colour by Dittmer’s reagent (Fig. 3). Following TLC, the structures of the lipids were assigned by MS. R. equi produced only α-mycolic acid. The carbon-chain lengths of mycolic acid species were around C30–44, and the predominant mycolic acids were C32 : 0, C34 : 0 and C36 : 0. The molecular masses of TMM and TDM were measured by MALDI-TOF MS, and the main molecular-related ions were detected at m/z 871, 899 for TMM, and 1378, 1406 for TDM, respectively (Fig. 4a, b). These molecular masses completely matched the composition of mycolic acids. As for the CL, fatty acids were predominantly composed of C12 : 0, C14 : 0, C16 : 0, C18 : 0 and 10-methyl C18 : 0 (data not shown). The ES-MS spectrum of CL showed the m/z 1436 for [M−H]−, which was composed of the two C16 : 0 and two 10-methyl C18 : 0 fatty acids (Fig. 4c). Taken together, the structures of TMM, TDM and CL from R. equi ATCC 33701 are proposed in Fig. 5.
Fig. 3.
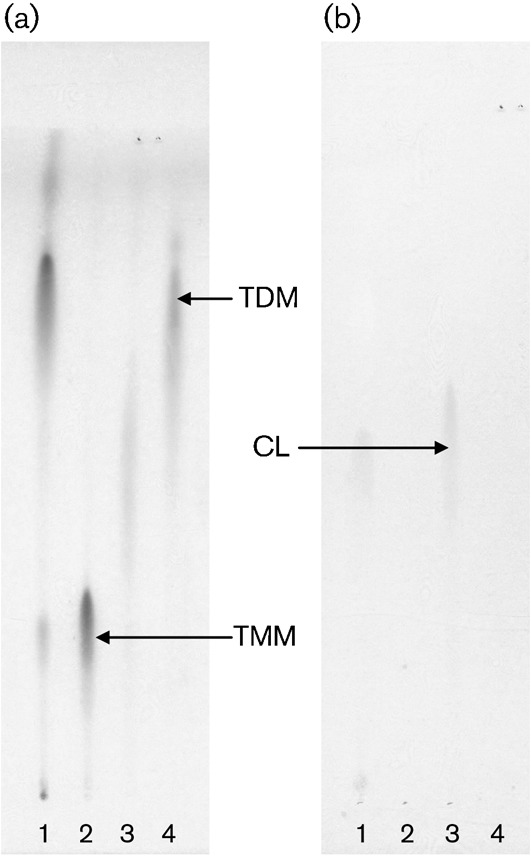
TLC analysis of purified TMM, TDM and CL derived from R. equi ATCC 33701. Unfractionated R. equi lipid (lane 1) and purified TMM, CL and TDM (lanes 2–4, respectively) were developed on TLC plates with the solvent system of chloroform/methanol/acetone/acetic acid (80 : 20 : 6 : 1, v/v), and detected with 20 % H2SO4 (a) or Dittmer’s reagent (b).
Fig. 4.

Mass spectra of TMM, TDM and CL derived from R. equi ATCC 33701. (a, b) R. equi TMM (a) and TDM (b) were acquired by MALDI-TOF MS using 10 mg 2,5-dihydroxybenzoic acid ml−1 in chloroform/methanol (1 : 1, v/v) as a matrix, and the molecular-related ions were detected as [M+Na]+ in positive mode. (c) R. equi CL was acquired by ES-MS and the molecular-related ions were detected as [M−H]− in negative mode. a.u., Arbitrary units.
Fig. 5.
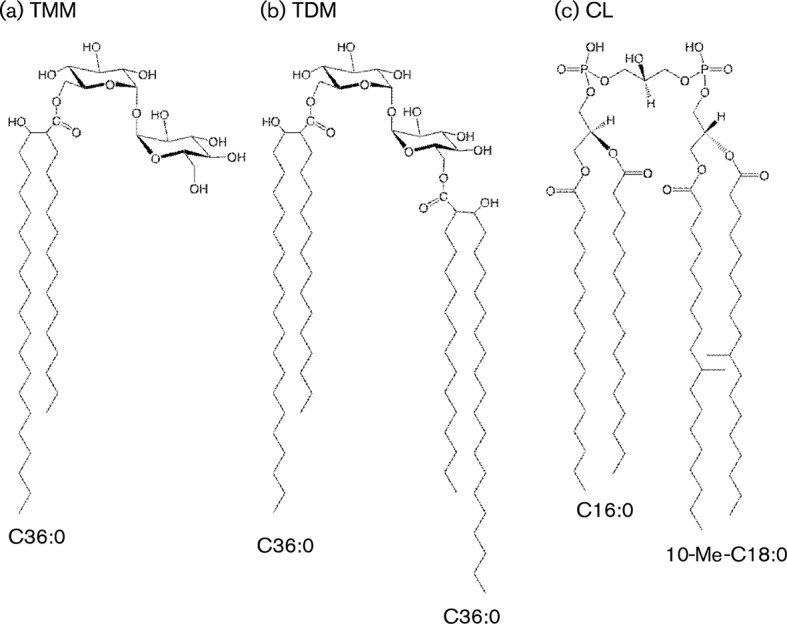
Proposed structures of R. equi TMM, TDM and CL. (a) R. equi TMM: trehalose 6-monomycolate, C48H92O13, exact mass 876.65 Da, molecular mass 877.24 Da. (b) R. equi TDM: trehalose 6,6′-dimycolate, C84H162O15, exact mass 1411.19 Da, molecular mass 1412.18 Da. (c) R. equi CL: cardiolipin, C79H154O17P2, exact mass 1437.07 Da, molecular mass 1438.01 Da.
