

Abstract
C. elegans has become an ideal model to study genetics of appetite control and energy metabolism because of its robust conservation in molecular mechanisms underlying appetite control and in regulation of the relevant feeding behavior. Satiety behavior in worms in particular shows striking similarities to that in mammals, as a worm becomes quiescent after a big meal, mimicking post-prandial sleep in mammals. Here we review our recent finding regarding the neuronal regulation of the behavior and the implication of the finding such as cyclicity of behavioral states. Based on the finding, we propose a rather speculative but intriguing view of how metabolism could link to post-prandial sleep.
Keywords: ASI, satiety quiescence, post-prandial sleep, antagonism between hunger and satiety, metabolic regulation of sleep
Appetite Control Circuit
In 1940, by introducing bilateral lesions in the hypothalamus of rats, Hetherington and Ransom found that removing a small part of the hypothalamus changes feeding behavior entirely, resulting in increase of food intake, body weight, and adiposity.1 They also observed that removing the adjacent part evokes the opposite effect: the rats don’t eat and starve to death. These results provided the first evidence of neuronal control of appetite. Later the locations were defined based on the specific molecular mechanisms and discrete neuronal pathways by which food intake is regulated. The ventromedial hypothalamus whose lesions resulted in hyperphagia (increase of feeding) contains pro-opiomelanocortin (POMC)-expressing neurons. The lateral hypothalamus whose lesion resulted in hypophasia (decrease of feeding) contains neuropeptide Y/agouti-related protein (NPY/AgRP)-expressing neurons. When POMC neurons get activated, they release POMC so that animals eat less. On the other hand, when NPY/AgRP neurons get activated, they release NPY/AgRP and animals eat more.2 Interestingly, when NPY/AgRP neurons get activated, they directly inhibit POMC neurons and if POMC neurons get activated, they inhibit NPY/AgRP neurons. This proximity of two loci of orexigenic and anorexigenic centers and their tight antagonism for each other seems to be the way brains have evolved to integrate food intake signals to control appetite.
To control appetite, brain receives signals originating from the gut. In their discovery of cholecystokinin (CCK) as a satiety signal, Gibbs and others found that ingested food did not evoke satiety unless it accumulated and was passed into the small intestine.3,4 Although the food was tasted and swallowed, the animal kept eating unless the stomach was full and the food reached the small intestine. This result shows that for the animal to feel satiated, nutrients have to reach the gut to be sensed. And then signal(s) sent to the brain relay the nutritional satisfaction, forming the satiety circuit.5
Currently one third of US adults are obese. Obesity is the main factor leading to serious health problems such as cardiovascular diseases and type 2 diabetes.6,7 As in all biological phenomena, multiple factors contribute to obesity. Nonetheless, studies from obese patients who are deficient in leptin8 or who carry mutations in melanocortin 4 receptors (MC4R)9 show that obesity can be a monogenic trait. Currently 11 genes are known as causes of obesity. Interestingly, all of them are involved in regulation of food intake, suggesting appetite control is the major way to regulate body weight and obesity.10
Appetite Control in C. elegans
C. elegans has been a powerful genetic model system to discover many complex yet evolutionarily conserved molecular pathways.11,12 Moreover, Ashrafi et al.13 provided genetic evidence that fat metabolism and energy expenditure are conserved between C. elegans and mammals. In addition, we discovered that the signaling and the behavior controlling food intake, which contributes directly to obesity, are also conserved between C. elegans and mammals; after a big meal, C. elegans become quiescent, mimicking behavioral sequence of satiety in mammals.14 This finding of behavior has given us a chance to use C. elegans to study genetics of food intake, and therefore, potential genetic contribution to obesity. (A note: herein, “worm” means C. elegans.)
In a recent study, we analyzed satiety quiescence behavior further using a newly developed tracking system combined to a Hidden Markov Model analysis. We monitored an individual worm’s locomotive activity throughout refeeding period and analyzed the speed to find the particular behavioral state of the worm at the given time point during refeeding. From this method we found several interesting new aspects of the satiety quiescence. (1) Under our analysis conditions, we could see worms show cyclicity among three distinct behavioral states (roaming, dwelling, and quiescent). (2) This cyclicity is dependent on the worms’ nutritional status and genotypes.15 (3) ASI, a pair of head sensory neurons, regulates the transition frequencies of between two states (quiescence and dwelling) mainly through regulating the TGFβ pathway.
