

Abstract
Spore photoproduct lyase (SPL), a member of the radical SAM superfamily, catalyzes the direct reversal of the spore photoproduct (SP), a thymine dimer specific to bacterial spores, to two thymines. SPL requires S-adenosyl-L-methionine (SAM) and a redox active [4Fe-4S] cluster for catalysis. Mössbauer analysis of anaerobically purified SPL indicates the presence of a mixture of cluster states with the majority (40%) as [2Fe-2S]2+ and a smaller amount (15%) as [4Fe-4S]2+ clusters. Upon reduction, the cluster content changes to primarily (60%) [4Fe-4S]+. The speciation information from Mössbauer data allowed us to deconvolute iron and sulfur K-edge X-ray absorption spectra to uncover electronic (XANES) and geometric (EXAFS) structural features of the Fe-S clusters, and their interactions with SAM. The Fe K-edge EXAFS provide evidence for elongation of a [2Fe-2S] rhomb of the [4Fe-4S] cluster upon binding SAM on the basis of an Fe…Fe scatterer at 3.0 Å. The XANES spectra of reduced SPL in the absence and presence of SAM overlay, indicating that SAM is not undergoing reductive cleavage. The XAS data for SPL samples and data for model complexes from literature allowed for the deconvolution of contributions from [2Fe-2S] and [4Fe-4S] clusters to the sulfur K-edge XANES spectra. The analysis of pre-edge features revealed electronic changes in the Fe-S clusters as a function of SAM presence. The spectroscopic findings were further corroborated by density functional theory calculations that provided insights into structural and electronic perturbations that can be correlated by considering the role of SAM as a catalyst or substrate.
Keywords: Spore photoproduct lyase, radical SAM, iron-sulfur cluster, Mössbauer spectroscopy, X-ray absorption spectroscopy, density functional theory
Introduction
Members of the radical S-adenosyl-L-methionine (SAM) superfamily utilize S-adenosyl-L-methionine (SAM) and a redox active [4Fe-4S] cluster to carry out diverse radical reactions including rearrangements, sulfur insertions and oxidations. They also function in DNA precursor, vitamin, cofactor, and antibiotic biosynthesis pathways, as well as the assembly of complex metallocofactors [6–8]. Enzymes in this superfamily typically contain a conserved CX3CX2C sequence that coordinates three of the iron atoms in the cluster. Electron-nuclear double resonance and X-ray crystallography of several members of the superfamily have shown that the unique iron is coordinated by the amino and carboxylate moieties of SAM [2–5, 9–15]. Mechanistic studies of these enzymes suggest that an inner-sphere electron transfer from a reduced [4Fe-4S]+ cluster to the sulfonium of SAM leads to homolytic S-C(5′) bond cleavage to generate a 5′-deoxyadenosyl radical (dAdo•) intermediate, which abstracts a hydrogen atom from substrate to initiate a radical transformation [1, 10–12, 16, 17].
Spore photoproduct lyase (SPL) is a member of the radical SAM superfamily that catalyzes the direct reversal of a specific DNA photoproduct, 5-thyminyl-5,6-dihydrothymine (spore photoproduct or SP), back to two thymines (Fig. 1) [18–21]. The methylene-bridged thymine dimer SP is the primary photoproduct when bacterial spores are subjected to UV radiation, and is rapidly repaired upon spore germination [19, 20, 22, 23]. The mechanism of SP repair has been the subject of considerable interest. We have previously demonstrated that SP repair is initiated by abstraction of a H atom from C6 of SP [21, 24]. The SP(C6) substrate radical is thought to promote a radical-mediated β-scission of the C-C bond linking the two thymines; the resulting product radical then abstracts an H atom to generate repaired thymine. While it was initially proposed that the product radical directly abstracted an H atom from 5′-deoxyadenosine to generate dAdo• [21, 24, 25], more recent results support the involvement of intermediary amino acids in the regeneration of dAdo• [26–29]. Regardless of the pathway of the radical transfer involved in the final steps of the reaction, the proposed SPL mechanism suggests a catalytic role for SAM; such a catalytic role for SAM has been supported experimentally by the observation of tritium transfer from C6 of SP to SAM (not dAdo) and by the observation that a single SAM molecule can mediate the repair of hundreds of SP photoproducts [21, 24]. Other studies, however, have provided evidence that one SAM is cleaved to methionine and dAdo• for each SP repaired [18, 30, 31]. Most but not all of the studies showing evidence for SAM as a substrate utilize a defined dinucleotide or dinucleoside SP, rather than SP in intact DNA, suggesting the possibility that stoichiometric SAM cleavage is favored with non-optimal substrates.
Fig. 1.
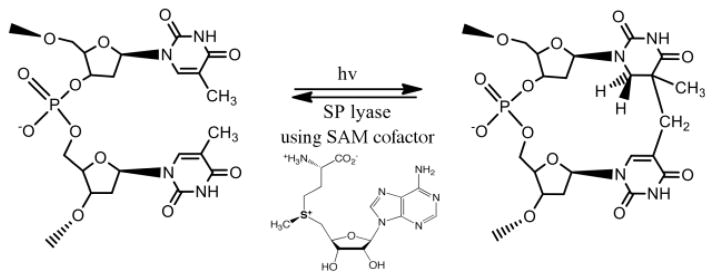
Formation and repair of SP by SPL
The Carell laboratory has published a crystal structure of SPL from Geobacillus thermo-denitrificans in complex with an SP lesion that illustrates the topological problem associated with direct H-atom abstraction from dAdo• by the product radical, with a distance between these two species of approximately 10 Å [28]. Between these two species in the crystal structure; however, are the conserved cysteine as well as a tyrosine residue, and a recent report from the Carell lab supports a role for a tyrosyl radical in catalysis [29]. The stereospecificity of SP repair by SPL has also been of considerable interest, and the enzyme has now been shown to repair only the 5R diastereomer of SP [32–35], despite earlier reports pointing to the opposite stereospecificity [36, 37].
Spectroscopic studies of SPL have demonstrated the presence of a single iron-sulfur cluster [18] in a mixture of cluster states when the enzyme is purified in vitro [24, 33, 37, 38]. The UV-visible absorbance spectrum of purified SPL shows a broad shoulder with a maximum of 413 nm, similar to other members of the radical SAM superfamily [33, 39, 40], with an additional broad feature around 600 nm that may indicate some [2Fe-2S] cluster content in the enzyme [41]. Upon reduction in the presence of dithiothreitol (DTT) and 5-deazariboflavin, the UV-visible spectral features decrease in intensity resulting in a single broad peak at ~410 nm. The changes in the UV-visible spectral absorbance indicate a redox-active Fe-S cluster, but do not address in detail the speciation of clusters present. As-purified SPL exhibits a nearly isotropic electron paramagnetic resonance spectroscopy (EPR) signal centered at g = 1.99 that is characteristic of a [3Fe-4S]1+ cluster, but which accounts for only 3 – 4% of the total iron [33]. Reduction of the enzyme results in a nearly axial signal with g values of 2.03, 1.93 and 1.92, and is consistent with a [4Fe-4S]+ cluster [33]. This signal accounts for 32–45 % of the total iron, indicating either incomplete reduction (leaving some in an EPR-silent state) and/or sub-stoichiometric Fe-S cluster in the purified protein. Addition of SAM to the reduced enzyme changes the g-values to 2.03, 1.92 and 1.82 and results in significantly reduced signal intensity [33]. These spectral changes are specific for the cluster/SAM interaction, as similar molecules (5′-deoxyadenosine, methionine and S-adenosyl-L-homocysteine) do not yield equivalent EPR spectral changes when added to reduced SPL [33]. Further evidence for coordination of SAM to the Fe-S cluster of SPL, in a manner similar to other radical SAM enzymes, has been provided by 2D-pulsed EPR technique, HYSCORE [38].
In this study, we carried out a combined biochemical, Mössbauer, and X-ray absorption spectroscopic (XAS) study quantitatively addressing the nature of cluster types present in the as-purified and reduced forms of SPL in the absence and presence of SAM. Mössbauer studies were essential in identifying and quantifying the speciation of cluster types. These speciation results and the rich literature of Fe-S model clusters were used as constraints in fitting and deconvoluting the X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) features at the Fe and S K-edges. Electronic and geometric information were obtained from XAS results about the cluster states and the interaction of SAM with the Fe-S clusters. The experimental structural insights and biochemical composition data were further analyzed by density functional theory-based (DFT) modeling. Although within the given study we do not provide direct molecular level description of the reaction mechanism of SPL, we utilized the wealth of structural information from our work and from the literature to generate experimentally testable hypothesis for potentially predicting a priori whether SAM cofactor will act as a catalyst or a substrate.
Materials and methods
All chemicals and other materials were obtained from commercial sources and were of the highest purity available, unless indicated otherwise.
SPL Expression and purification
The N-terminal His6-tagged SPL from Clostridium acetobutylicum (C.a.) was expressed using E. coli Tuner(DE3)pLysS cells transformed with a pET14b expression plasmid, containing the splB gene, grown in minimal media and purified anaerobically by Ni-HisTrap HP chromatography and FPLC as previously described [33]. To prepare selenomethionine (SeMet) labeled SPL, the N-terminal His6-tagged SPL from C.a. was expressed using E. coli B834(DE3)pLysS cells transformed with a pET14b expression plasmid, containing the splB gene, grown in SeMet media base (Molecular Dimensions/AthenasES) and purified anaerobically by Ni-HisTrap HP chromatography and FPLC, as previously described [33]. Following anaerobic dialysis in 20 mM sodium phosphate, 350 mM NaCl, 5% glycerol, pH 7.5, the enzyme was concentrated using an Amicon concentrator fitted with YM-10 membrane to a final concentration of 0.49 – 0.67 mM. All XAS samples were prepared with 30 volume % glycerol to avoid ice formation in an MBRAUN glove box maintained at less than 1 ppm O2 and immediately frozen in liquid N2 after removal from the antechamber. Protein, iron and sulfide assays were performed as previously described [42–44]. SAM was synthesized as previously described [10].
Preparation of Mössbauer samples
57Fe was purchased from Cambridge Isotope Laboratories, Inc., and dissolved in hot concentrated hydrochloric acid. The pH was adjusted to 7.0 – 7.2 with NaOH. SPL as-purified 57Fe samples were prepared from 10 L cultures using the defined minimal medium previously described with 57Fe substituted at the same molar concentrations for 56Fe. 57Fe (191 μM) was also added at induction with IPTG. SPL was anaerobically purified, dialyzed and concentrated as described above. Reduced samples were prepared by adding 5 mM DTT, 50 mM tris(hydroxymethyl)aminomethane, 100 μM solution of 5-deazariboflavin and illuminating the sample with a 300 W halogen lamp for 1 hr in an EPR tube placed in an ice-water bath. SAM was added to appropriate samples at a final concentration of 3 mM as a final step in preparation. Protein samples (640 μM) were loaded into 450 μL cups and immediately stored under liquid nitrogen.
