

Abstract
AIM: To determine the overall prevalence of H pylori and CagA positive H pylori infection and the prevalence of other bacterial and viral causes of chronic infection in patients with coronary heart disease (CHD), and the potential role of anti-heat-shock protein 60 (Hsp60) antibody response to these proteins in increasing the risk of CHD development.
METHODS: Eighty patients with CHD and 160 controls were employed. We also compared the levels of anti-heat-shock protein 60 (Hsp60) antibodies in the two groups. The H pylori infection and the CagA status were determined serologically, using commercially available enzyme-linked immunosorbent assays (ELISA), and a Western blotting method developed in our laboratory. Systemic antibodies to Hsp60 were determined by a sandwich ELISA, using a polyclonal antibody to Hsp60 to sensitise polystyrene plates and a commercially available human Hsp60 as an antigen.
RESULTS: The overall prevalence of H pylori infection was 78.7% (n = 63) in patients and 76.2% (n = 122) in controls (P = 0.07). Patients infected by CagA-positive (CagA+) H pylori strains were 71.4% (n = 45) vs 52.4% of infected controls (P = 0.030, OR = 2.27). Systemic levels of IgG to Hsp60 were increased in H pylori-negative patients compared with uninfected controls (P < 0.001) and CagA-positive infected patients compared with CagA-positive infected controls (P = 0.007).
CONCLUSION: CagA positive H pylori infection may concur to the development of CHD; high levels of anti-Hsp60 antibodies may constitute a marker and/or a concomitant pathogenic factor of the disease.
Keywords: H pylori, Coronary heart disease, CagA protein, Heat shock protein 60, Antibody response
INTRODUCTION
Atherosclerosis-related diseases -particularly coronary heart disease (CHD)- are a leading cause of death and disability in most developed countries. Many epidemiological studies have shown a strong relationship between CHD and chronic bacterial and viral infections, suggesting a primary role of inflammatory diseases in the pathogenesis of vascular cardiac disorders[1,2]. Infectious agents may cause a spectrum of systemic effects and induce atherosclerosis in several different ways. For instance, by increasing the production of circulating cytokines (interleukin-1 [IL-1] and interleukin-6 [IL-6]), through the generation of acute-phase reactants (white blood cells and C reactive protein) and the stimulation of immune-mediated responses, such as the production of antibodies targeted to the invading pathogens, etc[3]. Several authors[4] have also reported that infections might stimulate smooth muscle cell proliferation and migration and lipid accumulation; apoptosis of endothelial cells can be inhibited and many procoagulant effects could be produced[4].
H pylori infection is one of the most widely spread infectious diseases in human[5]. This microorganism infects half the world population and causes chronic gastritis. The disease usually lasts for the entire host’s life and constitutes a main risk determinant of peptic ulcer and gastric neoplasia[6,7]. The infection elicits a chronic humoral and cellular inflammatory response, stimulates an increase of polymorphs and basophils[8] and elevates the local and systemic concentrations of vasoactive cytokines[9], whose effects may not be confined to the digestive tract[10].
Recent epidemiological surveys have indicated that H pylori infection may be associated with atherosclerotic vascular diseases[11], although it is still disputed whether this infection increases the risk of CHD[12-14]. Some studies have shown an increased risk of CHD in patients with a systemic immune response to heat shock proteins (Hsps)[15]. Hsps are families of highly conserved proteins that share wide homologies of sequence among different species, ranging from bacteria to human beings[16,17]. They are induced or up-regulated in cells exposed to sudden elevations in temperature, but are also synthesized in large numbers when cells are exposed to stressful stimuli such as inflammation, infections, mechanical stress, hypoxia and oxidizing agents[16,17]. They play a fundamental role in the growth of bacteria at all temperatures and their production could represent an essential mechanism of cell protection against different noxae[17,18]. H pylori produces two main Hsps, a groEs-like HspA with a mass of 13 kDa, and a groEL-like HspB with a mass of 54-60 kDa[19,20]. Both proteins stimulate a specific systemic antibody response and, due to the high sequence homology of Hsps, it is highly possible that they can trigger an autoimmune response directed against the bacterial proteins and also to human tissues expressing Hsps, including vascular endothelial cells[20,21]. The aim of the present study was to determine the prevalence of anti-Hsp antibodies in patients with CHD and controls and to identify the potential role of an antibody response to these proteins in increasing the risk of CHD development. We tested serum samples for the overall prevalence of H pylori and CagA positive H pylori infection, and for antibodies to the other bacterial and viral causes of chronic infection that are recognised determinants of CHD risk development. Our results suggest that CagA positive H pylori infection may concur to the development of CHD and that high levels of anti-Hsp antibodies may constitute a marker and/or a pathogenic factor of the disease.