The three purified lipid fractions were used to pulse MHC class I-mismatched target cells in the CTL assay as described above for unfractionated R. equi lipid. In all three horses, there was statistically significant lysis of the TMM- and CL-pulsed PBACs (Fig. 6), which was similar to the level of the R. equi lipid-pulsed positive control, indicating that they were recognized by CTLs. Lysis of the TDM-pulsed PBACs was indistinguishable from the non-pulsed negative control. The lack of CTLs from the TDM-pulsed samples indicates that the lysis is specific to individual R. equi lipid fractions, rather than being a generalized occurrence to R. equi cell-wall lipids. An additional CTL assay using purified MMM, GMM, TDM, FMM and TMM from the non-pathogenic bacterium R. terrae failed to produce statistically significant lysis in a similar system, further confirming that the cytotoxicity was specific for R. equi lipids (data not shown). The TDM and TMM of R. equi and R. terrae are not identical lipids and have different carbon-chain lengths of their mycolic acids (Natsuhara et al., 1990).
Fig. 6.
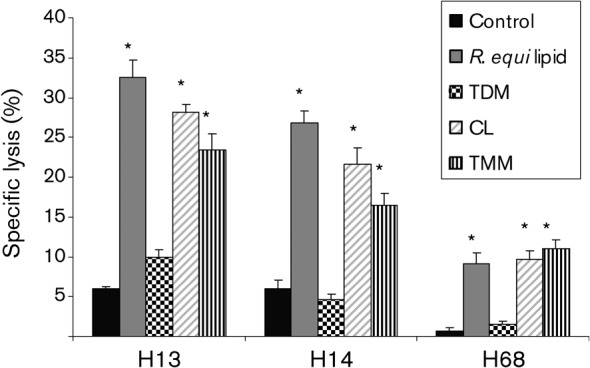
CTLs from immune horses recognize and lyse macrophages pulsed with R. equi TMM and CL, but not TDM. PBMCs (from horses #H13, H14 and H68) were stimulated for 5 days with live, virulent R. equi, then rested for 2 days. Target cells were pulsed with unfractionated R. equi lipid or fractionated lipid antigens for 9 h prior to the addition of rested effectors at an E : T ratio of 30 : 1. An asterisk indicates statistically significant lysis of target cells (>3 sem compared with the corresponding uninfected, non-antigen-pulsed control). These graphs represent a single experiment repeated in three animals.
Cytokine production by PBMCs in response to R. equi lipid antigen
A predominant method by which T lymphocytes protect against intracellular pathogens is the production of IFN-γ, which functions in macrophages to activate phagolysosomal fusion and produce reactive oxygen intermediates. To determine whether immune recognition of R. equi lipid antigens contributes to IFN-γ production in immune horses, PBMCs from adult horses were stimulated with R. equi cell-wall lipids or the fractionated lipid components TMM, TDM and CL. Transcription of IFN-γ was determined by real-time PCR. Because T-helper 2 (TH2) responses, as defined by high IL-4 and low IFN-γ transcription, are associated with an inability to clear R. equi infection and the production of characteristic lesions, IL-4 transcription was also measured. Identical cell cultures utilizing live, virulent R. equi were performed simultaneously as positive controls. The total numbers of IFN-γ and IL-4 transcripts in PBMCs from each horse were normalized against GAPDH, a constitutively expressed housekeeping gene. Data are displayed as fold increase in copy number compared with transcription of IFN-γ and IL-4 in unstimulated cells (medium alone) (Fig. 7). In all three immune adult horses, stimulation of PBMCs with R. equi lipid increased the transcription of IFN-γ to a level that matched the positive controls (i.e. the increase matched the increase in IFN-γ produced by infection of PBMCs with live, virulent R. equi). Increased transcription of IFN-γ occurred in response to all three lipid fractions, including TDM. Subsequent statistical analysis confirmed that the 100–1000-fold increase in IFN-γ is probably relevant. Each data point in Fig. 7 reflects the mean of triplicates from a single experiment, and was run across a population of three horses. After normalization of the copy number against GAPDH, the data points were transformed logarithmically and the means of the horses were analysed in a one-way ANOVA; the ANOVA with IFN-γ had P = 0.035. Subsequently, t-tests were run to determine differences between the lipid antigens and the controls. The t-tests demonstrated that, for IFN-γ, the live R. equi (positive control) and all of the lipids were different from the medium alone (negative control), with P<0.05, and none of the lipids were different from the live R. equi. None of the antigen treatments increased transcription of IL-4 significantly relative to the controls, and an ANOVA confirmed that lack of difference, with P = 0.935. To determine whether the production of IFN-γ was specific to R. equi lipids, a similar assay was done using GMM and FMM cell-wall lipids from R. terrae. Although there was significant production of IFN-γ, the increase compared with baseline was <10 % of the increase produced by the R. equi cell-wall lipid control (data not shown). Again, there was no significant increase in the transcription of IL-4.
