

Abstract
AIM: To examine the influence of lipoprotein lipase (LPL) gene polymorphism in ulcerative colitis (UC) patients.
METHODS: Peripheral blood was obtained from 131 patients with UC and 106 healthy controls for DNA extraction. We determined LPL gene polymorphisms affecting the enzyme at Ser447stop, as well as HindIII and PvuII polymorphisms using PCR techniques. PCR products were characterized by PCR-RFLP and direct sequencing. Polymorphisms were examined for association with clinical features in UC patients. Genotype frequencies for LPL polymorphisms were also compared between UC patients and controls.
RESULTS: In patients with onset at age 20 years or younger, C/G and G/G genotypes for Ser447stop polymorphism were more prevalent than C/C genotype (OR = 3.13, 95% CI = 0.95-10.33). Patients with H+/- or H-/- genotype for HindIII polymorphism also were more numerous than those with H+/+ genotype (OR = 2.51, 95% CI = 0.85-7.45). In the group with H+/+ genotype for HindIII polymorphism, more patients had serum triglyceride concentrations over 150 mg/dL than patients with H+/- or H-/- genotype (P < 0.01, OR = 6.46, 95% CI = 1.39-30.12). Hypertriglycemia was also more prevalent in patients with P+/+ genotypes for PvuII polymorphism (P < 0.05, OR = 3.0, 95% CI = 1.06-8.50). Genotype frequency for LPL polymorphism did not differ significantly between UC patients and controls.
CONCLUSION: Ser447stop and HindIII LPL polymorphisms may influence age of onset of UC, while HindIII and PvuII polymorphisms influence serum triglyceride in UC patients.
Keywords: Ulcerative colitis, Lipoprotein lipase, Lipid metabolism, Triglyceride
INTRODUCTION
Inflammatory bowel disease (IBD), characterized by chronic recurrent inflammation of the intestinal tract, includes two common forms: Crohn’s disease (CD) and ulcerative colitis (UC). For both forms, the etiology is unclear, but likely to be multifactorial. Factors that may affect IBD include diet, infantile environment and immune defense abnormalities limited to the intestinal tract. Recently, genetic factors have been examined in IBD, but many studies seeking susceptibility genes for IBD have not produced a consensus. We presently investigated possible influence of the lipoprotein lipase (LPL) gene in UC. LPL plays a critical role in lipid metabolism. Dietary triacylglycerol (TAG) exists in the human circulation as macromolecules (TAG-rich lipoprotein) that are too large to pass through the endothelium of most capillaries. LPL catalyses conversion of TAG-rich lipoprotein to triglyceride (TG), very low-density lipoprotein (VLDL), and chylomicrons (CM), all of which circulate and can enter tissues more readily to serve energy source[1,2]. Our interest in LPL in IBD was provoked by identifying high lipid intake as a risk factor for IBD. Shoda et al[3] reported associated intake of n-6 polyunsaturated fat and animal fat with development of CD in Japanese patients, while Geering et al[4] reported high intake of mono- and polyunsaturated fats to be a risk factor for the UC. Furthermore, the LPL gene has been localized to chromosome 8p22 near N-acetyltransferase2 (NAT2) gene, while Machida et al[5] reported an association between NAT2 gene haplotype NAT2*7B and CD. Considering these various findings, we suspected that LPL gene polymorphisms could influence characteristics and incidence of UC. These issues were examined in the present molecular genetic investigation.
MATERIALS AND METHODS
Subjects
We studied 131 patients with UC (75 males and 56 females) and 106 healthy controls (53 males and 53 females). Diagnosis of UC was based on conventional clinical, radiologic, endoscopic, and pathologic criteria. Characteristics of UC patients are shown in Table 1. We investigated the effect of LPL polymorphism on clinical features as shown in this table. To examine LPL polymorphisms in terms of influence on UC incidence, we compared LPL genotype frequencies in UC patients with those in controls.
Table 1.
Characteristics of patients with ulcerative colitis
| Characteristic | UC (n = 131) |
| Age of onset (yr) | 36.8 ± 15.3 |
| Colitis duration (yr) | 8.8 ± 7.7 |
| Extension | |
| Proctitis | 22 (16.8%) |
| Left-sided | 59 (45.0%) |
| Pancolitis | 50 (38.2%) |
| Type of clinical course | |
| First episode | 14 (10.7%) |
| Chronic relapse | 79 (60.3%) |
| Chronic persistent | 38 (29.0%) |
| Severity | |
| Mild | 40 (30.5%) |
| Moderate | 61 (46.6%) |
| Severe | 30 (22.9%) |
DNA extraction
Blood samples were obtained from patients and controls after they had given informed consent to sampling and analyses. This study was approved by the Fujita Health University Ethics Committee. DNA was extracted from blood samples using a PUREGENE DNA isolation kit (Gentra Systems, Inc, Minneapolis, USA).