MATERIALS AND METHODS
Patients and controls
We studied 80 consecutive patients with stable angina; their mean age was 65 years (range 45 to 75 years). Patients were admitted to this Institute for evaluation by clinical history, physical examinations, heart echography, and basal and exercise ECGs. Patients were enrolled if they showed signs or symptoms of angina at exercise ECG; an ST segment depression more than 2 mm was considered positive. As control, we enrolled 160 age- and gender-matched patients, who came from the same socio-economic background and were hospitalised in the same Institute for diseases other than CHD, vascular diseases, dyspeptic and liver disorders, hematological diseases, and thyroid abnormalities. Their mean age was 64.5 years (range 43 to 75 years). Patients and controls had not taken antibiotics potentially active against H pylori in the last three months. Both patients and controls gave their written informed consent.
Determination of H pylori infection and CagA status
The H pylori infectious status was determined serologically using a commercially available enzyme-linked immunosorbent assay with a sensitivity and specificity of 96% ca. (Helicobacter pylori IgG, Diesse, Monteriggioni, Siena, Italy). H pylori infectious status was confirmed by Western blotting (WB). WB was also used to detect antibodies to H pylori CagA. Briefly, a whole cell suspension of H pylori CCUG 17874 (a CagA-positive and cytotoxic strain) was denatured in Laemmli’s buffer at 100°C for 5 min and electrophoresed in a 10% polyacrylamide gel with sodium dodecylsulphate. The resolved proteins were transferred electrophoretically onto nitrocellulose membranes, and the free sites were saturated with 3% skim milk in phosphate buffered saline (PBS) pH 7.4 containing 0.1% Triton X (PMT). Afterward, strips were cut and immunoblotted with serum samples diluted 1:100 in PMT for immunoglobulin G (IgG). After overnight incubation at room temperatures, strips were washed three times with PMT, and a peroxidase labelled antibody to human IgG, diluted in PMT 1:2000 (Sigma Che. Co., Milan), was added and incubated at room temperatures for 90 min. Strips were washed three times with PMT, once with PBS-Triton X, and twice with Tris buffer 0.05 mol/L pH 6.8. The reaction was visualised by addition of the substrate (H2O2 in a solution of 4-chloro-1-naphthol in Tris buffer 0.05 M pH 6.8). The reaction was stopped with water. The presence of more than six bands of reaction indicated an infection. As positive controls, anti-CagA and anti-Hsp rabbit polyclonal antibodies (kindly given by R. Rappuoli, Novartis, Siena) were used.