Fig. 7.
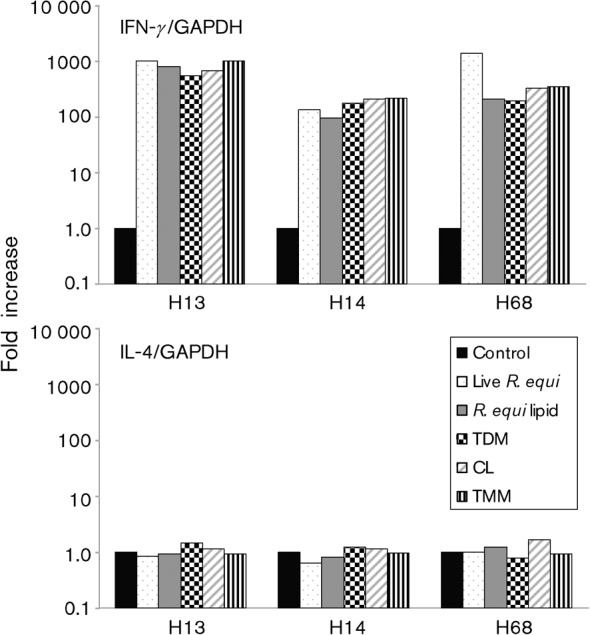
Lipid stimulation of PBMCs from immune horses induces IFN-γ, but not IL-4. Real-time PCR is shown for IFN-γ and IL-4 following 20 h stimulation with medium alone, live R. equi bacteria, unfractionated R. equi lipid or R. equi cell-wall lipids TMM, CL and TDM. IFN-γ and IL-4 have been normalized against GAPDH. Following antigen stimulation, all animals (horses #H13, H14 and H68) showed 100–1000-fold increases in the transcription of IFN-γ. These graphs represent a single experiment repeated in three animals.
Discussion
R. equi is a facultative intracellular bacterium related closely to M. tuberculosis (Rahman et al., 2003). Genome sequencing has confirmed the phylogenetic relationship between the two pathogens and shown that, like M. tuberculosis, a significant percentage of R. equi genes are involved in lipid metabolism (Rahman et al., 2003). The characteristic cell-wall lipids of these organisms have a number of critical functions and, importantly, are the end products of complex biochemical pathways (Brennan & Nikaido, 1995; Willcox et al., 2007). As a result, the unique lipids that are produced by these bacteria do not lend themselves to antigen variation in the same way that surface proteins of many pathogens can (Bricard & Porcelli, 2007).
Previous research in both mouse models and horses suggests strongly that immune clearance of R. equi requires secretion of IFN-γ, primarily by antigen-specific CD4+ TH1 lymphocytes. However, CD8+ T lymphocytes also appear to be involved (Hines et al., 2003; Kanaly et al., 1993, 1995, 1996). Although CD8+ T lymphocytes may contribute to the overall production of IFN-γ, a more important role is probably recognition and lysis of R. equi-infected cells, as has been demonstrated for M. tuberculosis (Canaday et al., 2001; Serbina et al., 2000; Skinner et al., 1997). In recent work from our laboratory, T lymphocytes from the blood and lungs of immune adult horses were examined for R. equi-specific cytolytic activity (Patton et al., 2004). In all experiments, T lymphocytes isolated from immune horses and stimulated with R. equi antigen ex vivo effectively lysed R. equi-infected target cells. Depletion studies showed that these CTLs were predominantly CD2+CD3+CD8+CD4− T lymphocytes and that antigen stimulation (which was required) increased the percentage of CD8+ cells in the effector population. Importantly, the recognition and killing of R. equi-infected cells were not restricted by ELA-A molecules. This lack of classical MHC class I restriction has been universal – easily demonstrated in every immune adult horse tested to date.
A subsequent study confirmed the presence of R. equi-specific, MHC class I-unrestricted CTLs in foals (Patton et al., 2005). However, CTLs were not present during the first few weeks of life – the period at which foals are considered most susceptible and probably first infected. Cytolytic cells then appeared between 3 and 8 weeks of age, a time frame consistent with the expected environmental exposure to R. equi and development of adaptive immune responses. The universal presence of MHC class I-unrestricted CTLs in immune adult horses and their development early in life in association with changes in age-associated susceptibility suggest that these effector cells are an important component of the protective immune response. Inducing R. equi-specific CTLs earlier in life may be vital to an immunization strategy designed to close or narrow the window of vulnerability for foals.