Cyclicity of behavioral states
When we first discovered satiety quiescence, which resembles post-prandial sleep in mammals, one of the most intriguing questions was whether the behavior was cyclic. Using our new method, we found worms cycle three distinct behavioral states (Fig. 1). In addition, the percent time worms spend at each state is determined by the food quality, feeding history, or genotypes; worms fed with poor quality food spent little time quiescent. On the contrary, worms fasted and fully refed with high-quality food spent most of their time quiescent. Mutants of egl-4, which encodes a worm cGMP-dependent protein kinase, fail to show quiescence and behave similar to worms fed with poor quality food.16 These results show that our new automated system not only validated our previous results but also measures satiety quiescence in a quantitative manner. In addition, we gained a new level of information: transition rates among states. Transition rates are a measurement of how frequently a worm comes in and out of each state. It is different from total time worms spend in each state. For instance, sleeping total 6 h, alternating sleep and being awake every 30 min for a 12 h span is behaviorally different from sleeping total 6 h without waking up and then staying awake for another 6 h, even if in both cases the sum of the hours of sleeping and the hours of being awake are the same. Transition rates among states provide us a chance to find different type of mutants, the mutants that would not “stay” quiescence even if they spend the same percent time quiescent as wild-type. In these mutants, quiescence state is not solidified (so that worms do not stay quiescent) but fragmented. We found ASI neurons regulate the total time in quiescence state by regulating the solidification of quiescence state.

Figure 1. Cyclicity of behavioral states. Locomotive activity for 30 min during the course of refeeding suggests three behavioral states: quiescence, roaming, and dwelling. For this simple illustration to show the behavioral pattern, states were determined subjectively based on the average speed for a certain span of time. Quiescence state (blue box) was shown for a period of no locomotive activity, dwelling state (green box) with medium speed, and roaming state (red box) with increased speed for a prolonged period.
ASI neurons regulate the transition frequencies of among states mainly through the TGFβ pathway
When we examined a TGFβ ligand mutant (daf-7), they were less quiescent and increased dwelling with no change in roaming. The HMM analysis shows this is due to daf-7 mutants switching from quiescence to dwelling more frequently and from dwelling to quiescence less frequently than wild-type worms (Fig. 2). This increased dwelling time indeed leads to increased food intake and fat storage.16 All downstream components of the TGFβ signaling pathway, such as a receptor and SMADs, are necessary for intact satiety quiescence, showing the whole pathway regulates satiety behavior. Additionally, a forward genetic screen for satiety quiescence mutants yielded a new mutation in the receptor, daf-1, again confirming TGFβ role in satiety. The transcription of DAF-7 is significantly upregulated after fasting and subsequent full-refeeding, suggesting that during refeeding DAF-7 protein level is increased to mediate satiety quiescence.

Figure 2. DAF-7 regulates transition rates from dwelling (D) to quiescence (Q) after fasting and full-refeeding. The transition rates among each state were calculated from the HMM analysis15 and normalized to the wild-type transition rate from Q to D to make the comparison easy. Wild-type has a higher tendency to enter quiescence than to exit quiescence as the ratio between two transition rates is 1.55 to 1. This higher tendency to stay quiescence results that wild-type worms spend approximately 60% of their time quiescent (1.55/2.55 × 100 = 60.78%). On the contrary, daf-7 mutants have 3-fold higher tendency to exit quiescence than that of wild-type. In addition, the rate of exiting quiescence is 4-fold higher than the rate of entering quiescence, resulting approximately 20% of their time quiescent. This shows DAF-7 mutants are defective in quiescence not because they are unable to enter quiescence state but because they exit quiescence a lot more frequently than wild-type. The defect of DAF-7 is similar to the defect caused by ASI ablation, suggesting DAF-7 action in ASI.