Determination of anti-Hsp60 antibodies
Antibodies to Hsp60 were determined by an ELISA, using a commercially available human Hsp60 (Sigma Che. Co., Milan, Italy). In preliminary tests, we determined the working concentrations of Hsp60 with the aid of a pool of human serum samples, which contained antibodies to H pylori HspB (54-60 kDa), as detected on WB. Briefly, we sensitised each well of polystyrene microtiter plates with 150 μL of an anti-polyclonal H pylori HspB antibody raised in rabbits, diluted 1:50 in PBS pH 7.4. After one hour of incubation at 37°C, we washed the plates three times with PBS containing 0.05% Tween 20 (PBST) and 2% bovine serum albumin (BSA). Then, we added to each well 2.5 μg of Hsp contained in 100 μL of PBS-BSA (this amount of Hsp was determined in preliminary tests). After one hour of incubation at 37°C and three washes with PBS-T-BSA, we added 100 μL of each serum samples, both from patients and controls, diluted 1:50 in PBS-BSA. Plates were incubated at 37°C for one hour, then they were washed three times and 100 μL of an anti-human immunoglobulin G (IgG) (Sigma Che. Co., Milan, Italy), labelled with peroxidase, diluted 1:2000, was added to each well. After incubation, we added to each well 50 μL of the substrate (a solution of tetramethylbenzidine dihydrochloride in 0.1 mol/L phosphate-citrate buffer pH 5.0, containing 0.03% sodium perborate and 0.01% of 30% hydrogen peroxide). Incubation was carried out at room temperatures in the dark for 30 min. The reaction was stopped by the addition of 50 μL of 2 mol/L sulphuric acid and read spectrophotometrically at 450 nm. All tests were performed in triplicate and levels of antibodies were expressed in optical density (OD). As a control, we used an anti-Hsp60 polyclonal serum raised in rabbits tested at several dilutions. The intra-tests and inter-tests deviations were lower than or equal to 10%, and lower than or equal to 15%, respectively.
Determination of other pathogens
Antibodies against other putative causes of chronic infections were determined by the following commercially available kits: Chlamydia pneumoniae (CFT-MAT Chlamydia, Diesse, Monteriggioni, Italy); Mycoplasma pneumoniae (CFF-MAT Mycoplasma, Diesse, Monteriggioni, Italy); cytomegalovirus (ENZIGNOST CMV IgG, Behering, Milan, Italy); herpes simplex virus (ENZIGNOST HSV IgG, Behering, Milan, Italy); E-B virus (ENZIGNOST HSV IgG, Behering, Milan, Italy).
Statistical analysis
The differences in the prevalence of infection by the various pathogens were compared using the chi-square test with the Yates’s correction. The mean levels of anti-urease antibodies were compared using the t-test for independent samples, utilising the software Primit.Exe, version 3.0.1. P values < 0.05 were considered significant.
RESULTS
Overall prevalence of H pylori infection
We determined the prevalence of H pylori infection between patients and controls to verify the hypothesis that such an infection could increase the risk of CHD. The mean age of patients was 65 years (range 45 to 75 years); controls had a similar mean age of 64.5 years (range 43 to 75 years). The overall prevalence of H pylori infection was 78.7% (n = 63) in patients and 76.2% (n = 122) in controls (P = 0.07).
Determination of CagA status in infected patients and controls
Recent studies have shown that infection by strains that express CagA protein induces increased levels of local and systemic cytokines that could contribute to the damage of the cardiovascular system. We therefore determined the seroprevalence of CagA seropositivity in patients and controls. Patients infected by CagA-positive (CagA+) H pylori strains were 71.4% (n = 45) vs 52.4% of infected controls (P = 0.030, OR = 2.27; 95% CI 1.0-5.1) (Figure 1).
Figure 1.
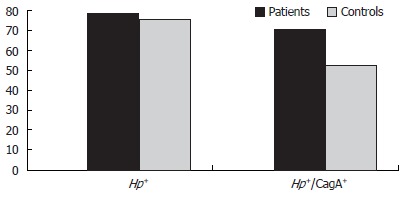
Prevalence of overall H pylori and CagA+ H pylori infection in patients and controls.
Prevalence of infections by pathogens other than H pylori
Since it is well-known that many pathogens could contribute to the genesis of a chronic systemic inflammatory status, we determined the seroprevalence of the most common infectious agents that might increase the risk of CHD. We found that the majority of both patients and controls were seropositive for C. pneumoniae, cytomegalovirus, herpes simplex virus and Epstein-Barr virus, while 46.2% of patients and 38.7% of controls had anti-M. pneumoniae antibodies (Table 1). No statistically significant difference was found in the prevalence of infections by the different pathogens in patients and controls (data not shown).
Table 1.