Preparation of XAS samples
Iron K-edge XAS samples were prepared in two different ways. The samples were first prepared using SPL protein (0.59 mM) expressed in Tuner(DE3)pLysS E. coli cells as described above. As-purified samples were prepared from the anaerobically purified/dialyzed enzyme in the absence and presence of SAM (3 mM). Reduced samples were prepared by the method described above and SAM (3 mM) was added to the photo-reduced enzyme as a final step in preparation. A second set of samples was prepared mainly for S K-edge XAS measurements using SeMet labeled SPL (0.49 or 0.67 mM) and equilibrated by Zn-dust reduced methyl viologen (3 mM) for about 1 hour. SAM (3 mM) was added to appropriate samples. On separate synchrotron data collection trips the Fe K-edge XAS data were also collected to confirm that presence of SeMet does not alter the structure of the Fe-S clusters. About 100 μL aliquot of each sample was loaded into a XAS Delrin pinhole cell sealed with thin (~5 μm) iron- and sulfur-free Kapton tape (Certispec) and frozen in liquid N2.
Mössbauer spectroscopy
Mössbauer spectra were recorded on a Mössbauer spectrometer equipped with a Janis 8DT variable temperature cryostat and operated at a constant acceleration mode in transmission geometry. The zero velocity refers to the centroid of a room-temperature spectrum of a metallic iron foil. Analysis of the spectra was performed with WMOSS (WEB Research).
X-ray absorption spectroscopy
The X-ray absorption spectroscopic measurements were carried out at Stanford Synchrotron Radiation Lightsource (SSRL) under storage ring (SPEAR3) condition of 3 GeV and current of 80–200 mA. Data for the X-ray Absorption Near-Edge Structure (XANES) and Extended X-ray Absorption Fine-Structure (EXAFS) analyses at the iron K-edge were collected at the 20-pole, 2 T Wiggler beamline 7–3 (BL7-3) equipped with a Si(220) downward reflecting, double-crystal monochromator. A 30-element Ge solid-state detector windowed at the Fe Kα emission line was used to collect data as fluorescence excitation spectra. The scattered beam intensity was attenuated by Soller slits and a few μm think Mn “Z-1” filter. The energy was calibrated using the first peak of the first derivative of an iron foil assigned to 7111.2 eV. During the measurement, the sample was in a liquid helium cryostat at a constant temperature of ~10 K. The beamline parameters were optimized at 8000 eV.
The sulfur K-edge XAS were collected on BL6-2. The measurements utilized a 54-pole wiggler beamline operating in high field mode of 10 kG with a Ni-coated harmonic rejection mirror and a fully tuned Si(111) crystal monochromator. The energy was calibrated using the first peak of thiosulfate powder assigned to 2472.0 eV. During the measurement, the sample was cooled by a liquid helium cryo-flow to an approximate temperature of 100 K. Details of the beamline optimization for S Kedge XAS studies have been published elsewhere [45].
The data is the average of at least five scans before background subtraction and normalization. Background correction and normalization of Fe and S K-edge XANES were performed using ADRP (Automated Data Reduction Protocol) [46]. The background removal for EXAFS was performed using AUTOBK algorithm [47], implemented with ATHENA [48] as the graphical interface. FEFFIT [49] and FEFF 8.4 [50] implemented in ARTEMIS [48] were used to refine and model the EXAFS features, respectively. Due to data collection issues, the XANES region of the reduced SPL samples in the presence of SAM had to be reconstructed as an average of all available spectra. This procedure introduced a less than 5% intensity variation along the rising edge and had a negligible effect for the EXAFS in the k = 0–3 Å−1 region. Due to the presence of multiple Fe-S clusters forms, the structural models for amplitude and phase information calculation were derived from combination of average Fe…Fe, Fe-S(sulfide), and Fe-S(thiolate) distances of reduced and oxidized FeS model compounds [51]. A non-linear least-squares minimization is used to vary the model and optimize the calculated fit to the observed EXAFS [49]. The mixed cluster speciation of the samples can readily lead to various acceptable fits with R(fit) < 15%, thus all fits were strictly constrained to speciation information from biochemical and Mössbauer data.
The quantitative analysis of the sulfur K-edge pre-edge features was accomplished by considering the concentration of sulfur absorbers in different chemical states. A normalized spline function fit to the post-edge region at 2480 eV was then renormalized by taking into account the number of sulfide ions per protein subunit from the sulfide assay, the number of cysteine residues per protein unit in a SeMet background from the amino acid sequence, protein concentrations, and the concentration of SAM added. These all contribute to the height of the edge jump; however, the pre-edge intensities will only be affected by the presence of Fe-S bonds. The pre-edge intensities were further corrected by the total number Fe centers present in the S-bound form as defined by the Mössbauer analysis.
Density functional theory-based modeling
The 2.77 Å resolution crystal structure of the pyruvate formate-lyase activating enzyme (PFL-AE) in complex with SAM [2] was used to generate the initial computational model for the follow up evaluation of the SAM interaction with the [4Fe-4S] cluster expressed by the Fe…Fe distances. In the time since we completed the computational analysis, a crystal structure for SPL at 2 Å resolution was published [28]. The comparison of the SAM position relative to the [4Fe-4S] cluster as well as the orientation of the three Cys residues coordinated to the [4Fe-4S] cluster (Supporting Information - Fig. S1) showed that the cluster and its inner sphere coordination environment of these two enzymes align very closely and no significant differences are expected to the computational results discussed here using the newer structure. Furthermore, the 2 Å crystal structure resolution limits the accuracy of the atomic positions of the iron and sulfur centers of the redox active site; however, the orientation of the three cysteine residues anchoring the Fe-S clusters can be considered chemically meaningful. Therefore, in the computational model, both the α- and β-C atoms of the coordinating cysteines were kept frozen to simulate the maximum protein strain imposed onto the Fe-S cluster. The electronic structure of the computational cluster model was carefully evaluated by considering all possible combinations of ferro- and antiferromagnetically coupled [2Fe-2S] rhombs with formally Fe2.5+ ions using the GIFA approach [52] (see electronic supporting information). Regardless of the initial electronic structure, the optimization state, or even the presence and nature of a ligand at the unique Fe site, the lowest energy spin coupling scheme was obtained when the Fe1 and Fe2 centers (PFL-AE structure 3CB8 [2]) were coupled ferromagnetically giving a MS = +9/2 spin state and this [2Fe-2S] rhomb was antiferromagnetically coupled with the ferromagnetically coupled Fe3 and Fe4 centers with MS = −9/2. The Fe1 is the coordinatively unique iron of the [4Fe-4S] cluster in PFL-AE, and the Fe2, Fe3, Fe4 centers are connected to the protein matrix via Cys29, Cys33, and Cys36 residues for PFL and Cys90, Cys94, and Cys97, for SPL; respectively. For a representative orientation, the rhomb formed by the Fe1 and Fe2 can be placed on top with Cys29 or Cys90 pointing the opposite direction to SAM. The second rhomb formed by Fe3 and Fe4 at the bottom has the Cys33 or Cys94 ligands in the back and the Cys36 or Cys97 in the front for PFL and SPL structures, respectively. The Cys residues were modeled as ethylthiolate ligands. All other atoms, including the computational model for SAM, where the nucleotide moiety was truncated to a H atom, were optimized at the BP86/TZVP level [53–55]. The electrostatic environment of the protein matrix was approximated using a water-based polarizable continuum model [56–58] with a commonly used dielectric constant of ε = 10 for polar protein environment [59]. The effect of the specific value of the dielectric constant in going from 10 to 40 units for the relative energies is expected to be at most 0.2 eV or 20 kJ/mol [60].
Results and analysis
Mössbauer spectroscopy
Mössbauer spectra of 57Fe-enriched SPL in four different states of the enzyme are presented in Figure 2: anaerobically purified SPL (A), as-purified enzyme with SAM (B), photo-reduced enzyme (C), and reduced enzyme with SAM (D). Their spectra were recorded at 4.2 K in a magnetic field of 50 mT, applied parallel to the γ radiation. The complexity of spectral features indicates that all four states of the enzyme contain multiple Fe species (Table 1). However, the spectra of the as-purified enzyme in the absence (A) and presence (B) of SAM are very similar, comprising a central quadrupole doublet, accounting for 55% of the total Fe absorption, and an ill-defined broad magnetic spectrum in the range of ±6 mm/s. The central quadrupole doublet can be further decomposed into two unresolved doublets. The major doublet (red lines in A and B), accounts for 40% of the Fe absorption and can be simulated as a superposition of two equal-intensity quadrupole doublets with parameters (ΔEQ(1) = 0.75 mm/s, δ(1) = 0.27 mm/s, and linewidth(1) = 0.27 mm/s; ΔEQ(2) = 0.46 mm/s, δ(2) = 0.27 mm/s, and linewidth(2) = 0.27 mm/s). These spectroscopic parameters indicate the presence of [2Fe-2S]2+ cluster [61]. The minor doublet (blue lines in A and B) accounts for 15% of the Fe absorption, and can be simulated as a superposition of two equal-intensity quadrupole doublets with parameters (ΔEQ(1) = 1.00 mm/s, δ(1) = 0.47 mm/s, and linewidth(1) = 0.31 mm/s; ΔEQ(2) = 1.20 mm/s, δ(2) = 0.48 mm/s, and linewidth(2) = 0.32 mm/s). These are typical for [4Fe-4S]2+ clusters [61]. As the Fe absorption percentages for the [2Fe-2S]2+ and [4Fe-4S]2+ clusters remain the same after addition of SAM, it appears that SAM has no major effects on the composition and oxidation states of the Fe-S clusters in the purified enzyme. However, as discussed later XAS data suggest a fine-tuning Fe-S electronic structure upon binding. The broad magnetic spectrum is attributed to high-spin ferric complexes, representing likely adventitious Fe bound to the N-terminal His6 tag on SPL that is utilized for purification.
Fig. 2.

Mössbauer spectra of as-purified SPL: (A) as-purified with 3 mM SAM (B), photo-reduced with 5-deazariboflavin in the presence of 5 mM DTT (C), and photo-reduced sample with 3 mM SAM (D). The spectra (hatched marks) were recorded at 4.2 K in a magnetic field of 50 mT applied parallel to the γ radiation. The solid lines plotted above the data are simulated spectra for the [2Fe-2S]2+ clusters (red), [4Fe-4S]2+ clusters (blue), [4Fe-4S]+ clusters (cyan), N/O-coordinated Fe2+ (green), S- coordinated Fe2+ (magenta), and fast relaxing Fe3+ (orange). The black lines overlaid with the experimental spectra in C and D are composite spectra. Parameters used for the simulations are given in the text
Table 1.