In this study, using methods by which mycobacterial lipid antigens were first identified, we demonstrate that R. equi antigens recognized by MHC class I-unrestricted CTLs are resistant to protease digestion and present in a whole-bacterial lipid extract. Notably, protease digestion of SRA did not diminish its ability to stimulate R. equi-specific, peripheral blood CTLs ex vivo. Likewise, CTLs stimulated with live R. equi recognized and killed ELA-A-mismatched equine macrophages pulsed with bacterial lipid. Further characterization of the R. equi cell-wall lipid demonstrated that it consisted of three principal fractions: trehalose lipids TMM and TDM, and CL. In CTL assays, only TMM and CL resulted in statistically significant cytotoxicity.
Trehalose lipids are glycosylated mycolates found in bacteria belonging to the order Actinomycetales (e.g. Mycobacterium, Rhodococcus, Gordonia and Nocardia). Trehalose consists of two d-glucose residues joined by their anomeric carbons. TDM, commonly known as ‘cord factor’, comprises trehalose esterified through its 6- and 6′-hydroxyl groups to two mycolic acid residues (Fig. 5). Generally, bacterial mycolic acids are α-alkyl, β-hydroxy fatty acids. In M. tuberculosis, the mycolic acids are 70–90 carbons, with 20–25 carbons in the alkyl branch and with one or two groups in the main chain as double bonds or cyclopropane rings (Tonge, 2000). These mycolic acids are considerably longer than those of both non-pathogenic environmental mycobacterium and R. equi (Fig. 5), and more complex than those of R. equi (Moody et al., 2002). TMM has a single mycolic acid chain and is involved in the transfer of mycolic acid into the bacterial cell wall. In M. tuberculosis, TDM and TMM are among the main glycolipids of the bacterial cell wall. Previous studies have demonstrated T-cell recognition of mycobacterial GMM (Moody et al., 1999). Likewise, CD1b-restricted T-cell lines from humans respond to either free mycolate or GMM without cross-reactivity – reflecting the role of T-cell specificity (conveyed through their TCR) for their hydrophilic caps. However, human antigen-presenting cells (APCs) were not able to cleave TDM at its most labile linkages to yield GMM or other mycolates that could be recognized by the T-cell lines. These studies indicate that there are probably limits on the abilities of APCs to process bacterial lipids to forms that are recognized by T lymphocytes. Therefore, the inability of the equine macrophages to convert TDM enzymically or chemically to an antigenic form may be an important reason that pulsing with TDM did not result in cytolysis in our CTL assay. In contrast, TMM might be bound and presented directly. Of course, processing and presentation of bacterial lipids may also be different in cells infected with live bacteria compared with cells pulsed with lipid.
A limit in the ability of equine macrophages to process microbial lipids may also account for the reduced ability of equine APCs to present antigens from the environmental species R. terrae. The carbon-chain length of R. terrae mycolic acids is C52–62, which is considerably longer than R. equi mycolic acid chains (Fig. 5), and there are three or four double bonds in R. terrae (Kurano et al., 1987). These differences in carbon-chain length and double bonds have led to a proposed reclassification of R. terrae as Gordonia terrae (Nishiuchi et al., 2000). Assuming that the glycosylated mycolates of R. terrae are presented by CD1, trimming of the mycolic acid chain so that it fits in the CD1-binding groove might be required. The lipid antigen-processing capabilities of equine APCs are unknown, as is the role of T-cell specificity for different hydrophilic head groups.
In contrast to TDM, target cells pulsed with CL were recognized by R. equi-specific CTLs and lysed. CL is a well-known and abundant component of certain bacterial membranes, notably in the mycolata; however, it has not previously been shown to be recognized by T lymphocytes in any species. CL was originally described as a constituent of the mitochondrial envelope in eukaryotes, notably in the heart muscles of mammals – which is how it got its common name. This unique phospholipid has a dimeric structure and four alkyl chains (Fig. 5), suggesting that processing may be required. Structural similarity of an R. equi lipid antigen (CL) to a mammalian lipid may not be surprising, for several reasons. First, much of the cellular machinery for processing antigens (including the MHC class I and II systems) is thought to have evolved for more basic cell functions, such as degradation of normal cell products. The pathways were then appropriated by the immune system for the breakdown of foreign molecules. Lipid and glycolipid processing may be at least partially limited to antigens that share structural features with normally metabolized self lipids (Porcelli, 2001). The seemingly limited ability of APCs to process complex microbial lipids like mycobacterial TDM to simpler antigens that can be presented to T lymphoyctes has been postulated to indicate that some components of the mycobacterial cell wall are resistant to the enzymes normally found in eukaryotic cells. Processing and presentation of a microbial phospholipid resembling a host lipid are understandable in this light. Although mycobacterial CL has not previously been shown to be presented by CD1 or recognized by T lymphocytes, it can be cleaved by lysosomal enzymes present in mammalian macrophages (Fischer et al., 2001).