We have identified a dozen head neurons that express a cGMP-gated ion channel subunit, tax-4, which are critical for satiety quiescence.14 Among them, ASI is the only known neuron that expresses daf-7. ASI regulates other food-related behavior such as lifespan extension by calorie restriction and the decision to enter the dauer diapause during scarcity of food.17-19 Thus, we hypothesized that the ASI neuron is sensing and conveying signals about the worm’s nutritional state to promote satiety quiescence. Calcium imaging results indicate that nutrients indeed activates ASI, showing ASI’s role in sensing nutrition. In addition, genetic ablation (as well as operational ablation) of ASI reduces the time worms spent quiescent, mimicking the quiescence defect of the TGFβ mutants. Finally, the ASI-minus worms are defective in satiety quiescence because they cannot stay quiescent; they exit quiescence a lot more frequently than worms with ASI intact, again mimicking the defect of the TGFβ mutants (Fig. 2). Taken together, these results demonstrate that TGFβ in ASI is necessary to promote satiety quiescence. It also suggests that ASI promotes satiety quiescence through the TGFβ pathway by consolidating the quiescence state to maintain it.
To identify the downstream neurons of ASI, we tested several transgenic strains with DAF-1 (the TGFβ receptor) expression targeted into various groups of neurons.20 Expressing DAF-1 in RIM (ring motor neurons) and RIC (ring interneurons) completely restores satiety behavior in daf-1 mutants, consistent with previous results that RIM and RIC are the action sites for the TGFβ pathway in fat storage and egg laying behavior.20 It has been suggested that DAF-7 binds to DAF-1 on RIM and RIC to inhibit the neurons and this lack of inhibition is the reason for the increased fat storage and egg laying defect of daf-1 mutants. Ablating RIM and RIC in daf-1 mutants by laser rescues the daf-1 mutants, restoring normal quiescence in daf-1 worms. This suggests that DAF-7 from ASI suppresses RIM and RIC activities to promote satiety quiescence. RIM and RIC, on the other hand, when they are active, might release one or more signals that inhibit quiescence.
Food for Thought
Antagonism between hunger and satiety
Based on the result that ASI inhibits RIM and RIC, we suggest potential antagonism between ASI and RIM + RIC to regulate satiety in worms. It is reasonable to think that animals need an orexigenic center and an anorexigenic center to integrate signals to evaluate constantly changing nutritional status. Indeed in hypothalamus, NPY-expressing neurons and POMC-expressing neurons are located next each other to communicate each other. Our finding, combined with previously known function of ASI, shows that ASI inhibits the activity of RIM and RIC through the TGFβ pathway. However, interesting questions such as how suppressing RIM and RIC evokes satiety, what are the hunger signals that prevent quiescence released from RIM and RIC, and whether the signal released from RIM and RIC can antagonize ASI as negative feedback still remains.
Sleep and metabolism: An ancient tie?
The last step of behavioral sequence of satiety is sleep (or quiescence) in both mammals and worms. It is curious to think what the relationship between “nutritional satisfaction” and “sleep” is. Do the animals sleep as a consequence of nutritional satisfaction? Or does sleep coincide with fullness for another reason or for no particular reasons? The purpose of sleep is still mysterious, thus we can only speculate what is the role of metabolic satisfaction to induce sleep. Intriguingly, however, several reports show that sleep deprivation increases food intake through decrease of leptin and increase of ghrelin21 and that sleep deprivation highly correlates with obesity,22-24 suggesting more than pure coincidental relationship between metabolic satisfaction and sleep. Moreover, a neuropeptide orexin/hypocretin regulates both sleep and feeding, providing a strong molecular evidence for the link between feeding and sleep.25,26
The three signals we found to regulate satiety quiescence in worms also mediate another nutrition-related developmental decision; lack of any of the three signals induces worms to become a dauer, a dormant form to endure a harsh environment. This suggests that sleep in worms also seems to be regulated by the signals that sense environment and nutritional well-being. As mentioned above, satiety doesn’t simply come from a full stomach; filling up the stomach with water doesn’t evoke satiety or its behavioral sequence. Therefore, it is safe to assume that satiety behavior and its behavioral sequence comes with fulfilling metabolic needs. Why would it end up causing sleep? It would be dangerous to sleep after an animal is full. Already less mobile after full feeding, if an animal falls asleep, it will make an escape from predators almost impossible. Yet, nutritional satisfaction often evokes emotional happiness and physical sleepiness.