Genotype
LPL polymorphisms were typed using polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) methods to detect Ser447stop, HindIII and PvuIIpolymorphisms[6]. The primer sets were as follows: for Ser447stop, 5´-GATGTGGCCTGAGTGTGACAG-3´ (forward) and 5´-TCCCTTAGGGTGCAAGCTCAG-3´ (reverse); for HindIII, 5´-GATGTCTACCTGGATAATCAAAG-3´(forward) and 5´-CTTCAGCTAGACATTGCTAGTGT-3´ (reverse); and for PvuII, 5´-GAGACACAGATCTCTCTTAAGAC-3´ (forward) and 5’-ATCAGGCAATGCGTATGAGGTAA-3’ (reverse). PCR was carried out in a 30-μL aliquot containing 50 ng of genomic DNA, 12 pmol of each primer, 3.0 μL of 10× buffer solution, 20 nmol/μL of dNTP, and 1U of Taq polymerase. PCR conditions for Ser447stop included initial denaturation at 95°C for 5 min, followed by 35 amplification cycles, each cycle containing denaturation at 95°C for 30 s, primer annealing at 62°C for 30 s, and extension at 72°C for 30 s. PCR conditions for HindIII and PvuII included initial denaturation at 95°C for 5min, followed by 35 amplification cycles, each cycle containing denaturation at 95°C for 30 s, primer annealing at 61°C for 30 s, and extension at 72°C for 30 s. PCR products subjected to overnight digestion with MnlI, HindIII, and PvuII were examined by electrophoresis on 20 g/L agarose gels. The strategy for the detection of the G allele (Ser447stop) is shown in Figure 1. A 315-bp segment of exon 9 of the lipoprotein lipase gene was amplified by the PCR. In the presence of the G allele, the amplified DNA contains a new site for MnlI, but does not contain a site in the presence of the C allele. However, there is a restriction site for MnlI in the forward primer sequence regardless of G/C mutation. After digestion, the PCR product yielded 248- and 67-bp fragments. The PCR product containing a HindIII restriction site yielded 210- and 140-bp fragments, while the product containing a PvuII restriction site yielded 222- and 209-bp fragments. Some products after digestion were examined on polyacrylamide gels (GeneGel Excel 12.5/24 kit from GE Healthcare Bio-sciences, Tokyo) and stained with DNA silver staining kit (GE Healthcare Bio-sciences, Tokyo), e.g. PvuII digestion-cases. Electrophoresed patterns of LPL polymorphisms are shown in Figure 2.
Figure 1.
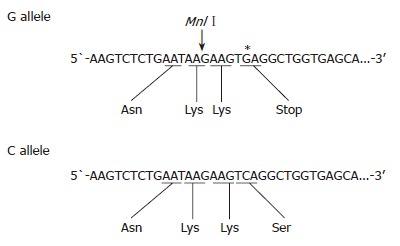
Strategy for detection of the C/G allele (Ser/stop) in exon 9 of the lipoprotein lipase by PCR-RFLP analysis. Introduction of G (*) is in place of C creates a new MnlI site.
Figure 2.
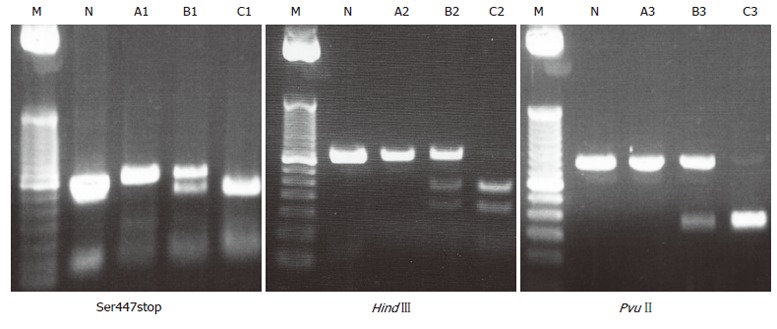
Electrophoresed pattern of LPL polymor-phism. M: Marker, N: No digestion. In Ser447stop, A1: C/C genotype (315 bp); B1: C/G genotype; C1: G/G genotype. In HindIII, A2: H+/+genotype; B2: H+/-genotype (350 bp); C2: H-/-genotype. In PvuII, A3: P+/+ genotype (431 bp); B3: P+/-genotype; C3: P-/-genotype.