Iron speciation in the various states of SPL form Mössbauer spectroscopy for two samples with iron and sulfur concentrations of 2.3/3.1±0.2 and 2.4/3.6±0.2 per monomer, respectively
| State of SPL | [2Fe-2S]2+ | [4Fe-4S]2+ | [4Fe-4S]+ | Fe2+-S | Fe2+-N/O | Fe3+ |
|---|---|---|---|---|---|---|
| as-purified | 40 | 15 | - | - | - | 45% |
| as-purified + SAM | 40 | 15 | - | - | - | 45% |
| photoreduced | - | - | 60 | 10 | 15 | 15% |
| reduced + SAM | - | - | 45 | 13 | 12 | 25% |
The spectra of the reduced enzyme in the absence (C) and presence (D) of SAM are also quite similar, but very different than the as-purified samples (A) and (B). These spectra indicate the presence of numerous chemically distinct iron environments. The solid black lines overlaid with the experimental spectra shown in C and D are the results of decomposing the spectra into four components. They are the composite of simulated spectra of (1) spin S = 1/2 [4Fe-4S]+ cluster (cyan lines: plotted at 60% of total Fe absorption in C and 45% in D), (2) and (3) two Fe2+ species (green lines: Fe2+ coordinated with N/O ligands in 10 and 13% in C and D, respectively; magenta lines: Fe2+ coordinated with S ligands in 15 and 12 % in C and D, respectively), and (4) fast relaxing Fe3+ species (orange line: 15% in C and 25% in D). The S=1/2 [4Fe-4S]+ cluster was simulated using the parameters obtained for the 4Fe-ferredoxin from Bacillus stearothermophilus [62]. The N/O-coordinated Fe2+ component was simulated using the parameters ΔEQ = 2.92 mm/s, and δ = 1.32 mm/s. The simulation of the sulfur coordinated Fe2+ component was done by ΔEQ = 3.26 mm/s, and δ = 0.72 mm/s as typical values for FeS4 site in rubredoxin [61]. The spectrum of the fast relaxing Fe3+ (orange line) was simulated with a broad quadrupole doublet with ΔEQ = 1.09 mm/s, δ = 0.59 mm/s, and a broad linewidth of 0.58 mm/s. The reduction of the reduced [4Fe-4S]+ cluster content in D compared to C may be interpreted as electron transfer to SAM and consequent reductive cleavage of the S-C bond; however, 15% difference does not appear to be in the form of oxidized [4Fe-4S]2+ cluster. Furthermore, the presence of the sulfonium S-C σ* feature at the sulfur K-edge spectra and the rising-edge inflection points of the Fe K-edge spectra clearly indicate the absence of SAM turnover reactivity.
Two independently prepared, anaerobically purified SPL samples were found to contain iron (2.3 and 3.1±0.2 per monomer) and acid-labile sulfide (2.4 and 3.6±0.2 per monomer) with content dependent upon preparation. Since SPL requires a [4Fe-4S] cluster for catalysis, the iron and sulfide contents indicate the presence of incompletely assembled [4Fe-4S] cluster. Furthermore, the Mössbauer data presented in Fig. 2 and Table 1 also demonstrate the presence of a mixture of cluster states and composition in the anaerobically prepared SPL. The incomplete cluster content and distribution of cluster states observed in SPL are likely artifacts due to the in vitro purification procedure, which is not unusual for radical SAM enzymes. It is important to note that upon photo-reduction the only cluster species present in SPL is the catalytically active [4Fe-4S]+ state.
Fe K-edge X-ray absorption spectroscopy - EXAFS analysis
XAS spectra of two independently prepared SPL samples (0.59 and 0.67 mM) were recorded at the Fe K-edge. The Fe K-edge spectra were reproducible for independently prepared protein samples and for data collected during different data collection beamtimes.
The results of EXAFS analysis for the Fe Kedge spectra of the same four SPL states characterized by Mössbauer are presented in Fig. 3. The Fourier transforms (FT) of Fe EXAFS data in Fig. 3B show two well-resolved peaks in the range of 1.5–3.2 Å, which are indicative of at least two different Fe…backscatter shells. The major peak at 2.25 Å corresponds to the Fe-S scattering path, where the S originates either from the sulfide and thiolate (Cys) ligands. The momentum (k) space sampling of up to 15 Å−1 and the loss of data quality at around 12.5 Å−1, in addition to the Fe-S cluster inhomogeneity, limit the resolution of the two different scattering paths. The as-purified and reduced samples show another intense and well-resolved peak at 2.7 Å that can be related to Fe…Fe scattering path. The EXAFS of as-purified SPL in the presence of SAM shows an additional shoulder on the Fe…Fe scattering peak at ~3.0 Å. Modeling of this feature with various Fe-X scatterers as shown in Fig. 4A suggests that it arises from a longer Fe…Fe scattering path on the basis of the similarity of phase and amplitude functions of the back-transformed FTIR window as indicated in the lower inset in Fig. 4A.
Fig. 3.

Fe K-edge EXAFS of as-purified (black), as-purified in the presence of SAM (red), reduced SPL (blue), and reduced SPL sample in the presence of SAM (green). (B) FT of the EXAFS spectra shown in (A). (B) Inset: The [4Fe-4S] cluster of HydE (PDB code: 3IIZ [1]) with Fe-S distances at 1.6 Å resolution
Fig. 4.

Fourier Transform of Fe K-edge EXAFS of as-purified (A) and reduced (B) SPL in the presence of SAM with the EXAFS spectra shown as upper insets (black: data; red: fits). Inset in (A): back-transformation of R range of 2.9–3.3 Å for comparison of various Fe-X scattering paths
Other features of the Fe K-edge EXAFS data are consistent with the experimental speciation results (Table 1). While the Mössbauer derived, Fe speciation data have limitations in inferring structural information for EXAFS analysis, chemically reasonable assumptions can be used to deconvolute the major spectral features and gain structure information for mixtures. As an example, the 45% ill-defined paramagnetic ferric species in the as-purified sample that is likely associated with the His-tag, and the 15–25% fast relaxing ferric species of the photoreduced sample can be modeled as hexacoordinate Fe site with O/N ligands. Similarly, the N/O coordinated ferrous component of the Mössbauer spectrum was modeled in EXAFS with a hexacoordinate complex and Fe…O/N scatterers at around 2.0 Å. The less then 25% contribution from a rubredoxin type ferrous absorber would show up under the main scattering feature at around 2.25 Å in Figs. 3 and 4 and cannot be resolved from Fe-S cluster features. However, the sulfur Kedge analysis will differentiate between Fe-S(thiolate) and Fe-S(sulfide) bond.
Fe K-edge X-ray absorption spectroscopy - XANES analysis
The Fe K-edge XANES spectra for as-purified and reduced SPL in the absence and presence of SAM are presented in Fig. 5B. The spectra for FeCl2, FeCl3, FeF2 and FeF3 are provided in Fig. 5A as a reference for the energy shift of the rising-edge position as a function of effective Fe oxidation states or the iron effective nuclear charge seen by the Fe core 1s electrons. The Kedge features are the result of the core 1s→valence np exitations. Thus, they are sensitive to changes in the absorber’s effective nuclear charge (Zeff). As the oxidation state increases and ligand environment becomes less covalent, the position of the edge shifts up in energy due to the increase in Zeff(Fe). This is well demonstrated by the spectral changes in Fig. 5A for a series of iron fluoride and chloride compounds. In going from a formally ferrous to ferric state, the edge positions shift by ~3 and ~5 eV higher for the chloride and fluoride salts, respectively. Changes in spectral features as a function of the ligand environment are also apparent. The more ionic fluoride ligand shifts up in energy the Fe K-edge rising-edge position compared to the chloride salts with more covalent Fe-Cl bonds.
Fig. 5.

Normalized Fe K-edge XANES of FeCl2, FeCl3, FeF2 and FeF3 (A) and as-purified and reduced SPL in the absence and presence of SAM (B). Samples were prepared using 0.59 mM C.a. SPL. SAM was added to a final concentration of 3 mM Inset: Pre-edge region of Fe K-edge XANES
By applying the same concepts to analysis of the protein data (Fig. 5B), the reduction of SPL from the [4Fe-4S]2+ to [4Fe-4S]+ cluster state is accompanied by a shift of ~1.2 eV to lower energy. The energy shift is smaller than in the iron chlorides (~2 eV) due to the reduction being delocalized over four irons. In addition, the sulfur ligands further reduce the magnitude of the change, since they are involved in the redox process. The slight spectral variations in the rising-edge inflection points among the oxidized (black and red spectra, Fig. 5B) and separately among the reduced SPL samples (green and blue spectra, Fig. 5B) are essentially at the resolution limit of Fe K-edge XAS data at beamline 7–3 of SSRL. However, the practically identical XANES spectra for the reduced SPL in the presence (green trace) and absence (blue trace) of SAM indicates that the reduced [4Fe-4S]+ cluster has not cleaved SAM, since the average effective oxidation state of Fe absorbers remains the same. This was further ensured by sequentially adding SAM after the illumination of deazaflavin had ceased. The presence of intact SAM cofactor less the hydrolytic decomposition was also supported by HPLC measurements [33].
The change in the pre-edge features is indicative of electronic structure perturbations upon SAM binding that are discussed in more detail for the S K-edge XANES analysis. The pre-edge features are a result of the 1s→3d quadrupole allowed transitions that gain intensity upon 3d-4p mixing due to the non-centrosymmetric coordination environment [63, 64]. The reference iron halides have octahedral crystal fields with negligible 3d-4p mixing, and thus the weak pre-edge intensity is due only to the weak 1s→3d excitations [63]. The higher intensity pre-edge features of SPL (Fig. 5B, inset) are a result of increased 3d-4p mixing, as expected for tetrahedral Fe ions. The change in the redox state of the iron centers is also well represented by the energy position of the pre-edge features. It is notable that upon SAM binding, the pre-edge feature intensities increase for the as-purified form, which can be interpreted as the emergence of a less centrosymmetric and/or a more tetrahedral Fe coordination environment. These structural changes are further explained by the analysis of S K-edge pre-edge features.
S K-edge X-ray Absorption Spectroscopy – XANES analysis
The XANES region of the sulfur K-edge spectra of the free ligands are presented in Fig. 6. The sulfur K-edge spectra of reference salts Na2S, NaSH, and NaSEt are shown in Fig. 6A. The rising-edge region shows a shift in the most reasonable edge position indicated by the energy positions of S 4p excitations to higher energies in the order of sulfide, hydrosulfide, and thiolate sulfur absorbers. As discussed earlier [65], the positive energy shift is due to increased energy differences between the sulfur 1s and 4p orbitals, which correlates with increase in the sulfur effective nuclear charge, Zeff(S).
Fig. 6.