Prevalence of infection by pathogens other than H pylori in patients and controls n (%)
| Group | C. pneumoniae | CMV | HSV-1 | EBV | M. pneumoniae |
| Patients (80) | 63 (78.7) | 75 (93.5) | 77 (96.2) | 74 (92.5) | 37 (46.2) |
| Controls (160) | 110 (68.7) | 145 (90.6) | 140 (87.5) | 148 (92.5) | 62 (38.7) |
Determination of anti-Hsp60 antibodies in patients and controls
Hsps are a family of well-conserved proteins and Hsp60, in particular, is widely shared by H pylori and eukaryotic cells. As antibodies to Hsps are found at high titers in cardiovascular disorders, we compared the levels of anti-Hsp60 antibodies in patients and controls. Levels of antibodies to Hsp60 were significantly increased in H pylori-negative (Hp-) patients, compared with those in H pylori-negative controls (341.5 ± 159.6 vs 197.6 ± 44.4; P < 0.001, 95% CI 66.4-221.3) (Figure 2); levels of antibodies to Hsp60 in CagA+ patients were higher than in CagA+ controls (418.8 ± 144.2 vs 317.2 ± 175.6; P = 0.007, 95% CI 28.8-174.3), but were not significantly higher than in Hp+/CagA- patients (350.2 ± 169.1; P = 0.110) and in Hp- patients (341.5 ± 159.6; P = 0.072) (Figure 2). Levels of antibodies to Hsp60 in CagA+ controls (317.2 ± 175.6) and Hp+/CagA- controls (297.1 ± 80.82) were higher than in Hp- controls (197.6 ± 44.42) (P = 0.006, and P < 0.001, respectively) (Figure 2).
Figure 2.
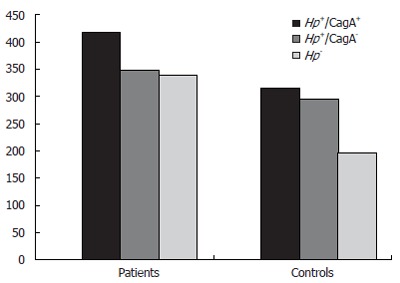
Mean levels (in optical density) of anti-Hsp60 systemic antibodies in patients and controls.
DISCUSSION
In the present study we have observed an increased prevalence of CagA+ H pylori infection, as well as increased levels of antibodies to Hsp60 in patients with CHD, compared with controls. Because of the notion that atherosclerosis may be regarded as an inflammatory process and that the presence of a chronic systemic inflammatory status is a strong risk factor for the development of coronary artery disease and ischemic stroke, we need to identify the cause of inflammation. In our study, almost all patients and controls were seropositive for pathogens other than H pylori, suggesting that these agents of chronic infection have a minor role (if any), at least in our area, in the development of cardiovascular disorders. H pylori is the logical and most important candidate for this role as a cause of the low-grade, persistent inflammatory stimulation induced by the infection, i.e. the vascular damage could be an indirect effect of systemic inflammatory mediators stimulated by the local mucosal inflammation, which may affect homeostasis. Recent epidemiological surveys have indicated that H pylori infection may be associated with atherosclerosis in different districts, but it is still disputed whether this infection increases the risk of CHD[11,12,14,21]. The increased levels of cytokines induced by infections can lead to changes in endothelial cell function by recruiting monocytes and T cell lymphocytes into the vessel walls; as a consequence, the local inflammatory responses may be exacerbated, even in the absence of resident pathogens. In addition to promoting atherosclerosis, infections can also trigger acute coronary events, such as plaque rupture[12]. Mendall et al[11] reported an association between H pylori infection and CHD, however, such an observation is still questioned[12-14,22]. A possible reason for contradictory results may include the variable circulation in different areas of H pylori strains endowed with an increased inflammatory potential, i.e. those possessing a genomic insertion called cag[23]. Patients infected by highly virulent H pylori strains can be easily identified, because CagA, the protein encoded by cagA, one of the cag genes, is always expressed[23,24]. CagA is strongly immunogenic and specific systemic antibodies can be determined by simple methods. H pylori is not a clonal pathogen. Although strains from unrelated cases of infection are all genomically diverse, most clinical isolates can be grouped into two types, according to whether they possess or not the pathogenicity island cag, a genomic region that encodes proteins involved in virulence such as cagA[23]. The importance of the infection by H pylori carrying the cag insertion has recently been confirmed by the observation that patients with CHD are more likely infected by CagA positive strains and have a more severe clinical picture of the disease[1,2,25]. The increased risk of CHD, to which patients infected by CagA-positive H pylori are exposed, may be attributed to the intensified inflammatory potential of such organisms[23]. Thus, the infectious status could determine elevated systemic levels of tumour necrosis factor-alfa, IL-1 beta, IL-6 and interleukin-8 and these cytokines may exert a deleterious activity against vascular endothelial cells. The chronic local inflammatory response to H pylori infection may also have repercussions for the whole organism[8,9]. Systemic indices of inflammation, such as levels of polymorphs and basophils, can be increased in individuals infected by H pylori and many vasoactive substances like cytokines, produced locally to fight the bacteria, may reach the blood stream and promote a chronic systemic inflammatory status of low degree, which may contribute to injury of other organs, different and far from the stomach[8-11].