Results of PCR-RFLP were confirmed by direct sequencing. DNA was extracted from agarose gels using an extraction kit (QIAGEN, Hilden, Germany). Then genotype was confirmed by sequence analysis using an auto sequencer (data not shown).
Statistical analysis
All data were analyzed by Excel 2000 and STASTISCA software. Clinical features of UC, allele frequency, and genotype distribution were evaluated by using χ2 test. A P value less than 0.05 was considered statistically significant.
Allelelic and genotype frequencies were determined from observed genotype counts, and the extensions of Hardy-Weinberg equilibrium were evaluated by using χ2 test.
RESULTS
LPL polymorphisms and risk of UC
The allele frequency of LPL polymorphism and the genotype distribution are shown in Table 2. The observed genotype data were consistent with Hardy-Weinberg equilibrium (Ser447stop: χ2 = 0.48, P = 0.49; HindIII:χ2 = 1.73, P = 0.19; PvuII: χ2 = 0.88, P = 0.35). In the patient group, one patient was not able to identify with genotype of HindIII polymorphism. In the control group, two patients were not able to identify with genotype of Ser447stop polymorphism. The allele frequency of Ser447stop polymorphism showed no difference between UC patients and controls. Frequencies of C/C, C/G and G/G genotypes of Ser447stop polymorphism in the control group were 78.8%, 18.3% and 2.9%, respectively, while these were not significantly different in patients. In the control group, frequencies of H+/+, H+/- and H-/- genotypes for HindIIIpolymorphism were 60.4%, 26.4% and 13.2%, respectively, while frequencies of P+/+, P+/- and P-/- genotype for PvuII polymorphism were 40.6%, 47.1% and 12.3%, respectively. In UC patients, neither HindIII nor PvuII polymorphisms differed significantly from genotype and allele frequencies in controls.
Table 2.
Allele frequency and genotype distribution of LPL polymorphism in UC patients and controls
| Allele frequency | Genotype (%) | OR (95% CI) | |||
| Ser447stop | C | G | CC | CG+GG | |
| UC (n = 131) | 0.92 | 0.08 | 111 (84.7) | 19 + 1 (15.3) | 1.49 |
| Control (n = 104) | 0.88 | 0.12 | 82 (78.8) | 19 + 3 (21.2) | (0.76-2.91) |
| HindIII | H+ | H- | H+/+ | H+/- + H-/- | |
| UC (n = 130) | 0.80 | 0.20 | 86 (66.1) | 37 + 7 (33.9) | 1.28 |
| Control (n = 106) | 0.74 | 0.26 | 64 (60.4) | 28 + 14 (39.6) | (0.75-2.18) |
| PvuII | P+ | P- | P+/+ | P+/- + P-/- | |
| UC (n = 131) | 0.70 | 0.30 | 65 (49.6) | 53 + 13 (50.4) | 1.44 |
| Control (n = 106) | 0.64 | 0.36 | 43 (40.6) | 50 + 13 (59.4) | (0.86-2.42) |
LPL polymorphism and characteristics of UC patients
We sought to identify associations between characteristics of UC patients (age at onset, gender, nature of the clinical course, extent of lesions, severity of colitis) and LPL polymorphisms. The relationship between age of onset of UC patients and their LPL polymorphisms are summarized in Table 3. In patients with onset at age 20 years or younger, more patients had either C/G or G/G genotype for Ser447stop than a C/C genotype (OR = 3.13, 95% CI = 0.95-10.33). In this early-onset group, more patients had either an H+/- or H-/- genotype for HindIII polymorphism than an H+/+ genotype (OR = 2.51, 95% CI = 0.85-7.45). Other characteristics (gender, nature of the clinical course, extent of lesions,severity of colitis) showed no significant differences between polymorphism-defined groups. Details of treatment among UC patients contained 5-aminosalicylic acid agents only (35 cases), steroid therapy (65 cases), operation (20 cases), other therapy (plasma exchange in 31 cases, immunosuppressant in 4 cases). We classified UC patients into a steroid-effective group (remission with a conventional steroid dose) and a steroid-resistant group (lack of such a remission, requiring surgery or other therapy, such as plasma exchange). We did not find an effect of LPL polymorphism on steroid effectiveness of UC patients.