S K-edge XANES of free S-ligands (A) Na2S, NaSH, NaSEt; (B) NaSEt, methionine, cysteine; (C) cysteine, crystalline and solution samples of SAM
The sulfur K-edge spectra of relevant organic compounds of ethane thiolate, cysteine, and methionine are compared in Fig. 6B. Cysteine and methionine show similar spectral features as well as similar rising edge energies; however, they have a greater Zeff(S) as compared to thiolate from the energy positions of S 4p excitations. The rising-edge inflection point of the methionine spectrum is slightly lower in energy relative to cysteine, which is expected due to the inductive effects of a proton vs. an alkyl group. The positively charged SAM displays a high intensity feature at around 2475.5 eV due to allowed excitations into the three unoccupied C–S σ* orbitals (Fig. 6C). The S 4p transition for a positively charged sulfonium absorber in SAM is at higher energy in comparison to the neutral sulfhydryl group of cysteine. Due to the known limited stability of SAM, we have included in Fig. 6C both the freshly synthesized crystalline solid SAM and its solution spectra. The lower energy feature at around 2472 eV in the solution sample is due to the decomposition of SAM by acid hydrolysis [10]. The spectral differences among the crystalline and the decomposed SAM are important in analyzing the S K-edge spectra of the SPL samples with SAM present. The approximately 5 eV energy window at the sulfur K-edge into the electronic structures of various sulfur species (S2−, RS−, RSH/RSR and R3S+) well demonstrate the power of this XAS technique.
The sulfur K-edge XANES spectra for as-purified and reduced SPL in the absence and presence of SAM are presented in Fig. 7. These samples were prepared by substituting all methionine residues with SeMet and using methylviologen as reducing agent to minimize the number of sulfur absorbers. The samples were by purified by removing exogenous sulfur sources and dialyzed in the absence of DTT and dithionite. The use of SeMet was critical to mask out the spectral contribution from 11 methionine sulfur absorbers that would have washed out the pre-edge features due to their contribution to the edge-jump intensity only. Thus, the spectra in Fig. 7A are due to four Cys residues, three of which are coordinated to the [4Fe-4S] cluster, the Fe-bound sulfides, and SAM if present. The larger intensities of the renormalized spectra for the ‘as-purified+SAM’ (red) and ‘reduced+SAM’ (green) samples are due to the presence of about 6-fold excess of SAM in addition to the four S(Cys) and sulfide absorbers.
Fig. 7.

Renormalized S K-edge XANES of as-purified and reduced SPL in the absence and presence of SAM by considering total S content including excess SAM (A) and the normalized spectra after subtracting excess SAM using the crystalline solid SAM spectrum (B). Samples were prepared using 0.49 mM C.a. SPL and SAM was added to a final concentration of 3 mM
The intensity of the pre-edge feature at the ligand K-edge arises due to excitations of the ligand 1s core electrons into the unoccupied metal d-orbitals that are covalently mixed with the ligand p-orbitals. The S K-edge pre-edge features (insets in Fig. 7) originate from the Fe-S bonds of three thiolates and the four sulfides. In order to obtain meaningful spectral comparison, we need to consider the various contributors to the pre-edge features (only Fe-bound S species) and to the S K-edge jump (total S content). The biochemical characterization of SPL samples gave 2.3±0.2 Fe and 2.4±0.2 acid-labile S2− content per protein molecule with 4 S(Cys) absorbers in 0.49 mM concentration. In addition, there was also SAM added to the solution at 3 mM, thus defining the total S absorber concentration contributing to the edge jump to be about 6.1 mM, while the pre-edge features are from 2.6 mM sulfur absorbers. Furthermore, the differences between the rising-edge features at ~2474 (methionine feature but due to the SAM hydrolysis product 5′-methylthioadenosine) and ~2476 eV (SAM feature) can be correlated with increased hydrolytic decomposition of SAM in the as-purified versus the reduced SPL samples.
Due to the mixed cluster states in SPL, pre-edge peaks for sulfide and thiolate contributions are not well resolved. Instead, these features are observed as broad envelopes of S 1s→3p(Fe 3d) excitations between 2468 and 2472 eV. Despite the modest resolution, we can obtain reasonable fits to the spectra (Fig. 8) using previously well-documented fits to the S K-edge pre-edge features of [2Fe-2S] and [4Fe-4S] model complexes [45, 66, 67]. The fits in Fig. 8 were obtained by constraining the various Fe-S species to the Mössbauer speciation from Table 1. These Fe-S species were described by the specific linewidth, lineshape, and peak intensity parameters of the S K-edge pre-edge fits for Fe-S(thiolates), [2Fe-2S] and [4Fe-4S] clusters with thiolate coordination in their oxidized and reduced forms. Table S1 summarizes the final fit parameters used to obtain Fig. 8.
Fig. 8.
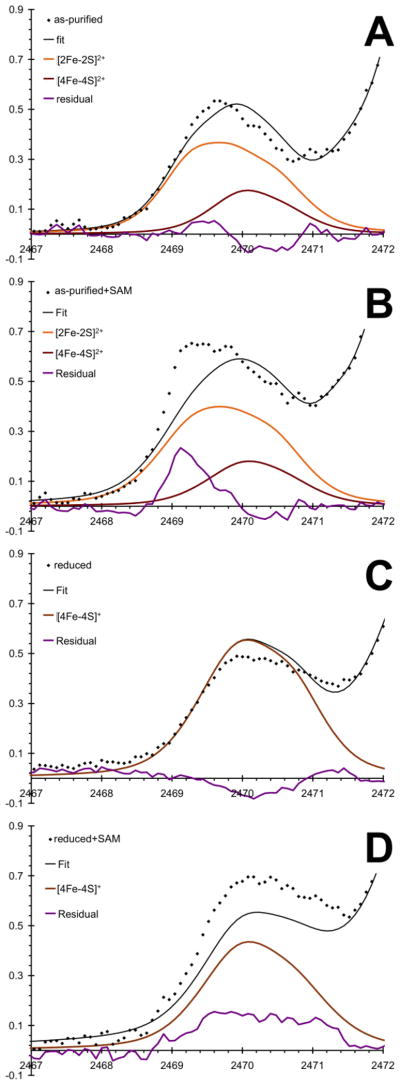
Representative fits of the Fe-bound sulfide and thiolate sulfur contributions to the pre-edge regions for as-purified (A) and reduced (C) SPL samples and with SAM (B and D, respectively). Dots represent the data points, black lines are the fitted data using Mössbauer speciation data, purple lines are the residuals, orange, dark red, and brown lines are spectral contributions from [2Fe-2S]2+, [4Fe-4S]2+, and [4Fe-4S]+ clusters, respectively.
Specifically, the oxidized and the reduced forms of the Fe-S clusters in SPL in the absence of SAM were fit using the S K-edge pre-edge features of [2Fe-2S] and [4Fe-4S] model complexes in relative ratios of 40:15. The peak linewidths and positions were allowed to vary within only ±0.3 eV and ±0.1 eV, respectively. Figs. 8A and C show the residuals (purple traces) that can be related to the difference of the Fe-S bonding in the protein-bound and the synthetic model clusters. The protein-bound Fe-S cluster in the as-purified form shows a slightly increased sulfide character (~2469.5 eV) at the expense of the thiolate character (~2470.5 eV), which can be rationalized by, for example, increased hydrogen bonding interactions involving the thiolates vs. the sulfide ligands. The quantitative comparisons of pre-edge intensities of the protein-bound and synthetic clusters are summarized in Table S2. It is important to recall (Table 1) that the spectral features at both Fe and S K-edges of the as-purified protein are primarily due to the [2Fe-2S] clusters with a small contribution from the [4Fe-4S] cluster. In the case of a protein-bound [2Fe-2S] cluster, the cluster-binding pocket of SPL would not be completely filled with the cluster and SAM, thus the [2Fe-2S] cluster can interact with water solvent or other small molecules, such as glycerol (see computational modeling below).
In the reduced form, the trends seen for the as-purified samples are reversed, as the residual function is negative at the sulfide features. This indicates that the additional electron upon reduction mainly affects the Fe-S(sulfide) bonding. It is also expected that the more reduced state of the cluster will enhance the effect of hydrogen bonding interactions. This is actually supported by the increased difference of Fe-S pre-edge intensity for the sulfide and thiolate features of the reduced synthetic model versus the protein-bound [4Fe-4S]+ cluster (Table S2). The reduced slope of the higher energy side of the pre-edge feature in Fig. 8B is also consistent with the likely presence of a rubredoxin type Fe2+ coordination environment, since the corresponding pre-edge feature is expected to be part of the rising edge as a shoulder adding intensity to the spectrum at around 2471.5 eV (Figs. 8C and D).
Addition of SAM to both as-purified and reduced forms of SPL greatly affects the lineshape and intensities of the S K-edge pre-edge features (Figs. 8B and D). The large spectral changes are in agreement with previous observations of significant effects of SAM binding on the structure of the [4Fe-4S]2+ cluster in BioB that indicate partial valence localization in the [2Fe-2S] rhomb involving the substrate binding site and no change in the other valence-delocalized [2Fe-2S] rhomb [68]. The more intense pre-edge feature and the lower energy shift of the sulfide features indicate a remarkable structural change relative to the purified form in the absence of SAM. Since most of the spectral intensity for the as-purified SPL samples originates from the [2Fe-2S] cluster, we can interpret this spectral change as an indication for alteration in the coordination environment. As discussed below for computational modeling, SAM coordination to one of the Fe sites (Fe2 with Cys29 in the model) of a [2Fe-2S] cluster is energetically and structurally feasible. For example, the presence of a less covalent amine or carboxyl ligand versus a cysteine thiolate can enhance the electron donation from the sulfide to Fe and thus result in an energetically shifted and more intense sulfide feature. The presence of a strongly perturbed coordination environment in the protein-bound cluster relative to its synthetic model is also represented by the increased reduction in the sulfide and thiolate spectral features (Table S2) relative to the as-purified form without SAM present. The less symmetric ligand environment is further supported by the more intense pre-edge feature at the Fe K-edge (Fig. 5B).
Mössbauer data suggest that the addition of SAM reduces the amount of [4Fe-4S] cluster present in the reduced SPL by about 15%. This is expected to give a lower pre-edge intensity (Fig. 8D) relative to the reduced form in the absence of SAM (Fig. 8C). The increased amount of rubredoxin like Fe coordination can account for the intensity increase at around 2471 and 2472 eV for the oxidized and reduced enzyme, but the sulfide feature at around 2470 eV should be lower or at least comparable intensity as in Fig. 8C. These inconsistencies may be resolved by considering again SAM coordination to incompletely assembled Fe-S clusters or mononuclear Fe sites with thiolate coordination as illustrated below in computational modeling.
Mapping of the [4Fe-4S]/SAM interaction
For further insights into the structural aspects of the [4Fe-4S] cluster of SPL and its interaction with SAM, we carried out density functional theory-based (DFT), relaxed potential energy surface mapping calculations. The distances between the sulfonium sulfur of SAM (S+) and the unique iron (Fe1) or the closest sulfide (S) were systematically varied and the potential energy diagrams for the constrained Fe1…S+ (brown trace and symbols) and S…S+ (orange trace and symbols) distances were determined (Fig. 9, panel A). These calculations indicate that Fe1…S+ and S…S+ distances can vary within an Å without significant energy change (within 5 kJ mol−1). As the constrained distances are decreased below approximately 3.2 Å for the S…S+ interaction or below 2.7 Å for the Fe1…S+ interaction, the potential energy increases sharply. These distances thus likely represent lower limits for the SAM sulfonium – cluster interaction in radical SAM enzymes. Direct orbital overlap between the sulfonium of SAM and the [4Fe-4S]+ cluster has been inferred based on electron-nuclear double resonance studies of PFL-AE [10], supporting a mechanism in which the [4Fe-4S]+ cluster of radical SAM enzymes reduces SAM via inner-sphere electron transfer. Such direct orbital overlap is plausible based on the potential energy curves shown in Fig. 9A; the sums of the van der Waals radii as a crude estimate for significant orbital overlap are 3.6 and 3.0 Å for two negatively charged sulfurs and a pair of negative sulfur and positive iron ions, respectively. Both distances are within the potential energy minimum region in Fig. 9A.