If we may accept that there is a relationship between sleep and feeding, followings might be the possible reasons. Feeding might induce sleep because sleep is required for metabolic processes after full feeding. This association can be suggested in snakes that feed on large prey with long-term intervals of meals. In these animals, the metabolic rate is increased after feeding more than 5-fold than that of fasted snakes, accompanying various changes including production of digestive enzymes and remodeling of the gut.27,28 Thus, it is possible that for these huge internal changes to digest a big sized meal, snakes cannot move but must rest and sleep.
Another possibility is that metabolic satisfaction from full feeding might serve as an indicator of safety: sleep is essential for life for many animals, but animals in nature could hardly sleep because of constant threats of predators. Secured food and full feeding might have been associated with sleep because that can be the best indication of a good environment to sleep safely.
The third possibility is that sleep is a default behavioral state when an animal is released from alert. Hungry animals explore to seek food with constant vigil. Nutritional satisfaction could relieve animals from this alert state and have them stop seeking food. This relief might induce sleep.
All these speculations are not mutually exclusive. Yet, sleep being a default state after released from alert is most intriguing. Certain vigilance signals to keep animals awake function as hunger signals; acetylcholine that keeps animals awake can function as a hunger signal in both mammals and worms.5,29,30 Orexin is required for consolidation of wakefulness and its level is increased when animals endure longer fasting.25 These facts suggest that while animals are seeking food, the low nutrition level keeps the animal awake by increasing the level of certain “alert” or “wakefulness” neurotransmitters: you have to find food to survive. If you are getting hungrier, you become more desperate to be awake. Once the nutritional needs are fulfilled, the alert signals go away and the opposite behavior that has been suppressed, i.e., sleep, follows. In this hypothetical scenario, the link between metabolism and sleep is apparent; sleep is the final stage of metabolic relief from the vigil to find food (Fig. 3).
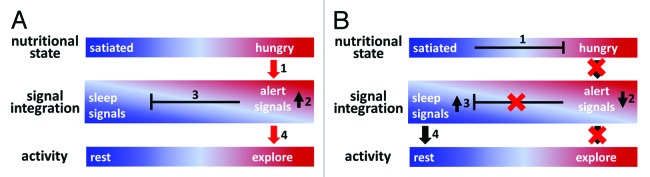
Figure 3. Hypothetical scenarios to link metabolic satisfaction to sleep. (A) When an animal is hungry, certain alert signals (or wakeful signals) are released to prevent sleep and promote food seeking. (B) Once the hunger signals go away, the suppression of sleep is released and animals fall asleep. (A) When an animal is hungry. (B) When an animal is satiated.
Conclusions
We found ASI neurons are activated by nutrients to induce satiety quiescence in worms. Disrupting ASI functions fragmented quiescence duration such that worms cannot stay quiescent. Based on our results, we suggest that RIM and RIC are downstream neurons of ASI to antagonize ASI function in satiety quiescence. Although we do not know the identities of the molecules released from RIM and RIC to convey hunger, the known function of ASI and RIM and RIC suggests that the similar cellular mechanisms, antagonism between orexigenic and anorexigenic to regulate appetite in mammals, exist in worms to regulate food intake and satiety quiescence. It is intriguing to think that the link between sleep and metabolism might have been established through the same two groups of molecules that regulate an antagonism between hunger and satiety.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by NIH grant R01DK083593.