Table 3.
Age of onset of UC patients and LPL polymorphisms
| Age of onset (yr) | Genotype | OR (95% CI) | |
| Ser447stop | C/G+G/G | C/C | |
| < 20 (n = 15) | 5 | 10 | 3.13 |
| ≥ 20 (n = 116) | 16 | 100 | (0.94-10.33) |
| HindIII | H+/- + H-/- | H+/+ | |
| < 20 (n = 15) | 8 | 7 | 2.31 |
| ≥ 20 (n = 115) | 36 | 79 | (0.84-7.45) |
| PvuII | P+/- + P-/- | P+/+ | |
| < 20 (n = 15) | 9 | 6 | 1.55 |
| ≥ 20 (n = 116) | 57 | 59 | (0.52-4.64) |
We also investigated serum total cholesterol in terms of LPL polymorphism in UC patients. First, 108 UC patients were classified into groups with total cholesterol below or above 220 mg/dL. LPL polymorphisms did not differ between these groups. Next, triglyceride concentration in 88 UC patients was divided into those below or above 150 mg/dL (Table 4). Patients with an H+/+ genotype for HindIII polymorphism were more likely to have triglyceride concentrations over 150 mg/dL than those with an H+/- or H-/- genotype (P < 0.01, OR = 6.46, 95% CI = 1.39-30.12). This was also true for patients with a P+/+ genotype for PvuII polymorphism compared with those with P+/- or P-/- genotype (P < 0.05, OR = 3.0, 95% CI = 1.06-8.50).
Table 4.
Triglyceride levels of UC patients and LPL polymorphisms
| Triglycerides (mg/dL) | Genotype | OR (95%CI) | |
| Ser447stop | C/C | C/G + G/G | |
| ≥ 150 (n = 20) | 19 | 1 | 4.93 (0.61-40.03) |
| < 150 (n = 68) | 54 | 14 | |
| HindIII | H+/+ | H+/- + H-/- | |
| ≥ 150 (n = 20) | 18b | 2 | 6.46 (1.39-30.12) |
| < 150 (n = 67) | 39 | 28 | |
| PvuII | P+/+ | P+/- + P-/- | |
| ≥ 150 (n = 20) | 13a | 7 | 3.0 (1.06-8.50) |
| < 150 (n = 68) | 26 | 42 | |
P < 0.05 vs P+/- + P-/- group;
P < 0.01 vs H+/- + H-/- group.
DISCUSSION
The LPL gene, located on chromosome 8p22[7], consists of 10 exons. As for the LPL polymorphisms investigated in this study, the Ser447stop polymorphism is within exon 9[8], involving substitution of G for C at nucleotide 1595. The HindIII polymorphism is located in intron 8[9,10], and PvuII in intron 6[9,11]. Abnormalities in LPL function have been associated with various diseases, those linked with LPL polymorphisms are cardiovascular disease[12], cerebrovascular disease[13], chylomycronemia[14], insulin resistance[15], Alzheimer´s disease[16] and various infections[17]. Life styles and habits also can influence many diseases, possibly including UC. We presently sought out the relationship between UC and the LPL gene.
This study is probably the first to report an association between characteristic of UC and LPL polymorphism, representing two important findings. The first finding was that HindIII and PvuII polymorphisms affected serum triglyceride concentrations in UC patients. Some studies reported that Ser447stop polymorphism activated LPL, decreasing triglycerides and increasing HDL cholesterol. As opposited to Ser447stop, HindIII polymorphism lowered LPL activity, and elevated triglycerides, and decreased HDL cholesterol. PvuII polymorphism has similar function[12,18]. Using a cutoff triglyceride value of 150 mg/dL, the above relationship for HindIII and PvuII polymorphisms held true in our UC patients. Patients homozygous for HindIII polymorphism were even more likely to have triglyceride above 150 mg/dL than those homozygous for PvuII polymorphism. HindIII polymorphism, therefore, would appear to suppress LPL activity more strongly than PvuII polymorphism. Among studies of associations between triglyceride in healthy individuals and LPL polymorphism, Ann et al[19] reported that HindIII polymorphism elevated triglycerides, while Chamberlain et al[18] pointed out some subjects with HindIII polymorphism showed normal concentrations. Since associations between elevated triglyceride in healthy individuals and HindIII polymorphism thus are a matter of some disagreement, our finding that UC patients with H+/+ genotype for LPL polymorphism were particularly likely to have elevated triglycerides, some part of the difference seen in our UC patients might be specific to UC.