Fig. 9.
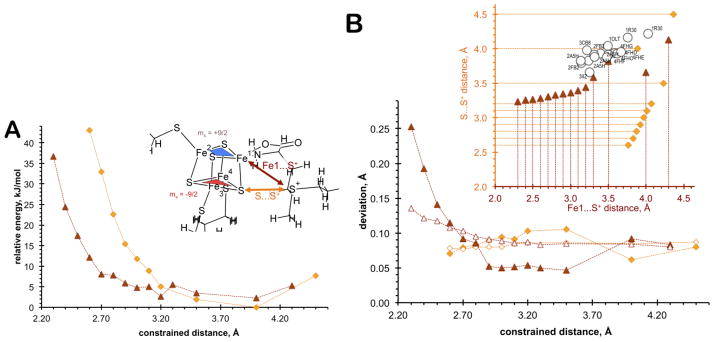
(A) Potential energy curves for the scanned Fe1…S+ (brown triangles) and S…S+ (orange diamonds) distances. Inset: Illustration of the scanned distances and the antiferromagnetically coupled [2Fe-2S] rhombs. (B) Deviation between Fe…Fe distances within ferromagnetically coupled [2Fe-2S] rhombs shown with blue and red shapes in inset of (A) (solid symbols) and between antiferromagnetically coupled [2Fe-2S] rhombs (hollow symbols) as a function of scanned Fe1…S+ (brown triangles) and S…S+ (orange diamonds) distances. Inset: Calculated and experimental correlation between Fe1…S+ and S…S+ distances as metric parameters for SAM and [4Fe-4S] interaction (orange diamonds and brown triangles represent optimized Fe1…S+ and S…S+ distances when S…S+ and Fe1…S+ distances were kept fixed, respectively; hollow circles represent experimental data with PDB codes (1OLT [3]; 1R30 [9]; 2ASH [14]; 2FB2 [4, 5]; 3CB8 [2]; 3IIZ [1])
Relevant to the interpretation of our EXAFS data, we evaluated the difference between the Fe…Fe distances as a function of the constrained Fe1…S+ and S…S+ distances. We compared the deviations between Fe…Fe distances within the ferromagnetically coupled [2Fe-2S] rhombs (top and bottom separately, solid symbols) and between the antiferromagnetically coupled [2Fe-2S] rhombs (between top and bottom, hollow symbols) as the constrained Fe1…S+ and S…S+ distances were varied (Fig. 9B). Using fixed crystallographic orientations of the Cys ligands, we found that the top MS = +9/2 [Fe1Fe2–2S] rhomb (blue triangle) prefers energetically to be antiferromagnetically coupled to the bottom MS = −9/2 [Fe3Fe4-2S] rhomb (red triangle), regardless of the nature of the ligand at the site differentiated Fe site (Fe1). Fig. 9B indicates that there can be more than 0.05 Å difference in Fe…Fe distances within a [2Fe-2S] rhomb, and close to 0.1 Å difference in Fe…Fe distances between the two ferromagnetically coupled [2Fe-2S] rhombs. These differences in Fe…Fe distances are large enough for being detected by EXAFS. Remarkably, as the SAM ligand approaches the [4Fe-4S] cluster, the magnitude of distortion in the Fe…Fe distances of the rhombs increases sharply, while it remains relatively unperturbed if the S…S+ distance is driven.
When the Fe1…S+ distances were driven from 2.3 to 3.2 Å with 0.1 Å stepsize (Fig. 9B, inset, horizontal axis and brown triangles), the corresponding optimized S…S+ distances also follow a nearly linear trend up to about 3.2 Å. If the Fe1…S+ distance is 3.3 Å and longer, the SAM ligand starts to detach from the unique Fe site of the [4Fe-4S] cluster and the optimized closest S…S+ distance no longer follows any trend. A similar discontinuity is observed at a slightly longer S…S+ distance (~3.5 Å) (vertical axis and orange diamonds).
The unique Fe…S(sulfonium) and shortest S(sulfonium)….S(sulfide) distances from crystal structures of SAM metalloenzymes (circles in Fig. 9B, inset) groups around this 3.2–3.5 Å Fe1…S+ and 3.6–4.0 Å S…S+ distance domain. Interestingly, the lowest resolution structure (3.4 Å for 1R30) corresponds to the most outlying data point to the above trend; while the data points corresponding to the highest resolution structure (1.62 Å for 3IIZ) lay closest to the lowest limits of the above domain. Thus, these two distances can be considered as shape parameters for the description of SAM interaction with the [4Fe-4S] cluster. There is always a potential concern with DFT-based optimized geometries, which is the accuracy of the level of theory and the completeness of the computational model; however the BP86/VTZ level of theory has been shown to give acceptable molecular structures for Fe-S clusters [52], the incorporation of a low dielectric environment captures most of the electrostatic effects by the protein environment, and the constrained α- and β-C positions of the thiolate ligands represent a steric strain imposed by the protein matrix.
Speciation dependent SAM/cluster interactions
In order to provide further support for the interpretation of the EXAFS spectra and the chemical adequacy of the fits, we carried out additional computational modeling by considering the oxidized and reduced states of the [4Fe-4S] and [2Fe-2S] clusters in the presence and absence of SAM as a ligand. The comprehensive summary of the EXAFS fits and the corresponding calculated structural parameters are shown in Table 2. The atomic coordinates of the optimized structures and their molecular graphical representations are provided in the Supporting Information.
Table 2.
Summary of experimental (labeled with EXAFS) and calculated Fe…Fe, Fe-S (in a rhomb), Fe..S (between rhombs) and Fe-O/N distances from EXAFS analysis (see above) and DFT calculations (average distances are given in Å, and n refers to coordination number or redundancy parameter in EXAFS fits; all calculations were completed with protein steric constraints on the Cys ligands from the PFL AE crystal structure 3CF8 [2] and with ε = 10 polarizable dielectric environment)
| sample | Fe…Fe | n | Fe-Ss | n | Fe..Ss | n | Fe-St | n | Fe…St | n | Fe-O/N | n | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| as-purified | EXAFS | 2.72 | 1.0 | 2.19 | 1.5 | 2.30 | 1.5 | 1.92 | 0.5 | ||||
| 2Fe(III) | 5.07 | 1.0 | 2.31 | 1.5 | 4.39 | 1.0 | 2.00 | 2.5 | |||||
| 6.03 | 0.5 | ||||||||||||
|
| |||||||||||||
| [2Fe-2S]2+ | 2.64 | 1.0 | 2.22 | 2.0 | 2.30 | 1.5 | 4.47 | 1.5 | 2.17 | 0.5 | |||
|
| |||||||||||||
| [4Fe-4S]2+ | 2.65 | 3.0 | 2.22 | 1.0 | 3.87 | 1.0 | 2.25 | 0.75 | 4.68 | 2.3 | 2.08 | 0.5 | |
| max. value | 2.70 | 2.32 | 2.0 | ||||||||||
|
| |||||||||||||
| as-purified + SAM | EXAFS | 2.75 | 0.5 | 2.20 | 2.0 | 2.25 | 1.5 | 1.93 | 0.5 | ||||
| 3.01 | 0.5 | ||||||||||||
|
| |||||||||||||
| 2Fe(III) | 3.59 | 0.5 | 2.31 | 1.5 | 3.82 | 1.0 | 2.06 | 3.0 | |||||
| 5.77 | 0.5 | ||||||||||||
| [2Fe-2S(S,N)]2+ | 2.69 | 0.5 | 2.23 | 2.0 | 2.31 | 1.5 | 4.53 | 1.5 | 2.17 | 0.5 | |||
| [2Fe-2S(S,N,O)]2+ | 2.71 | 0.5 | 2.27 | 2.0 | 2.32 | 1.5 | 4.47 | 1.5 | 2.23 | 0.5 | |||
| [2Fe-2S(N,O)]2+ | 2.47 | 0.5 | 2.28 | 2.0 | 2.35 | 1.5 | 4.69 | 0.5 | 2.12 | 0.5 | |||
|
| |||||||||||||
| [4Fe-4S]2+ | 2.72 | 3.0 | 2.23 | 1.0 | 3.90 | 1.0 | 2.27 | 0.75 | 4.72 | 2.3 | 2.19 | 0.5 | |
| max. value | 2.79 | 2.33 | 2.0 | ||||||||||
|
| |||||||||||||
| reduced | EXAFS | 2.72 | 2.0 | 2.23 | 2.5 | 2.29 | 1.0 | 1.94 | 0.75 | ||||
|
| |||||||||||||
| Fe(II)+Fe(III) | 4.65 | 0.5 | 2.31 | 1.5 | 4.43 | 1.0 | 2.02 | 2.5 | |||||
| 5.50 | 0.5 | ||||||||||||
|
| |||||||||||||
| 2Fe(II) | 4.32 | 0.5 | 2.36 | 1.5 | 4.53 | 1.0 | 2.05 | 2.5 | |||||
| 4.86 | 0.5 | ||||||||||||
|
| |||||||||||||
| [4Fe-4S]1+ | 2.64 | 3.0 | 2.26 | 1.0 | 3.88 | 1.0 | 2.28 | 0.75 | 4.70 | 2.3 | 2.17 | 0.25 | |
| max. value | 2.70 | 2.33 | 2.0 | ||||||||||
|
| |||||||||||||
| reduced +SAM | EXAFS | 2.78 | 1.0 | 2.25 | 3.0 | 2.24 | 0.75 | 1.91 | 0.5 | ||||
| 3.00 | 1.0 | ||||||||||||
| 3.18 | 1.0 | ||||||||||||
|
| |||||||||||||
| Fe(II)+Fe(III) | 3.62 | 0.5 | 2.35 | 1.5 | 4.02 | 1.0 | 2.09 | 3.0 | |||||
| 5.64 | 0.5 | ||||||||||||
|
| |||||||||||||
| 2Fe(II) | 3.10 | 0.5 | 2.25 | 1.5 | 3.70 | 1.0 | 2.08 | 3.0 | |||||
| 5.02 | 0.5 | ||||||||||||
|
| |||||||||||||
| [4Fe-4S]1+ | 2.71 | 3.0 | 2.27 | 1.0 | 3.92 | 1.0 | 2.29 | 0.75 | 4.75 | 2.3 | 2.25 | 0.5 | |
All experimental data shown in rows labeled ‘EXAFS’ of Table 2 were obtained using the speciation information from Mössbauer data (Table 1). The composition of the as-purified form of the SPL samples was identified to be dominantly [2Fe-2S] cluster (40%) and [4Fe-4S] in about a 3:1 ratio. The mononuclear Fe content was comparable to the amount of the [2Fe-2S] cluster. Thus, the observed spectral features both in the Fe K-edge EXAFS and the S K-edge XANES need to originate from both the mononuclear Fe site and the binuclear Fe cluster. The average Fe…Fe distance from EXAFS analysis appear to be about 0.08 Å longer than the calculated distance, while the Fe-S(sulfide) and Fe-S(thiolate) distances are remarkably close to the experimental value. The Fe-O/N distance shows an even a larger deviation between experiment and theory, indicating that the Fe-S cluster-based computational models did not capture this species well. A computational model of the His tag as a representation of mononuclear Fe (up to 40%, Table 1) with explicit solvent environment may provide a better explanation for the short distance relative to a solvated Fe ion as calculated.