Expansion and stimulation of CTLs using live R. equi allowed us to study the effector cells that are produced as a result of infection and, therefore, the cells that are probably most relevant to immunity in vivo. However, our initial design was to use lipid to stimulate PBMCs and then test those cells for the ability to kill either R. equi-infected or lipid-pulsed targets. The advantage of this initial approach was the potential to expand only CTLs that recognized R. equi lipids and to reduce any non-lipid and non-specific effects. Although we did observe killing in these preliminary assays, stimulation with unfractionated lipid also increased background killing to much higher levels. We believe that this occurred due to carry-over of lipid antigen that we were unable to wash out of cell cultures into the CTL assays. Now that we have identified specific R. equi lipids recognized by equine CTLs and have the potential means to isolate more, we hope to test their ability to expand effector cells from PBMCs. Purified lipid may also provide a means to clone CTLs from the peripheral blood of horses.
Because of the role of type I immune responses, notably CD4+ TH1 lymphocytes, in immune clearance of both R. equi and M. tuberculosis, we also assayed the effects of the purified R. equi lipids on transcription of signature TH1 and TH2 cytokines. Using PBMCs from the peripheral blood of immune adult horses, all three R. equi lipid components resulted in 100–1000-fold-increased transcription of IFN-γ, but not of IL-4 – consistent with a type I immune response. These results support our hypothesis that immune recognition of R. equi lipids and/or glycolipids by T lymphocytes plays an important role in immunity to R. equi. The upregulation of IFN-γ transcription in response to TDM was not expected, however, as TDM did not induce CTLs. A possible explanation is that the two culture systems select for different populations of T lymphocytes. In the CTL system, PMBCs are in culture for 7 days (5 days with antigen and 2 days at rest). The result is expansion of the CD8+ T lymphocytes, whilst the CD4+ T lymphocyte population remains at constant levels (Patton et al., 2004). In contrast, the cytokine assay is a more traditional short-term culture whereby PMBCs are stimulated for 18 h ex vivo. This second assay probably measures transcription by a mixture of T lymphocytes, notably CD4+ T cells and possibly NK T cells – rather than the CD8+ cells that are expanded using live R. equi in the 7 day cultures. It may be that equine CD4+ T cells and/or NK T cells are more likely to bear TCRs that recognize TDM. Additionally, although the 18 h time point was chosen based on previous time-course experiments, it is also possible that measuring cytokines, notably IL-4, at additional time points would have produced different results. Regardless, these data do not support our hypothesis that the inability of TDM to pulse target cells in the CTL assays reflected an inability of equine APCs to process and present R. equi TDM. Nevertheless, the cytokine response appeared to be specific to R. equi lipids. GMM and FMM from R. terrae resulted in only minor increases in IFN-γ transcription (<10 % of R. equi lipids) and no increase in IL-4 transcription.
Because of the early age at which foals would probably need to receive their first immunization and the inherent limitations of the developing immune system at this time of life, prevention of rhodococcal pneumonia via an immunization strategy represents a formidable challenge. On the other hand, the strong immunity that develops naturally in the vast majority of young horses suggests that successful immunization is possible. The data that we present here (and previously) suggest that bacterial lipids will be a component of any successful immunization strategy. This could be as part of a subunit vaccine or, which is perhaps more likely, in the form of an attenuated live vaccine. Among the required outcomes of an effective vaccine may be induction of CTLs that recognize R. equi lipid antigens and thereby lyse infected macrophages. In addition, the vaccine will almost certainly need to induce TH1 CD4+ T lymphocytes that produce IFN-γ – possibly including IFN-γ in response to R. equi lipids and/or glycolipids. NK T cells that recognize microbial lipids and/or self lipids (the latter following stimulation of Toll-like receptors) may also be important.
These experiments have begun to define more specifically the R. equi lipids that are recognized by T lymphocytes in immune horses. The ability to induce immune responses to these glycosylated mycolates (TMM and TDM) and phospholipids (CL) should probably be evaluated as new vaccines are tested in foals. For example, one of the few successful immunization experiments of foals to date utilized live, virulent R. equi delivered at high doses and on multiple days by an oral route (nasogastric tube) (Hooper-McGrevy et al., 2005). The formulation (virulent bacteria), dose and administration method are unacceptable for real-world use. Moreover, almost nothing is known about the T-lymphocyte responses that were induced. It would be interesting to see whether oral immunization stimulated responses that are now known to be associated with protective immunity to R. equi – including recognition of microbial lipids, production of IFN-γ and induction of MHC class I-unrestricted CTLs. Alternatively, R. equi lipids could be tested directly as immunogens, as done previously with mycobacterial lipids in guinea pigs (Dascher et al., 2003; Hiromatsu et al., 2002).
Although we suspect that presentation of R. equi lipid antigen occurs via the CD1 system, we have yet to prove this conclusively. Experiments using defined lipid, CD1-transfected target cells, T-cell lines and possibly blocking antibodies are required. In anticipation of this work, however, we are mapping and cloning the equine family of CD1 genes. Considering the role of CD1 in presentation of mycobacterial lipids and the importance of rhodococcal pneumonia as a naturally occurring disease of the horse, we expect that CD1 will play an important role in immunity to R. equi. Significantly, research on presentation and recognition of microbial lipids in the horse will also provide further information on the relative role of these immune responses in immunity to human pathogens. As mice lack group 1 CD1 genes (instead expressing only group 2 CD1d genes), there is a need for additional animal models, especially in reference to pathogens with unique cell-wall lipids (e.g. bacteria in the taxon ‘Mycolata’).