Previous studies have reported that serum triglyceride concentrations did not differ between UC patients and controls[20,21]. In this study, however, patients with H+/+ genotype had serum triglyceride concentrations over 150 mg/dL compared to patients with H+/- or H-/-, thereby suggesting that HindIII polymorphism may contribute to elevate triglyceride levels directly or may influence other gene-elevated triglyceride concentrations. Although few studies have examined associations between UC and LPL, lipid intake has been reported to influence risk of IBD. Many studies have reported relationship between lipid metabolism and inflammation. During various human inflammatory states, serum triglyceride concentrations increase because some cytokines responsible for inflammatory responses, including tumor necrosis factor (TNF)-α, interferon (IFN)-γ, inhibit LPL activity[22]. Other studies reported that lipopolysaccarides derived from Gram-negative bacteria reduced LPL activity in macrophages[23]. LPL also directly induces the expression of the TNF-α gene[24], which synergizes with IFN-γ in stimulating nitric oxide synthetase expression in macrophages[25]. Since LPL is strongly associated with cytokines, it may contribute to development of inflammation. In addition, De Sanctis et al[26] reported an association between natural killer (NK) cells and LPL polymorphism. Some studies have suggested that UC is associated with changes in humoral immunity[27,28] and cellular immunity[29], while UC may be associated with loss of immune tolerance in the intestine. Association between LPL and characteristic of UC may shed some light on mechanisms of onset of UC.
The second finding was that Ser447stop polymorphism and HindIII polymorphism might be associated with age of onset of UC patients. In patients with onset at 20 years or younger, more patients had either C/G or G/G genotype at Ser447stop than a C/C genotype, while more patients had either H+/- or H-/- genotype for HindIII polymorphism than H+/+ genotype. As for the significance of relatively early onset, the LPL gene might influence onset of disease directly or by acting upon another gene or factor. In addition to genotype, age may affect LPL activity, while lipid metabolism might influence onset of IBD. Hamilton et al[30] reported that older individuals had inefficient triglyceride metabolism and elevated serum triglycerides because of reduction of LPL activity in postural skeletal muscle during aging. Among UC patients in our study, 10% (2/20) of those aged 20 to 29 years and 15% (4/26) of those aged 30 to 39 years showed triglycerides over 150 mg/dL, while Arai et al[31] reported that, in a Japanese population sample, the mean triglyceride concentration was 83 mg/dL in the third, and 118 mg/dL in the fourth decade of life. Our finding that younger UC patients appeared more likely to have high triglyceride concentration requires further investigation.
In conclusion, our study indicates that LPL polymor-phism influences lipid metabolism in UC patients and age of onset of UC, and might contribute to onset and biologic behavior of UC.
Many studies have considered gene polymorphisms in UC, but few have suggested an influence of polymorphism on metabolism in UC patients. Further study on LPL and other gene polymorphisms in UC remains an important line of investigation.