Addition of SAM to the as-purified SPL sample does not change the speciation significantly; however, in EXAFS analysis a new Fe…Fe scatterer feature appears at around 3 Å that is absent for the sample without SAM. We evaluated the structural possibility of SAM coordination to a [2Fe-2S] cluster bound to the [4Fe-4S] active site pocket. The calculation indicates that the experimentally observed, elongated Fe…Fe distance cannot originate from the [2Fe-2S] cluster, since the longest Fe…Fe distance (2.71 Å) was shorter than that of the [4Fe-4S] cluster. The longest Fe…Fe scatterer distance (2.79 Å) in the oxidized [4Fe-4S] cluster with SAM approximates within 0.2 Å of the experimental 3 Å. This is a poor agreement; however, an increased protein strain effect that forces the SAM into the vicinity of the [4Fe-4S] cluster more than what is in the computational model based on SPL-AE structure could account for additional elongation in the Fe…Fe distance. Contrary, the calculated and experimental Fe-S distances are in good agreement, but the Fe-O/N distances were calculated to be longer than the EXAFS value.
We have also evaluated computationally the possibility of some of the EXAFS features originating from mononuclear Fe sites within the SAM cluster binding pocket where one of the sites were coordinated with Cys and three water molecules and the other with two Cys and two water molecules (see lines 2Fe(III), and Fe(II)+Fe(III)/2Fe(II) in Table 2 for mononuclear oxidized and reduced models, respectively). The atomic positional constraints on the Cys residue positions were maintained as in all other calculations. The considerable reduction of the Fe…Fe distance by 1.5 Å relative to those in Fe-S clusters is due to the bridging carboxyl oxygen (Ox) of the SAM bringing the two Fe sites together, while the amine group of SAM makes the coordination environment of the two Fe sites approximately similar ([ONSSOx] vs. [OxOOSS], see Supporting Information for molecular structures). The 3.59 Å Fe…Fe distance (row 2Fe(III) in Table 2 for as-purified with SAM model) is now too long to be considered as plausible source of the longer Fe…Fe scatterer.
The speciation problem for the reduced SPL samples is less convoluted due to the absence of additional Fe-S clusters other than the physiologically relevant [4Fe-4S]. However, we had to also consider the presence of mononuclear Fe sites due to their considerable percentage (40%) in the sample without SAM that increased by about 15% if SAM was present (Table 1). The EXAFS data interpretation and the calculated geometric data for the sample without SAM follow similar trends as discussed above for the as-purified form. Interestingly, addition of SAM not only alters the speciation by converting about 15% of the [4Fe-4S] cluster to mononuclear Fe sites (Table 1), but also results in the appearance of the 3 Å Fe…Fe scatter feature also observed in the as-purified enzyme, as well as another Fe…Fe scatterer at much longer distance (3.18 Å). The 3 Å scatterer likely results from SAM-induced cluster distortion resulting in longer Fe…Fe distances. The longer 3.18 Å scattering distance can be reasonably assigned to two mononuclear Fe sites that are bridged with a SAM (3.10 Å calculated).
Discussion
Mössbauer spectroscopy is a powerful tool to determine cluster states of Fe-S proteins, especially those states that are inaccessible by EPR spectroscopy. When combined with iron and sulfur K-edge XAS measurements, detailed information can be obtained regarding the geometric and electronic structures of the protein-bound Fe-S cluster(s). We describe here the use of this combined approach supplemented with careful biochemical characterization to provide important insights into the nature of the Fe-S cluster of SPL and its interaction with SAM.
Given that SPL is a member of the radical SAM protein superfamily with a common structural feature of a redox active [4Fe-4S] cluster and SAM to initiate radical catalysis, ideally SPL should have four irons and four sulfides per monomer. The observation that SPL purifies with sub-stoichiometric iron and sulfide content per monomer indicates incomplete incorporation of the [4Fe-4S] cluster, an observation that is not uncommon among radical SAM enzymes. Consistent with this, Mössbauer spectroscopy revealed a mixture of [2Fe-2S] (40% of total iron) and [4Fe-4S] (15% of total iron) cluster states present in the as-purified SPL. In addition, approximately 45% of the iron in as-purified SPL appears as adventitiously bound ferric species. The addition of SAM did not have a significant effect on the Mössbauer spectrum and thus on the cluster states of the as-purified enzyme. Due to the considerable amount of mononuclear Fe, and [2Fe-2S] cluster content, in our XAS analysis (discussed below) we had to consider the physiologically non-relevant, but chemically plausible SAM coordination modes.
Upon reduction, this mixture of clusters converts to primarily [4Fe-4S] clusters. Reductive cluster conversions from [3Fe-4S] and [2Fe-2S] states to [4Fe-4S]2+/+ have previously been reported for several radical SAM enzymes including biotin synthase [69–71], lipoyl synthase [72, 73], lysine 2,3-aminomutase [74, 75], and the activating enzymes of pyruvate formate lyase [76–79] and anaerobic ribonucleotide reductase [80], although the physiological relevance of these cluster conversions remain unknown. Reduced SPL was estimated to contain 60% [4Fe-4S]+ clusters as well as about 40% contribution from high-spin Fe2+ complexes. The latter was broken down into 10% N/O coordinated Fe2+ and 15% rubredoxin like FeS4 species, and 15% of fast relaxing Fe3+ species. Upon adding SAM to the reduced SPL, a 15% decrease in the [4Fe-4S]+ state is observed as compared to the reduced form in the absence SAM; however, this is unlikely to be due to the non-productive SAM cleavage since we see practically identical Fe K-edge spectra of the two reduced samples (discussed further below).
Our EXAFS results demonstrate the emergence of a new feature at ~3.0 Å as a shoulder on the Fe…Fe shell when SAM is added to both the as-purified as well as the reduced enzyme. This feature is observed only when SAM is present and remains when the parameters (range of k-space considered, shape of the Fourier Transform window, etc.) of EXAFS data analysis are varied. This shoulder feature fits well with an elongated Fe…Fe scatterer of 3.0 Å relative to the expected average Fe…Fe distance (2.72 Å) for a classical [4Fe-4S] cluster with tetrathiolate coordination. When modeled as a Fe…S(sulfonium) or Fe…C/N/O scatterer, the feature is clearly not in phase (lower inset in Fig. 4A). The intensity of this feature at 3.0 Å suggests this is due to elongation of two Fe…Fe distances per cluster. Distortion of the [4Fe-4S] cluster upon SAM coordination at the unique iron of the cluster would be expected, and our computational potential energy surface mapping study also indicated the distortion of the [4Fe-4S] cluster as a function of SAM…[4Fe-4S] cluster distance. The structural distortion observed by EXAFS, however, deviates from the changes expected due to stoichiometric 3:1 site differentiation. On the basis of computational results, we propose that upon SAM coordination, the Fe…Fe distance within the ferromagnetically coupled, Ms = +9/2 [2Fe-2S] rhomb containing the site-differentiated Fe site becomes considerably elongated relative to the other Ms = −9/2 rhomb (by about 0.1 Å at a SAM…[4Fe-4S] cluster separation of 2.7 Å). This increases up to 0.2 Å at around 2.4 Å separation, and drops to 0.05 Å above 3 Å. Concomitantly, the two other Fe…Fe distances involving the unique Fe site along the antiferromagnetic interaction between the two rhombs are also being elongated relative to the other Fe…Fe distances by about the same magnitude (0.12–0.09 Å). Thus, the SAM coordinated cluster can be described structurally as a 2:2 site-differentiated cluster.
To determine whether other members of the radical SAM superfamily share a structural distortion upon SAM binding, we compared the Fe…Fe bond distances of the [4Fe-4S] cluster in the crystal structures solved for members of this superfamily (Table 3). Detailed comparison of experimental Fe…Fe, Fe-S distances in radical SAM enzymes and aconitase are summarized in Table S3. Of the handful of structures solved for this superfamily [1–5, 9, 14, 28], only one example (MoaA, resolution 2.8 and 2.2 Å, Refs. [4, 5]) has structures both with and without SAM bound. In MoaA, the coordinatively unsaturated, unique Fe atom has slightly longer average Fe…Fe distances than the Cys coordinated Fe ions by 0.03 – 0.05 Å; however, the Fe…Fe distance increases upon SAM ligation by 0.07 – 0.22 Å. The modest resolution of MoaA structures may downplay the importance of the structural distortion especially below 0.1 Å; however crystal structures containing bound SAM cofactor with resolution down to 1.25 Å also support that the average Fe…Fe bond lengths are longer (~ 0.07 – 0.25 Å) for the unique Fe coordinated by SAM than for the three cysteine bound Fe atoms in the cluster, resulting in a slightly distorted cluster structure.
Table 3.
Variation of Fe…Fe distances in Å in MoaA (1TV7 at resolution 2.8 Å [4, 5]) upon SAM binding (1TV8 at resolution 2.2 Å [4, 5]). Cluster numbering as in PDB code 3CIW with the unique Fe labeled as Fe4
| Enzyme Cluster |
MoaA A |
MoaA+SAM A |
Difference | MoaA B |
MoaA+SAM B |
Difference |
|---|---|---|---|---|---|---|
| Fe1…Fe2 | 2.71 | 2.71 | 0.00 | 2.72 | 2.80 | 0.08 |
| Fe2…Fe3 | 2.63 | 2.63 | 0.00 | 2.66 | 2.47 | −0.19 |
| Fe3…Fe4 | 2.56 | 2.72 | 0.16 | 2.62 | 2.84 | 0.22 |
| Fe4…Fe1 | 2.80 | 2.74 | −0.06 | 2.76 | 2.82 | 0.06 |
| Fe1…Fe3 | 2.65 | 2.69 | 0.04 | 2.66 | 2.50 | −0.16 |
| Fe2…Fe4 | 2.79 | 2.88 | 0.09 | 2.76 | 2.83 | 0.07 |
The observation of Fe…Fe distances become elongated upon SAM binding agrees with our EXAFS results of SPL and further supports the idea that the [4Fe-4S] cluster of SAM enzymes can be described structurally as a 2:2 site differentiated cluster. The Fe3…Fe4 distance of the SAM-bound PFL-AE structure is also elongated to 2.88 Å compared to the other distances in the cluster [2], while the HydE SAM-bound structure appears to have 3 elongated Fe…Fe distances for the unique Fe [1]. These crystallographic results, together with our EXAFS results reported herein, raise the possibility for the functional significance of cluster distortion upon SAM binding. The distortion is expected to affect electronic properties and reactivity of the cluster, and thus it can play a role in modulating or gating electron transfer to the sulfonium of SAM. The significant distortion observed in our EXAFS studies of SPL, together with the fact that SPL utilizes SAM as a cofactor rather than a co-substrate, raises the question of whether the degree of cluster distortion upon SAM binding is correlated with the reversibility of the electron transfer from the cluster to SAM.