Another explanation for contradictory results in experimental and clinical studies, pointing to find out a real association between Hp and CHD, may reside in the different inclusion criteria of patients and controls employed in different studies and the strong association of H pylori infection with confounding factors, such as age and social class. In order to avoid these variables we have enrolled both patients and controls of the same gender, age and coming from the same social background. The infection by CagA-positive H pylori cannot directly induce coronary atherosclerosis and need, most probably, the presence of other co-factors capable of inducing the onset and evolution of ischaemic heart disease. Hsps are highly conserved, immunogenic molecules whose cellular levels are raised by heat, inflammatory mediators and other forms of physiological stress[16,17]. Their most important function is to enhance cellular survival under physiologically stressful conditions, since they have been recognized as molecular chaperones and helpful in correct protein folding and oligomeric assembly[16-18]. Although Hsps are normally intracellular proteins, marked stress-induced overexpression may lead to their presentation on the cell surface, stimulating an autoimmune reaction, and thereby representing an important marker of inflammation. The observation that almost all patients with gastric carcinoma have systemic antibodies to H pylori HspB may support such observations[26]. According to the hypothesis of Wick et al[27], the association between high levels of anti-Hsp antibodies and atherosclerotic vascular disease is due to an autoimmune reaction to endothelial cells that express high levels of Hsp in response to stressful stimuli, like oxyded LDL, free radicals, local infections, cytokines or hemodynamic stress. In the present study, we confirmed that the increased anti-Hsp60 immune response observed in patients cannot be attributed to chronic infections by pathogens other than H pylori, since their prevalence in patients was similar to that in controls. On the contrary, the infection by CagA+ H pylori strains increased the levels of anti-Hsp60 antibodies both in patients and controls; however, although in patients the difference was not statistically significant, in controls such a difference was significant, suggesting that a relationship between chronic H pylori infection and development of antibodies to Hsp60 cannot be excluded. Another explanation could consist in the possibility that the inflammatory response triggered by the infection, together with putative toxic substances secreted by bacteria, alters the epithelial Hsp to such a degree that the patient’s immune system loses the immune tolerance to self chaperon and starts producing autoantibodies that cross-react with H pylori Hsps. Latif et al[28] have identified a strong homology between cardiac myosin heavy chain and Hsp60, suggesting that a cross-reactivity between similar epitope motifs may contribute to autoimmunity; circulating anti-Hsp antibodies may be involved in an autoimmune reaction to myocytes or endothelial cells, respectively, that have expressed Hsps due to stress.
Considering the importance of CHD in the industrialized world as a main cause of illness and since H pylori infection (even by CagA-positive strains) can be easily eradicated by specific treatments, the accurate definition of this new risk factor may lead to novel strategies for the prevention of ischemic heart disease.