Footnotes
S- Editor Liu Y L- Editor Kumar M E- Editor Ma WH
References
- 1.Mead JR, Irvine SA, Ramji DP. Lipoprotein lipase: structure, function, regulation, and role in disease. J Mol Med (Berl) 2002;80:753–769. doi: 10.1007/s00109-002-0384-9. [DOI] [PubMed] [Google Scholar]
- 2.Merkel M, Eckel RH, Goldberg IJ. Lipoprotein lipase: genetics, lipid uptake, and regulation. J Lipid Res. 2002;43:1997–2006. doi: 10.1194/jlr.r200015-jlr200. [DOI] [PubMed] [Google Scholar]
- 3.Shoda R, Matsueda K, Yamato S, Umeda N. Epidemiologic analysis of Crohn disease in Japan: increased dietary intake of n-6 polyunsaturated fatty acids and animal protein relates to the increased incidence of Crohn disease in Japan. Am J Clin Nutr. 1996;63:741–745. doi: 10.1093/ajcn/63.5.741. [DOI] [PubMed] [Google Scholar]
- 4.Geerling BJ, Dagnelie PC, Badart-Smook A, Russel MG, Stockbrügger RW, Brummer RJ. Diet as a risk factor for the development of ulcerative colitis. Am J Gastroenterol. 2000;95:1008–1013. doi: 10.1111/j.1572-0241.2000.01942.x. [DOI] [PubMed] [Google Scholar]
- 5.Machida H, Tsukamoto K, Wen CY, Shikuwa S, Isomoto H, Mizuta Y, Takeshima F, Murase K, Matsumoto N, Murata I, et al. Crohn's disease in Japanese is associated with a SNP-haplotype of N-acetyltransferase 2 gene. World J Gastroenterol. 2005;11:4833–4837. doi: 10.3748/wjg.v11.i31.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narita S, Tsuchiya N, Wang L, Matsuura S, Ohyama C, Satoh S, Sato K, Ogawa O, Habuchi T, Kato T. Association of lipoprotein lipase gene polymorphism with risk of prostate cancer in a Japanese population. Int J Cancer. 2004;112:872–876. doi: 10.1002/ijc.20477. [DOI] [PubMed] [Google Scholar]
- 7.Sparkes RS, Zollman S, Klisak I, Kirchgessner TG, Komaromy MC, Mohandas T, Schotz MC, Lusis AJ. Human genes involved in lipolysis of plasma lipoproteins: mapping of loci for lipoprotein lipase to 8p22 and hepatic lipase to 15q21. Genomics. 1987;1:138–144. doi: 10.1016/0888-7543(87)90005-x. [DOI] [PubMed] [Google Scholar]
- 8.Hata A, Robertson M, Emi M, Lalouel JM. Direct detection and automated sequencing of individual alleles after electrophoretic strand separation: identification of a common nonsense mutation in exon 9 of the human lipoprotein lipase gene. Nucleic Acids Res. 1990;18:5407–5411. doi: 10.1093/nar/18.18.5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stocks J, Thorn JA, Galton DJ. Lipoprotein lipase genotypes for a common premature termination codon mutation detected by PCR-mediated site-directed mutagenesis and restriction digestion. J Lipid Res. 1992;33:853–857. [PubMed] [Google Scholar]
- 10.Heinzmann C, Ladias J, Antonarakis S, Kirchgessner T, Schotz M, Lusis AJ. RFLP for the human lipoprotein lipase (LPL) gene: HindIII. Nucleic Acids Res. 1987;15:6763. doi: 10.1093/nar/15.16.6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li SR, Oka K, Galton D, Stocks J. Pvu-II RFLP at the human lipoprotein lipase (LPL) gene locus. Nucleic Acids Res. 1988;16:2358. doi: 10.1093/nar/16.5.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thorn JA, Chamberlain JC, Alcolado JC, Oka K, Chan L, Stocks J, Galton DJ. Lipoprotein and hepatic lipase gene variants in coronary atherosclerosis. Atherosclerosis. 1990;85:55–60. doi: 10.1016/0021-9150(90)90182-i. [DOI] [PubMed] [Google Scholar]
- 13.Shimo-Nakanishi Y, Urabe T, Hattori N, Watanabe Y, Nagao T, Yokochi M, Hamamoto M, Mizuno Y. Polymorphism of the lipoprotein lipase gene and risk of atherothrombotic cerebral infarction in the Japanese. Stroke. 2001;32:1481–1486. doi: 10.1161/01.str.32.7.1481. [DOI] [PubMed] [Google Scholar]
- 14.Santamarina-Fojo S. The familial chylomicronemia syndrome. Endocrinol Metab Clin North Am. 1998;27:551–67, viii. doi: 10.1016/s0889-8529(05)70025-6. [DOI] [PubMed] [Google Scholar]
- 15.Simsolo RB, Ong JM, Saffari B, Kern PA. Effect of improved diabetes control on the expression of lipoprotein lipase in human adipose tissue. J Lipid Res. 1992;33:89–95. [PubMed] [Google Scholar]