Acknowledgements
This work was supported by Morris Animal Foundation grant numbers D04EQ-027 and D07EQ-056, the Washington State University Equine Research Fund, the Achievement Reward for College Scientists (ARCS), the Poncin Fellowship, and grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the Japan Health Sciences Foundation. Special thanks go to Drs Melissa T. Hines and Wendy C. Brown. We are grateful to Mr Hideki Nakagawa and Robert Nelson for providing exceptional technical assistance and to Emma Karel for her efforts in managing the equine herd.
Abbreviations:
- APC
antigen-presenting cell
- CL
cardiolipin
- CTL
cytotoxic T lymphocyte
- ELA
equine lymphocyte alloantigen
- FMM
fructose monomycolate
- GMM
glucose monomycolate
- IFN
interferon
- IL
interleukin
- MHC
major histocompatibility complex
- MMM
mannose monomycolate
- NK
natural killer
- PBAC
peripheral blood adherent cell
- PBMC
peripheral blood mononuclear cell
- SRA
soluble R. equi antigen
- TCR
T-cell receptor
- TDM
trehalose dimycolate
- TMM
trehalose monomycolate
Edited by:
A supplementary figure showing a silver-stained polyacrylamide gel demonstrating complete digestion of SRA following proteinase K and subtilisin treatment is available with the online version of this paper.
References
- Beckman E. M., Porcelli S. A., Morita C. T., Behar S. M., Furlong S. T., Brenner M. B. (1994). Recognition of a lipid antigen by CD1-restricted αβ+ T cells. Nature 372, 691–694. [DOI] [PubMed] [Google Scholar]
- Bhatt A., Fujiwara N., Bhatt K., Gurcha S. S., Kremer L., Chen B., Chan J., Porcelli S. A., Kobayashi K. & other authors (2007). Deletion of kasB in Mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc Natl Acad Sci U S A 104, 5157–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boom W. H., Canaday D. H., Fulton S. A., Gehring A. J., Rojas R. E., Torres M. (2003). Human immunity to M. tuberculosis: T cell subsets and antigen processing. Tuberculosis (Edinb) 83, 98–106. [DOI] [PubMed] [Google Scholar]
- Brennan P. J., Nikaido H. (1995). The envelope of mycobacteria. Annu Rev Biochem 64, 29–63. [DOI] [PubMed] [Google Scholar]
- Bricard G., Porcelli S. A. (2007). Antigen presentation by CD1 molecules and the generation of lipid-specific T cell immunity. Cell Mol Life Sci 64, 1824–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigl M., Brenner M. B. (2004). CD1: antigen presentation and T cell function. Annu Rev Immunol 22, 817–890. [DOI] [PubMed] [Google Scholar]
- Canaday D. H., Wilkinson R. J., Li Q., Harding C. V., Silver R. F., Boom W. H. (2001). CD4+ and CD8+ T cells kill intracellular Mycobacterium tuberculosis by a perforin and Fas/Fas ligand-independent mechanism. J Immunol 167, 2734–2742. [DOI] [PubMed] [Google Scholar]
- Catucci L., Depalo N., Lattanzio V. M., Agostiano A., Corcelli A. (2004). Neosynthesis of cardiolipin in Rhodobacter sphaeroides under osmotic stress. Biochemistry 43, 15066–15072. [DOI] [PubMed] [Google Scholar]
- Dascher C. C., Hiromatsu K., Xiong X., Morehouse C., Watts G., Liu G., McMurray D. N., LeClair K. P., Porcelli S. A., Brenner M. B. (2003). Immunization with a mycobacterial lipid vaccine improves pulmonary pathology in the guinea pig model of tuberculosis. Int Immunol 15, 915–925. [DOI] [PubMed] [Google Scholar]
- Dittmer J. C., Lester R. L. (1964). A simple, specific spray for the detection of phospholipids on thin-layer chromatograms. J Lipid Res 15, 126–127. [PubMed] [Google Scholar]
- Fischer K., Chatterjee D., Torrelles J., Brennan P. J., Kaufmann S. H. E., Schaible U. E. (2001). Mycobacterial lysocardiolipin is exported from phagosomes upon cleavage of cardiolipin by a macrophage-derived lysosomal phospholipase A2. J Immunol 167, 2187–2192. [DOI] [PubMed] [Google Scholar]
- Folch J., Lees M., Sloane Stanley G. H. (1957). A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226, 497–509. [PubMed] [Google Scholar]
- Grotzke J. E., Lewinsohn D. M. (2005). Role of CD8+ T lymphocytes in control of Mycobacterium tuberculosis infection. Microbes Infect 7, 776–788. [DOI] [PubMed] [Google Scholar]
- Hava D. L., Brigl M., van den Elzen P., Zajonc D. M., Wilson I. A., Brenner M. B. (2005). CD1 assembly and the formation of CD1–antigen complexes. Curr Opin Immunol 17, 88–94. [DOI] [PubMed] [Google Scholar]
- Hines M. T., Paasch K. M., Alperin D. C., Palmer G. H., Westhoff N. C., Hines S. A. (2001). Immunity to Rhodococcus equi: antigen-specific recall responses in the lungs of adult horses. Vet Immunol Immunopathol 79, 101–113. [DOI] [PubMed] [Google Scholar]