Footnotes
Supported by a grant from the University of Siena, PAR 2004 “H pylori infection, hosts’ aplotypes of inflammatory cytokines and the risk of ischemic heart disease”
S- Editor Wang J L- Editor Zhu LH E- Editor Ma WH
References
- 1.Pasceri V, Cammarota G, Patti G, Cuoco L, Gasbarrini A, Grillo RL, Fedeli G, Gasbarrini G, Maseri A. Association of virulent Helicobacter pylori strains with ischemic heart disease. Circulation. 1998;97:1675–1679. doi: 10.1161/01.cir.97.17.1675. [DOI] [PubMed] [Google Scholar]
- 2.Gunn M, Stephens JC, Thompson JR, Rathbone BJ, Samani NJ. Significant association of cagA positive Helicobacter pylori strains with risk of premature myocardial infarction. Heart. 2000;84:267–271. doi: 10.1136/heart.84.3.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epstein SE, Zhou YF, Zhu J. Infection and atherosclerosis: emerging mechanistic paradigms. Circulation. 1999;100:e20–e28. doi: 10.1161/01.cir.100.4.e20. [DOI] [PubMed] [Google Scholar]
- 4.Epstein SE. The multiple mechanisms by which infection may contribute to atherosclerosis development and course. Circ Res. 2002;90:2–4. [PubMed] [Google Scholar]
- 5.Graham DY. Campylobacter pylori and peptic ulcer disease. Gastroenterology. 1989;96:615–625. doi: 10.1016/s0016-5085(89)80057-5. [DOI] [PubMed] [Google Scholar]
- 6.Parsonnet J. Helicobacter pylori and gastric cancer. Gastroenterol Clin North Am. 1993;22:89–104. [PubMed] [Google Scholar]
- 7.Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet. 1991;338:1175–1176. doi: 10.1016/0140-6736(91)92035-z. [DOI] [PubMed] [Google Scholar]
- 8.Graham DY, Osato MS, Olson CA, Zhang J, Figura N. Effect of H. pylori infection and CagA status on leukocyte counts and liver function tests: extra-gastric manifestations of H. pylori infection. Helicobacter. 1998;3:174–178. doi: 10.1046/j.1523-5378.1998.08018.x. [DOI] [PubMed] [Google Scholar]
- 9.Perri F, Clemente R, Festa V, De Ambrosio CC, Quitadamo M, Fusillo M, Grossi E, Andriulli A. Serum tumour necrosis factor-alpha is increased in patients with Helicobacter pylori infection and CagA antibodies. Ital J Gastroenterol Hepatol. 1999;31:290–294. [PubMed] [Google Scholar]
- 10.Patel P, Mendall MA, Khulusi S, Northfield TC, Strachan DP. Helicobacter pylori infection in childhood: risk factors and effect on growth. BMJ. 1994;309:1119–1123. doi: 10.1136/bmj.309.6962.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mendall MA, Goggin PM, Molineaux N, Levy J, Toosy T, Strachan D, Camm AJ, Northfield TC. Relation of Helicobacter pylori infection and coronary heart disease. Br Heart J. 1994;71:437–439. doi: 10.1136/hrt.71.5.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ossei-Gerning N, Moayyedi P, Smith S, Braunholtz D, Wilson JI, Axon AT, Grant PJ. Helicobacter pylori infection is related to atheroma in patients undergoing coronary angiography. Cardiovasc Res. 1997;35:120–124. doi: 10.1016/s0008-6363(97)00090-4. [DOI] [PubMed] [Google Scholar]
- 13.Whincup P, Danesh J, Walker M, Lennon L, Thomson A, Appleby P, Hawkey C, Atherton J. Prospective study of potentially virulent strains of Helicobacter pylori and coronary heart disease in middle-aged men. Circulation. 2000;101:1647–1652. doi: 10.1161/01.cir.101.14.1647. [DOI] [PubMed] [Google Scholar]
- 14.Kowalski M, Konturek PC, Pieniazek P, Karczewska E, Kluczka A, Grove R, Kranig W, Nasseri R, Thale J, Hahn EG, et al. Prevalence of Helicobacter pylori infection in coronary artery disease and effect of its eradication on coronary lumen reduction after percutaneous coronary angioplasty. Dig Liver Dis. 2001;33:222–229. doi: 10.1016/s1590-8658(01)80711-8. [DOI] [PubMed] [Google Scholar]