- 16.Baum L, Chen L, Masliah E, Chan YS, Ng HK, Pang CP. Lipoprotein lipase mutations and Alzheimer's disease. Am J Med Genet. 1999;88:136–139. doi: 10.1002/(sici)1096-8628(19990416)88:2<136::aid-ajmg8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 17.Hardardóttir I, Grünfeld C, Feingold KR. Effects of endotoxin and cytokines on lipid metabolism. Curr Opin Lipidol. 1994;5:207–215. doi: 10.1097/00041433-199405030-00008. [DOI] [PubMed] [Google Scholar]
- 18.Chamberlain JC, Thorn JA, Oka K, Galton DJ, Stocks J. DNA polymorphisms at the lipoprotein lipase gene: associations in normal and hypertriglyceridaemic subjects. Atherosclerosis. 1989;79:85–91. doi: 10.1016/0021-9150(89)90037-3. [DOI] [PubMed] [Google Scholar]
- 19.Ahn YI, Kamboh MI, Hamman RF, Cole SA, Ferrell RE. Two DNA polymorphisms in the lipoprotein lipase gene and their associations with factors related to cardiovascular disease. J Lipid Res. 1993;34:421–428. [PubMed] [Google Scholar]
- 20.Capristo E, Mingrone G, Addolorato G, Greco AV, Gasbarrini G. Glucose metabolism and insulin sensitivity in inactive inflammatory bowel disease. Aliment Pharmacol Ther. 1999;13:209–217. doi: 10.1046/j.1365-2036.1999.00461.x. [DOI] [PubMed] [Google Scholar]
- 21.Iannello S, Cavaleri A, Milazzo P, Cantarella S, Belfiore F. Low fasting serum triglyceride level as a precocious marker of autoimmune disorders. MedGenMed. 2003;5:20. [PubMed] [Google Scholar]
- 22.Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45:1169–1196. doi: 10.1194/jlr.R300019-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.White JR, Chait A, Klebanoff SJ, Deeb S, Brunzell JD. Bacterial lipopolysaccharide reduces macrophage lipoprotein lipase levels: an effect that is independent of tumor necrosis factor. J Lipid Res. 1988;29:1379–1385. [PubMed] [Google Scholar]
- 24.Renier G, Skamene E, DeSanctis JB, Radzioch D. Induction of tumor necrosis factor alpha gene expression by lipoprotein lipase. J Lipid Res. 1994;35:271–278. [PubMed] [Google Scholar]
- 25.Renier G, Lambert A. Lipoprotein lipase synergizes with interferon gamma to induce macrophage nitric oxide synthetase mRNA expression and nitric oxide production. Arterioscler Thromb Vasc Biol. 1995;15:392–399. doi: 10.1161/01.atv.15.3.392. [DOI] [PubMed] [Google Scholar]
- 26.de Sanctis JB, Blanca I, Radzioch D, Bianco NE. Lipoprotein lipase expression in natural killer cells and its role in their cytotoxic activity. Immunology. 1994;83:232–239. [PMC free article] [PubMed] [Google Scholar]
- 27.Gionchetti P, Rizzello F, Venturi A, Brigidi P, Matteuzzi D, Bazzocchi G, Poggioli G, Miglioli M, Campieri M. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: a double-blind, placebo-controlled trial. Gastroenterology. 2000;119:305–309. doi: 10.1053/gast.2000.9370. [DOI] [PubMed] [Google Scholar]
- 28.Geng X, Biancone L, Dai HH, Lin JJ, Yoshizaki N, Dasgupta A, Pallone F, Das KM. Tropomyosin isoforms in intestinal mucosa: production of autoantibodies to tropomyosin isoforms in ulcerative colitis. Gastroenterology. 1998;114:912–922. doi: 10.1016/s0016-5085(98)70310-5. [DOI] [PubMed] [Google Scholar]
- 29.Makita S, Kanai T, Oshima S, Uraushihara K, Totsuka T, Sawada T, Nakamura T, Koganei K, Fukushima T, Watanabe M. CD4+CD25bright T cells in human intestinal lamina propria as regulatory cells. J Immunol. 2004;173:3119–3130. doi: 10.4049/jimmunol.173.5.3119. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton MT, Areiqat E, Hamilton DG, Bey L. Plasma triglyceride metabolism in humans and rats during aging and physical inactivity. Int J Sport Nutr Exerc Metab. 2001;11 Suppl:S97–104. doi: 10.1123/ijsnem.11.s1.s97. [DOI] [PubMed] [Google Scholar]
- 31.Arai H, Yamamoto A, Matsuzawa Y, Saito Y, Yamada N, Oikawa S, Mabuchi H, Teramoto T, Sasaki J, Nakaya N, et al. Serum lipid survey and its recent trend in the general Japanese population in 2000. J Atheroscler Thromb. 2005;12:98–106. doi: 10.5551/jat.12.98. [DOI] [PubMed] [Google Scholar]