- Hines S. A., Stone D. M., Hines M. T., Alperin D. C., Knowles D. P., Norton L. K., Hamilton M. J., Davis W. C., McGuire T. C. (2003). Clearance of virulent but not avirulent Rhodococcus equi from the lungs of adult horses is associated with intracytoplasmic gamma interferon production by CD4+ and CD8+ T lymphocytes. Clin Diagn Lab Immunol 10, 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiromatsu K., Dascher C. C., Leclair K. P., Sugita M., Furlong S. T., Brenner M. B., Porcelli S. A. (2002). Induction of CD1-restricted immune responses in guinea pigs by immunization with mycobacterial lipid antigens. J Immunol 169, 330–339. [DOI] [PubMed] [Google Scholar]
- Hondalus M. K. (1997). Pathogenesis and virulence of Rhodococcus equi. Vet Microbiol 56, 257–268. [DOI] [PubMed] [Google Scholar]
- Hooper-McGrevy K. E., Wilkie B. N., Prescott J. F. (2005). Virulence-associated protein-specific serum immunoglobulin G-isotype expression in young foals protected against Rhodococcus equi pneumonia by oral immunization with virulent R. equi. Vaccine 23, 5760–5767. [DOI] [PubMed] [Google Scholar]
- Kanaly S. T., Hines S. A., Palmer G. H. (1993). Failure of pulmonary clearance of Rhodococcus equi infection in CD4+ T-lymphocyte-deficient transgenic mice. Infect Immun 61, 4929–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaly S. T., Hines S. A., Palmer G. H. (1995). Cytokine modulation alters pulmonary clearance of Rhodococcus equi and development of granulomatous pneumonia. Infect Immun 63, 3037–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaly S. T., Hines S. A., Palmer G. H. (1996). Transfer of a CD4+ Th1 cell line to nude mice effects clearance of Rhodococcus equi from the lung. Infect Immun 64, 1126–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakousis P. C., Bishai W. R., Dorman S. E. (2004). Mycobacterium tuberculosis cell envelope lipids and the host immune response. Cell Microbiol 6, 105–116. [DOI] [PubMed] [Google Scholar]
- Kurano S., Sugimoto N., Sumi Y., Sawai H., Kato Y., Kaneda K., Yano I. (1987). Newly isolated glycolipids from Rhodococcus terrae cell wall and their granuloma forming activities. Yakugaku Zasshi 107, 46–52. [DOI] [PubMed] [Google Scholar]
- Li B., Rossman M. D., Imir T., Fusun Oner-Eyuboglu A., Wa Lee C., Biancaniella R., Carding S. R. (1996). Disease-specific changes in γδ T cell repertoire and function in patients with pulmonary tuberculosis. J Immunol 157, 4222–4229. [PubMed] [Google Scholar]
- Linder R. (1997). Rhodococcus equi and Arcanobacterium haemolyticum: two ‘coryneform’ bacteria increasingly recognized as agents of human infection. Emerg Infect Dis 3, 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez A. M., Hines M. T., Palmer G. H., Alperin D. C., Hines S. A. (2002). Identification of pulmonary T-lymphocyte and serum antibody isotype responses associated with protection against Rhodococcus equi. Clin Diagn Lab Immunol 9, 1270–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire T. C., Tumas D. B., Byrne K. M., Hines M. T., Leib S. R., Brassfield A. L., O'Rourke K. I., Perryman L. E. (1994). Major histocompatibility complex-restricted CD8+ cytotoxic T lymphocytes from horses with equine infectious anemia virus recognize Env and Gag/PR proteins. J Virol 68, 1459–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody D. B., Porcelli S. A. (2003). Intracellular pathways of CD1 antigen presentation. Nat Rev Immunol 3, 11–21. [DOI] [PubMed] [Google Scholar]
- Moody D. B., Reinhold B. B., Reinhold V. N., Besra G. S., Porcelli S. A. (1999). Uptake and processing of glycosylated mycolates for presentation to CD1b-restricted T cells. Immunol Lett 65, 85–91. [DOI] [PubMed] [Google Scholar]
- Moody D. B., Briken V., Cheng T.-Y., Roura-Mir C., Guy M. R., Geho D. H., Tykocinski M. L., Besra G. S., Porcelli S. A. (2002). Lipid length controls antigen entry into endosomal and nonendosomal pathways for CD1b presentation. Nat Immunol 3, 435–442. [DOI] [PubMed] [Google Scholar]
- Natsuhara Y., Yoshinaga J., Shogaki T., Sumi-Nishikawa Y., Kurano S., Kato Y., Kaneda K., Oka S., Yano I. (1990). Granuloma-forming activity and antitumor activity of newly isolated mycoloyl glycolipid from Rhodococcus terrae 70012 (Rt. GM-2). Microbiol Immunol 34, 45–53. [DOI] [PubMed] [Google Scholar]