Relevantly, similar cluster distortions have also been observed for aconitase, when the unique Fe atom in the [4Fe-4S] cluster is ligated by citrate [81]. In the coordinatively unsaturated form, the average Fe…Fe distances for the unique Fe atom are ~ 0.01 Å shorter than the protein-bound Fe atoms [82]. However, upon ligation by citrate, the average Fe…Fe distances for the unique Fe atom are 0.2 Å longer than the average Fe…Fe distances for the protein-bound Fe atoms, indicating a distortion of the [4Fe-4S] cluster. Several of the Fe…Fe distances increase in the S642A aconitase crystal structure in the presence of the citrate substrate (PDB ID: 1C96, Reference [81]). The Fe1…Fe2 distance increases by 0.17 Å, Fe3…Fe4 increases by 0.18 Å, the Fe4…Fe1 distance increases by 0.23 Å and the Fe2…Fe4 distance increases by a surprising 0.38 Å.
The Fe K-edge XANES in Fig. 5 shows two distinct oxidation states for the as-purified and reduced samples as observed from the different rising edge energy positions. The higher intensity of the pre-edge feature for as-purified SPL in the presence of SAM is also indicative of considerable distortion of the Fe-S cluster structures upon interacting with SAM as pre-edge features gain intensity when the symmetry is reduced and thus Fe 4p orbitals are allowed to mix with Fe 3d orbitals. A noteworthy observation is the small difference in the spectral features between the reduced sample and the reduced sample in the presence of SAM. EPR spectroscopy of the reduced enzyme in the presence of SAM shows a broadened [4Fe-4S]+ signal with a significant decrease in intensity [24, 33]. Previous studies have suggested the decreased EPR intensity is due to non-productive SAM cleavage indicating that SAM is reductively cleaved by the [4Fe-4S]+ cluster to produce methionine, 5′deoxyadenosine and a [4Fe-4S]2+ cluster without turnover of substrate to product occurring [18]. However, a recent study demonstrated little dAdo• production in the absence of dithionite and substrate [33]. Similarly, the Fe K-edge data indicate practically no oxidation of the reduced [4Fe-4S]+ cluster upon binding SAM. This parallels our previous observations that the interaction of SAM with the [4Fe-4S]+ cluster of SPL does not immediately trigger SAM cleavage and the accompanied oxidation of the cluster [33].
The sulfur K-edge spectra of free ligands (Fig. 6) demonstrate a shift in the edge position to higher energies, directly indicating an increase in the sulfur effective nuclear charge (Zeff) in the order of Na2S, NaSH, NaSEt, Cys/Met, and SAM. The high intensity feature for SAM ~2475.5 eV is due to the presence of three C-S σ* acceptor orbitals. The presence of a growing spectral feature in subsequent scans at around 2473.7 eV in the S K-edge spectrum of a SAM containing sample can be indicative of hydrolytic decomposition giving 5′-methylthioadenosine (MTA).
A detailed quantitative evaluation of the S Kedge XAS spectra for the SPL samples (Fig. 7) to an Fe-S bond basis [45, 66, 67] was hindered by the presence of a mixture of cluster states. However, combining the biochemical and spectroscopic measurements on iron and sulfide content, protein concentrations, known amount of SAM excess, and Mössbauer speciation allowed us to obtain comparative electronic structural information between synthetic Fe-S cluster models and the clusters of SPL. Using reference XAS data from literature of biomimetic compounds [45, 66, 67], we obtained a qualitative estimate for the reduction of Fe-S bond covalency for the protein-bound iron-sulfur clusters. From these analyses, we can detect reduction of sulfur electron donation in the [2Fe-2S] clusters relative to the biomimetic [2Fe-2S] clusters. The same reduction is considerably smaller in the [4Fe-4S] clusters, but remarkably larger when the reduced forms of SPL are considered. These changes correspond to chemically meaningful trends, since reduced form of an Fe-S cluster is expected to show stronger hydrogen bonding interactions due to the increased overall electron density. Also, the partially assembled [2Fe-2S] cluster in the SAM cluster binding pocket will likely manifest considerable solvent interaction and potentially coordinate small molecule/counter ion binding.
Upon SAM binding the spectral features change drastically indicating the formation of a rather different Fe coordination environment than in the as-purified or reduced Fe-S clusters without SAM present. The most striking spectral difference at the S K-edge was observed for the SAM binding to the as-purified form, while the Fe K-edge XAS features are practically identical. Changes in the pre-edge features correspond to increased Fe-sulfide and Fe-thiolate bond covalency. We can rationalize this by considering the structural changes for a [2Fe-2S] cluster when SAM coordinates. While this is physiologically likely non-relevant in SPL, it is a remarkable perturbation. The coordination of the SAM to the oxidized form of the [4Fe-4S] cluster appears to have minimal effect on Fe-S bond covalency; however, we see a reverse trend for the reduced [4Fe-4S] cluster upon SAM coordination as both the sulfide and the thiolate bond covalencies decrease. The considerably larger change in the sulfide may be rationalized by the reorganization of the enzyme’s cluster binding pocket, as the bulky SAM binds to the Fe site, the pocket becomes more crowded and protein strain may be induced. This increased strain effect can be indirectly seen from the distortion of the [4Fe-4S] cluster geometry. The larger crowdedness within the cluster/SAM binding pocket facilitates the strengthening of the interactions between the cluster and nearby dipoles or H-bonds. This in turn can result in the reduced Fe-S donation.
Overall the combined spectroscopic analysis of anaerobically purified SPL indicates that the interaction between the [4Fe-4S] cluster and SAM produces a distortion of the cluster resulting in elongation of Fe…Fe distances. This observation may extend to other members of the radical SAM superfamily and can be critical for reducing the cluster or initiating radical catalysis. Complementary computational studies suggest an essential role for the protein environment in fine-tuning the relative position of the redox active cluster and the SAM substrate. Increased protein strain that results in a closer packing of the bound SAM molecule to the [4Fe-4S] cluster would facilitate a rapid inner-sphere electron transfer from the cluster into the sulfonium S-C σ* orbital. The protein strain may also be important in providing a gating mechanism to prevent the electron back-transfer for radical SAM metalloenzymes where SAM is a substrate. In the enzymes such as SPL where SAM acts to reversibly generate a dAdo• intermediate, the protein strain may also stabilize the radical/oxidized [4Fe-4S] pair relative to the sulfonium/reduced [4Fe-4S] pair, thus allow for a catalytic processes to take place.
Supplementary Material
Acknowledgments
This work has been supported by the NIH (GM67804 and GM54608 to JBB) and NSF (0744820 to RKS). The Astrobiology Biogeocatalysis Research Center is funded by the NASA Astrobiology Institute grant NNA08C-N85A. Portions of this research were conducted at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Footnotes
Electronic supplementary material The online version of this article (doi: to be specified) contains supplementary material, which is available to authorized users. Full access to XAS and DFT data are provided at computational.chemistry.montana.edu/SI.
Contributor Information
Sunshine C. Silver, Email: mrssils@gmail.com.
David J. Gardenghi, Email: dgardenghi@gmail.com.
Sunil G. Naik, Email: naikravi7@gmail.com.
Eric M. Shepard, Email: shepardem@gmail.com.
Boi Hanh Huynh, Email: vhuynh@physics.emory.edu, Department of Physics, Emory University, Atlanta, Georgia 30322.
