
Figure 2.
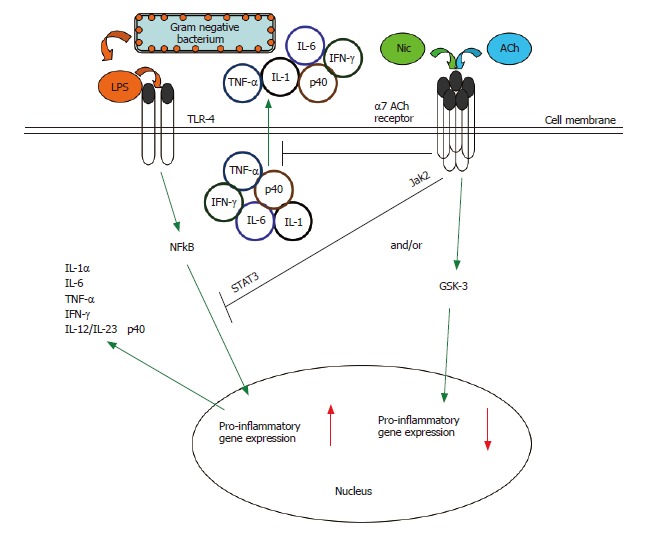
The cholinergic anti-inflammatory pathway. Multiple inflammatory stimuli activate the NFκB system and lead to the release of pro-inflammatory cytokines from innate immune cells. For example, interaction of bacterial LPS with Toll-like receptors (TLRs) on the monocyte surface induces a pro-inflammatory response characterized by the production and release of several key pro-inflammatory cytokines[88]. The α7 nAChR-dependent cholinergic anti-inflammatory pathway, triggered endogenously by acetylcholine or exogenously by nicotine, can suppress the production of several pro-inflammatory cytokines in activated monocytic cells (see Figure 4)[21,22,32,39,45]. Such nicotine-mediated suppression of TNF in vivo protects mice from endotoxic shock[32,46]. The cholinergic anti-inflammatory pathway acts at both the transcriptional and post-translational levels. Engagement of the α7 nAChR results in the rapid suppression of the release of pre-formed pro-inflammatory cytokines[32,39,45-47]. Engagement of the α7 nAChR also results in inactivation of the NFκB system, preventing the upregulation of pro-inflammatory gene activity[32]. There is a need to further explore the signaling within the cholinergic anti-inflammatory pathway. In macrophages, the nicotine-induced suppression of pro-inflammatory cytokine release involves recruitment of the tyrosine kinase Jak2 to the α7 nACh receptor, the subsequent phosphorylation of the transcription factor STAT3, and the activation of STAT3 and SOC3 signaling cascade[61]. We have shown the potential convergence of the nicotinic anti-inflammatory and an endogenous, GSK-3-dependent anti-inflammatory pathway[88] in monocytes (see Figure 5).