- Nishiuchi Y., Baba T., Yano I. (2000). Mycolic acids from Rhodococcus, Gordonia, and Dietzia. J Microbiol Methods 40, 1–9. [DOI] [PubMed] [Google Scholar]
- Patton K. M., McGuire T. C., Fraser D. G., Hines S. A. (2004). Rhodococcus equi-infected macrophages are recognized and killed by CD8+ T lymphocytes in a MHC class I-unrestricted fashion. Infect Immun 72, 7073–7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton K. M., McGuire T. C., Hines M. T., Mealey R. H., Hines S. A. (2005). Rhodococcus equi-specific cytotoxic T lymphocytes in immune horses and development in asymptomatic foals. Infect Immun 73, 2083–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M. G. V., Vassilev T., Kemmerly S. A. (2002). Rhodococcus equi infection in transplant recipients: a case of mistaken identity and review of the literature. Transpl Infect Dis 4, 52–56. [DOI] [PubMed] [Google Scholar]
- Porcelli S. A. (2001). Cutting glycolipids down to size. Nat Immunol 2, 191–192. [DOI] [PubMed] [Google Scholar]
- Rahman M. T., Herron L. L., Kapur V., Meijer W. G., Byrne B. A., Ren J., Nicholson V. M., Prescott J. F. (2003). Partial genome sequencing of Rhodococcus equi ATCC 33701. Vet Microbiol 94, 143–158. [DOI] [PubMed] [Google Scholar]
- Rao V., Fujiwara N., Porcelli S. A., Glickman M. S. (2005). Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med 201, 535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roura-mir C., Moody D. B. (2003). Sorting out self and microbial lipid antigens for CD1. Microbes Infect 5, 1137–1148. [DOI] [PubMed] [Google Scholar]
- Serbina N. V., Liu C.-C., Scanga C. A., Flynn J. L. (2000). CD8+ CTL from lungs of Mycobacterium tuberculosis-infected mice express perforin in vivo and lyse infected macrophages. J Immunol 165, 353–363. [DOI] [PubMed] [Google Scholar]
- Siliciano R. F., Keegan A., Dintzis R., Dintzis H., Shin H. (1985). The interaction of nominal antigen with T cell antigen receptors. I. Specific binding of multivalent nominal antigen to cytolytic T cell clones. J Immunol 135, 906–914. [PubMed] [Google Scholar]
- Skinner M. A., Yuan S., Prestidge R., Chuk D., Watson J. D., Tan P. L. J. (1997). Immunization with heat-killed Mycobacterium vaccae stimulates CD8+ cytotoxic T cells specific for macrophages infected with Mycobacterium tuberculosis. Infect Immun 65, 4525–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenger S., Modlin R. L. (1999). T cell mediated immunity to Mycobacterium tuberculosis. Curr Opin Microbiol 2, 89–93. [DOI] [PubMed] [Google Scholar]
- Szereday L., Baliko Z., Szekeres-Bartho J. (2003). γ/δ T cell subsets in patients with active Mycobacterium tuberculosis infection and tuberculin anergy. Clin Exp Immunol 131, 287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai S. (1997). Epidemiology of Rhodococcus equi infections: a review. Vet Microbiol 56, 167–176. [DOI] [PubMed] [Google Scholar]
- Tonge P. J. (2000). Another brick in the wall. Nat Struct Biol 7, 94–96. [DOI] [PubMed] [Google Scholar]
- Torres-Tortosa M., Arrizabalaga J., Villanueva J. L., Gálvez J., Leyes M., Valencia M. E., Flores J., Peña J. M., Pérez-Cecilia E. & other authors (2003). Prognosis and clinical evaluation of infection caused by Rhodococcus equi in HIV-infected patients. Chest 123, 1970–1976. [DOI] [PubMed] [Google Scholar]
- Ueda S., Fujiwara N., Naka T., Sakaguchi Y., Ozeki I., Yano T. (2001). Structure–activity relationship of mycoloyl glycolipids derived from Rhodococcus sp. 4306. Microb Pathog 30, 91–99. [DOI] [PubMed] [Google Scholar]
- Ulrichs T., Moody D. B., Grant E., Kaufmann S. H. E., Porcelli S. A. (2003). T-cell responses to CD1-presented lipid antigen in humans with Mycobacterium tuberculosis infection. Infect Immun 71, 3076–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox B. E., Willcox C., Dover L., Besra G. S. (2007). Structures and functions of microbial lipid antigens presented by CD1. Curr Top Microbiol Immunol 314, 73–110. [DOI] [PubMed] [Google Scholar]
- Zhang W., Lonning S. M., McGuire T. C. (1998). Gag protein epitopes recognized by ELA-A-restricted cytotoxic T lymphocytes from horses with long-term equine infectious anemia virus infection. J Virol 72, 9612–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinner S. H. (1999). Changing epidemiology of infections in patients with neutropenia and cancer: emphasis on Gram-positive and resistant bacteria. Clin Infect Dis 29, 490–494. [DOI] [PubMed] [Google Scholar]