- 15.Prohászka Z, Duba J, Horváth L, Császár A, Karádi I, Szebeni A, Singh M, Fekete B, Romics L, Füst G. Comparative study on antibodies to human and bacterial 60 kDa heat shock proteins in a large cohort of patients with coronary heart disease and healthy subjects. Eur J Clin Invest. 2001;31:285–292. doi: 10.1046/j.1365-2362.2001.00819.x. [DOI] [PubMed] [Google Scholar]
- 16.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 17.Xu Q, Wick G. The role of heat shock proteins in protection and pathophysiology of the arterial wall. Mol Med Today. 1996;2:372–379. doi: 10.1016/s1357-4310(96)10034-4. [DOI] [PubMed] [Google Scholar]
- 18.Winfield JB, Jarjour WN. Stress proteins, autoimmunity, and autoimmune disease. Curr Top Microbiol Immunol. 1991;167:161–189. doi: 10.1007/978-3-642-75875-1_10. [DOI] [PubMed] [Google Scholar]
- 19.Fayet O, Ziegelhoffer T, Georgopoulos C. The groES and groEL heat shock gene products of Escherichia coli are essential for bacterial growth at all temperatures. J Bacteriol. 1989;171:1379–1385. doi: 10.1128/jb.171.3.1379-1385.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suerbaum S, Thiberge JM, Kansau I, Ferrero RL, Labigne A. Helicobacter pylori hspA-hspB heat-shock gene cluster: nucleotide sequence, expression, putative function and immunogenicity. Mol Microbiol. 1994;14:959–974. doi: 10.1111/j.1365-2958.1994.tb01331.x. [DOI] [PubMed] [Google Scholar]
- 21.Pérez-Pérez GI, Thiberge JM, Labigne A, Blaser MJ. Relationship of immune response to heat-shock protein A and characteristics of Helicobacter pylori-infected patients. J Infect Dis. 1996;174:1046–1050. doi: 10.1093/infdis/174.5.1046. [DOI] [PubMed] [Google Scholar]
- 22.Lamb DJ, El-Sankary W, Ferns GA. Molecular mimicry in atherosclerosis: a role for heat shock proteins in immunisation. Atherosclerosis. 2003;167:177–185. doi: 10.1016/s0021-9150(02)00301-5. [DOI] [PubMed] [Google Scholar]
- 23.Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiang Z, Censini S, Bayeli PF, Telford JL, Figura N, Rappuoli R, Covacci A. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect Immun. 1995;63:94–98. doi: 10.1128/iai.63.1.94-98.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Figura N, Palazzuoli A, Faglia S, Lenzi C, Borrello F, Palazzuoli V, Nami R, Dal Canto N, De Regis F, Vaira D, et al. Infection by CagA-positive Helicobacter pylori strains in patients with ischemic heart disease: prevalence and association with exercise-induced electrocardiographic abnormalities. Dig Dis Sci. 2002;47:831–836. doi: 10.1023/a:1014708520885. [DOI] [PubMed] [Google Scholar]
- 26.Iaquinto G, Todisco A, Giardullo N, D'Onofrio V, Pasquale L, De Luca A, Andriulli A, Perri F, Rega C, De Chiara G, et al. Antibody response to Helicobacter pylori CagA and heat-shock proteins in determining the risk of gastric cancer development. Dig Liver Dis. 2000;32:378–383. doi: 10.1016/s1590-8658(00)80256-x. [DOI] [PubMed] [Google Scholar]
- 27.Wick G, Knoflach M, Kind M, Henderson B, Bernhard D. Heat shock proteins and stress in atherosclerosis. Autoimmun Rev. 2004;3 Suppl 1:S30–S31. [PubMed] [Google Scholar]
- 28.Latif N, Taylor PM, Khan MA, Yacoub MH, Dunn MJ. The expression of heat shock protein 60 in patients with dilated cardiomyopathy. Basic Res Cardiol. 1999;94:112–119. doi: 10.1007/s003950050133. [DOI] [PubMed] [Google Scholar]