Robert K. Szilagyi, Email: szilagyi@montana.edu, NAI Astrobiology Biogeocatalysis Research Center, Department of Chemistry & Biochemistry, Montana State University, Bozeman, MT, 59718
Joan B. Broderick, Email: jbroderick@chemistry.montana.edu, NAI Astrobiology Biogeocatalysis Research Center, Department of Chemistry & Biochemistry, Montana State University, Bozeman, MT, 59718
References
- 1.Nicolet Y, Amara P, Mouesca JM, Fontecilla-Camps JC. Proc Natl Acad Sci USA. 2009;106:14867–14871. doi: 10.1073/pnas.0904385106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. Proc Natl Acad Sci USA. 2008;105:16137–16141. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Layer G, Moser J, Heinz DW, Jahn D, Schubert WD. EMBO J. 2003;22:6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hänzelmann P, Schindelin H. Proc Natl Acad Sci USA. 2004;101:12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hänzelmann P, Schindelin H. Proc Natl Acad Sci USA. 2006;103:6829–6834. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shepard EM, Broderick JB. In: Comprehensive natural products ii: Chemistry and biochemistry. Mander LN, Liu HW, editors. Elsevier Press; Oxford U.K: 2010. pp. 625–662. [Google Scholar]
- 8.Frey PA, Hegeman AD, Ruzicka FJ. Crit Rev Biochem Mol Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 9.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Science. 2004;303:76–79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:3143–3151. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 11.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 12.Walsby CJ, Ortillo D, Yang J, Nnyepi M, Broderick WE, Hoffman BM, Broderick JB. Inorg Chem. 2005;44:727–741. doi: 10.1021/ic0484811. [DOI] [PubMed] [Google Scholar]
- 13.Chen D, Walsby C, Hoffman BM, Frey PA. J Am Chem Soc. 2003;125:11788–11789. doi: 10.1021/ja036120z. [DOI] [PubMed] [Google Scholar]
- 14.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. Proc Natl Acad Sci USA. 2005;102:13819–13824. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicolet Y, Rubach JK, Posewitz MC, Amara P, Mathevon C, Atta M, Fontecave M, Fontecilla-Camps JC. J Biol Chem. 2008;283:18861–18872. doi: 10.1074/jbc.M801161200. [DOI] [PubMed] [Google Scholar]
- 16.Henshaw TF, Cheek J, Broderick JB. J Am Chem Soc. 2000;122:8331–8332. [Google Scholar]
- 17.Dey A, Peng Y, Broderick WE, Hedman B, Hodgson KO, Broderick JB, Solomon EI. J Am Chem Soc. 2011;133:18656–18662. doi: 10.1021/ja203780t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rebeil R, Nicholson WL. Proc Natl Acad Sci USA. 2001;98:9038–9043. doi: 10.1073/pnas.161278998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munakata N, Rupert CS. J Bacteriol. 1972;111:192–198. doi: 10.1128/jb.111.1.192-198.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munakata N, Rupert CS. Mol Gen Genet. 1974;130:239–250. doi: 10.1007/BF00268802. [DOI] [PubMed] [Google Scholar]
- 21.Cheek J, Broderick JB. J Am Chem Soc. 2002;124:2860–2861. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- 22.Varghese AJ. Biochem Biophys Res Commun. 1970;38:484–490. doi: 10.1016/0006-291x(70)90739-4. [DOI] [PubMed] [Google Scholar]
- 23.Donnellan JE, Jr, Setlow RB. Science. 1965;149:308–310. doi: 10.1126/science.149.3681.308. [DOI] [PubMed] [Google Scholar]
- 24.Buis JM, Cheek J, Kalliri E, Broderick JB. J Biol Chem. 2006;281:25994–26003. doi: 10.1074/jbc.M603931200. [DOI] [PubMed] [Google Scholar]
- 25.Mehl RA, Begley TP. Org Lett. 1999;1:1065–1066. doi: 10.1021/ol9908676. [DOI] [PubMed] [Google Scholar]
- 26.Chandor-Proust A, Berteau O, Douki T, Gasparutto D, Ollagnier-de-Choudens S, Fontecave M, Atta M. J Biol Chem. 2008;283:36361–36368. doi: 10.1074/jbc.M806503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang L, Lin G, Nelson RS, Jian Y, Telser J, Li L. Biochemistry. 2012;51:7173–7188. doi: 10.1021/bi3010945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benjdia A, Heil K, Barends TRM, Carell T, Schlichting I. Nucleic Acids Res advance online publication. 2012 doi: 10.1093/nar/gks603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kneuttinger AC, Heil K, Kashiwazaki G, Carell T. Chem Commun online. 2012 doi: 10.1039/c2cc37735g. [DOI] [PubMed] [Google Scholar]
- 30.Pieck JC, Hennecke U, Pierik AJ, Friedel MG, Carell T. J Biol Chem. 2006;281:36317–36326. doi: 10.1074/jbc.M607053200. [DOI] [PubMed] [Google Scholar]
- 31.Yang L, Lin G, Liu D, Dria KJ, Telser J, Li L. J Am Chem Soc. 2011;133:10434–10447. doi: 10.1021/ja110196d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chandra T, Silver SC, Zilinskas E, Shepard EM, Broderick WE, Broderick JB. J Am Chem Soc. 2009;131:2420–2421. doi: 10.1021/ja807375c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silver SC, Chandra T, Zilinskas E, Ghose S, Broderick WE, Broderick JB. J Biol Inorg Chem. 2010;15:943–955. doi: 10.1007/s00775-010-0656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mantel C, Chandor A, Gasparutto D, Douki T, Atta M, Fontecave M, Bayle PA, Mouesca JM, Bardet M. J Am Chem Soc. 2008;130:16978–16984. doi: 10.1021/ja805032r. [DOI] [PubMed] [Google Scholar]
- 35.Heil K, Kneuttinger AC, Schneider S, Lischke U, Carell T. Chem Eur J. 2011;17:9651–9657. doi: 10.1002/chem.201100177. [DOI] [PubMed] [Google Scholar]
- 36.Friedel MG, Berteau O, Pieck JC, Atta M, Ollagnier-de-Choudens S, Fontecave M, Carell T. Chem Commun. 2006:445–447. doi: 10.1039/b514103f. [DOI] [PubMed] [Google Scholar]
- 37.Chandor A, Berteau O, Douki T, Gasparutto D, Sanakis Y, Ollagnier-de-Choudens S, Atta M, Fontecave M. J Biol Chem. 2006;281:26922–26931. doi: 10.1074/jbc.M602297200. [DOI] [PubMed] [Google Scholar]
- 38.Chandor A, Douki T, Gasparutto D, Gambarelli S, Sanakis Y, Nicolet Y, Ollagnier-de-Choudens S, Atta M, Fontecave M. C R Chimie. 2007;10:756–765. [Google Scholar]
- 39.Broderick JB, Henshaw TF, Cheek J, Wojtuszewski K, Smith SR, Trojan MR, McGhan RM, Kopf A, Kibbey M, Broderick WE. Biochem Biophys Res Comm. 2000;269:451–456. doi: 10.1006/bbrc.2000.2313. [DOI] [PubMed] [Google Scholar]
- 40.Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, Jr, Marletta MA. Biochemistry. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- 41.Ugulava NB, Sacanell CJ, Jarrett JT. Biochemistry. 2001;40:8352–8358. doi: 10.1021/bi010463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 43.Beinert H. Methods Enzymol. 1978;54:435–445. doi: 10.1016/s0076-6879(78)54027-5. [DOI] [PubMed] [Google Scholar]
- 44.Beinert H. Anal Biochem. 1983;131:373–378. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- 45.Solomon EI, Hedman B, Hodgson KO, Dey A, Szilagyi RK. Coord Chem Rev. 2005;249:97–129. [Google Scholar]
- 46.Gardenghi DJ. Automated data reduction protocol. 2011 http://computational.chemistry.montana.edu/ADRP.
- 47.Newville M, Līviņš P, Yacoby Y, Rehr JJ, Stern EA. Phys Rev B. 1993;47:14126–14131. doi: 10.1103/physrevb.47.14126. [DOI] [PubMed] [Google Scholar]
- 48.Ravel B, Newville M. J Synchrotron Radiat. 2005;12:537–541. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- 49.Newville M, Ravel B, Haskel D, Rehr JJ, Stern EA, Yacoby Y. Physica B. 1995;208:154–156. [Google Scholar]
- 50.Ankudinov AL, Nesvizhskii AI, Rehr JJ. Phys Rev B. 2003;67:115120–115126. [Google Scholar]
- 51.Rao PV, Holm RH. Chem Rev. 2004;104:527–559. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 52.Szilagyi RK, Winslow MA. J Comput Chem. 2006;27:1385–1397. doi: 10.1002/jcc.20449. [DOI] [PubMed] [Google Scholar]
- 53.Becke AD. Phys Rev A. 1988;38:3098– 3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 54.Perdew JP. Phys Rev B. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 55.Schaefer A, Huber C, Ahlrichs R. Chem Phys. 1994;100:5829–5835. [Google Scholar]
- 56.Tomasi J, Persico M. Chem Rev. 1994;94:2027–2094. [Google Scholar]
- 57.Cossi M, Barone V, Cammi R, Tomasi J. Chem Phys Lett. 1996;255:327–335. [Google Scholar]
- 58.Barone V, Cossi M, Tomasi J. J Chem Phys. 1997;107:3210–3221. [Google Scholar]
- 59.Schutz CN, Warshel A. Proteins-Structure Function and Genetics. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 60.Rokhsana D, Howells AE, Dooley DM, Szilagyi RK. Inorganic Chemistry. 2012;51:3513–3524. doi: 10.1021/ic2022769. [DOI] [PubMed] [Google Scholar]
- 61.Huynh BH, Kent TA. In: Advances in inorganic biochemistry. Eichhorn GL, Marzilli LG, editors. Elsevier Scientific; 1984. pp. 164–223. [Google Scholar]
- 62.Middleton P, Dickson DPE, Johnson CE, Rush JD. Eur J Biochem. 1978;88:135–141. doi: 10.1111/j.1432-1033.1978.tb12430.x. [DOI] [PubMed] [Google Scholar]
- 63.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:6297–6314. [Google Scholar]
- 64.Shulman RG, Yafet Y, Eisenberger P, Blumberg WE. Proc Natl Acad Sci USA. 1976;73:1384–1388. doi: 10.1073/pnas.73.5.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shadle SE, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1995;117:2259–2272. doi: 10.1021/ja002183v. [DOI] [PubMed] [Google Scholar]
- 66.Glaser T, Hedman B, Hodgson KO, Solomon EI. Acc Chem Res. 2000;33:859–868. doi: 10.1021/ar990125c. [DOI] [PubMed] [Google Scholar]
- 67.Dey A, Glaser T, Couture MMJ, Eltis LD, Holm RH, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2004;126:8320–8328. doi: 10.1021/ja0484956. [DOI] [PubMed] [Google Scholar]
- 68.Cosper MM, Jameson GNL, Davydov R, Eidsness MK, Hoffman BM, Huynh BH, Johnson MK. J Am Chem Soc. 2002;124:14006–14007. doi: 10.1021/ja0283044. [DOI] [PubMed] [Google Scholar]
- 69.Ugulava NB, Gibney BR, Jarrett JT. Biochemistry. 2000;39:5206–5214. doi: 10.1021/bi9926227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duin EC, Lafferty ME, Crouse BR, Allen RM, Sanyal I, Flint DH, Johnson MK. Biochemistry. 1997;36:11811–11820. doi: 10.1021/bi9706430. [DOI] [PubMed] [Google Scholar]
- 71.Tse Sum Bui B, Florentin D, Marquet A, Benda R, Trautwein AX. FEBS Lett. 1999;459:411–414. doi: 10.1016/s0014-5793(99)01300-9. [DOI] [PubMed] [Google Scholar]
- 72.Busby RW, Schelvis JPM, Yu DS, Babcock GT, Marletta MA. J Am Chem Soc. 1999;121:4706–4707. [Google Scholar]
- 73.Ollagnier-de Choudens S, Fontecave M. FEBS Lett. 1999;453:25–28. doi: 10.1016/s0014-5793(99)00694-8. [DOI] [PubMed] [Google Scholar]
- 74.Lieder K, Booker S, Ruzicka FJ, Beinert H, Reed GH, Frey PA. Biochemistry. 1998;37:2578–2585. doi: 10.1021/bi972417w. [DOI] [PubMed] [Google Scholar]
- 75.Petrovich RM, Ruzicka FJ, Reed GH, Frey PA. Biochemistry. 1992;31:10774–10781. doi: 10.1021/bi00159a019. [DOI] [PubMed] [Google Scholar]
- 76.Broderick J, Duderstadt R, Fernandez D, Wojtuszewski K, Henshaw T, Johnson M. J Am Chem Soc. 1997;119:7396–7397. [Google Scholar]
- 77.Krebs C, Broderick WE, Henshaw TF, Broderick JB, Huynh BH. J Am Chem Soc. 2002;124:912–913. doi: 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- 78.Yang J, Naik SG, Ortillo DO, Garcia-Serres R, Li M, Broderick WE, Huynh BH, Broderick JB. Biochemistry. 2009;48:9234–9241. doi: 10.1021/bi9010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krebs C, Henshaw TF, Cheek J, Huynh BH, Broderick JB. J Am Chem Soc. 2000;122:12497–12506. [Google Scholar]
- 80.Ollagnier S, Meier C, Mulliez E, Gaillard J, Schuenemann V, Trautwein A, Mattioli T, Lutz M, Fontecave M. J Am Chem Soc. 1999;121:6344–6350. [Google Scholar]
- 81.Lloyd SJ, Lauble H, Prasad GS, Stout CD. Protein Sci. 1999;8:2655–2662. doi: 10.1110/ps.8.12.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robbins AH, Stout CD. Proc Natl Acad Sci USA. 1989;86:3639–3643. doi: 10.1073/pnas.86.10.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.