References
- 1.Hetherington AW, Ranson SW. Hypothalamic lesions and adiposity in the rat. Anat Rec. 1940;78:149–72. doi: 10.1002/ar.1090780203. [DOI] [Google Scholar]
- 2.Schwartz MW, Woods SC, Porte D, Jr., Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–71. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 3.Gibbs J, Young RC, Smith GP. Cholecystokinin elicits satiety in rats with open gastric fistulas. Nature. 1973;245:323–5. doi: 10.1038/245323a0. [DOI] [PubMed] [Google Scholar]
- 4.Gibbs J, Young RC, Smith GP. Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol. 1973;84:488–95. doi: 10.1037/h0034870. [DOI] [PubMed] [Google Scholar]
- 5.You YJ, Avery L. Appetite Control: worm’s-eye-view. Animal Cells Syst (Seoul) 2012;16:351–6. doi: 10.1080/19768354.2012.716791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoyert DL, Xu J. Deaths: Preliminary Data for 2011. Natl Vital Stat Rep. 2012;61:1–52. [PubMed] [Google Scholar]
- 7.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee Executive summary: heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:143–52. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 8.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–8. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 9.Farooqi IS, Yeo GS, Keogh JM, Aminian S, Jebb SA, Butler G, Cheetham T, O’Rahilly S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106:271–9. doi: 10.1172/JCI9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mutch DM, Clément K. Unraveling the genetics of human obesity. PLoS Genet. 2006;2:e188. doi: 10.1371/journal.pgen.0020188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han M, Sternberg PW. let-60, a gene that specifies cell fates during C. elegans vulval induction, encodes a ras protein. Cell. 1990;63:921–31. doi: 10.1016/0092-8674(90)90495-Z. [DOI] [PubMed] [Google Scholar]
- 12.Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–9. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- 13.Ashrafi K, Chang FY, Watts JL, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature. 2003;421:268–72. doi: 10.1038/nature01279. [DOI] [PubMed] [Google Scholar]
- 14.You YJ, Kim J, Raizen DM, Avery L. Insulin, cGMP, and TGF-beta signals regulate food intake and quiescence in C. elegans: a model for satiety. Cell Metab. 2008;7:249–57. doi: 10.1016/j.cmet.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallagher T, Bjorness T, Greene R, You YJ, Avery L. The geometry of locomotive behavioral states in C. elegans. PLoS One. 2013;8:e59865. doi: 10.1371/journal.pone.0059865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gallagher T, Kim J, Oldenbroek M, Kerr R, You YJ. ASI regulates satiety quiescence in C. elegans. J Neurosci. 2013;33:9716–24. doi: 10.1523/JNEUROSCI.4493-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bargmann CI, Horvitz HR. Control of larval development by chemosensory neurons in Caenorhabditis elegans. Science. 1991;251:1243–6. doi: 10.1126/science.2006412. [DOI] [PubMed] [Google Scholar]
- 18.Ren P, Lim CS, Johnsen R, Albert PS, Pilgrim D, Riddle DL. Control of C. elegans larval development by neuronal expression of a TGF-beta homolog. Science. 1996;274:1389–91. doi: 10.1126/science.274.5291.1389. [DOI] [PubMed] [Google Scholar]
- 19.Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–9. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- 20.Greer ER, Pérez CL, Van Gilst MR, Lee BH, Ashrafi K. Neural and molecular dissection of a C. elegans sensory circuit that regulates fat and feeding. Cell Metab. 2008;8:118–31. doi: 10.1016/j.cmet.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spiegel K, Leproult R, L’hermite-Balériaux M, Copinschi G, Penev PD, Van Cauter E. Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol, and thyrotropin. J Clin Endocrinol Metab. 2004;89:5762–71. doi: 10.1210/jc.2004-1003. [DOI] [PubMed] [Google Scholar]
- 22.Hasler G, Buysse DJ, Klaghofer R, Gamma A, Ajdacic V, Eich D, Rössler W, Angst J. The association between short sleep duration and obesity in young adults: a 13-year prospective study. Sleep. 2004;27:661–6. doi: 10.1093/sleep/27.4.661. [DOI] [PubMed] [Google Scholar]
- 23.Taheri S, Lin L, Austin D, Young T, Mignot E. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med. 2004;1:e62. doi: 10.1371/journal.pmed.0010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spaeth AM, Dinges DF, Goel N. Effects of Experimental Sleep Restriction on Weight Gain, Caloric Intake, and Meal Timing in Healthy Adults. Sleep. 2013;36:981–90. doi: 10.5665/sleep.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–85. doi: 10.1016/S0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 26.Tsujino N, Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev. 2009;61:162–76. doi: 10.1124/pr.109.001321. [DOI] [PubMed] [Google Scholar]
- 27.Secor SM, Stein ED, Diamond J. Rapid upregulation of snake intestine in response to feeding: a new model of intestinal adaptation. Am J Physiol. 1994;266:G695–705. doi: 10.1152/ajpgi.1994.266.4.G695. [DOI] [PubMed] [Google Scholar]
- 28.Secor SM, Diamond J. Adaptive responses to feeding in Burmese pythons: pay before pumping. J Exp Biol. 1995;198:1313–25. doi: 10.1242/jeb.198.6.1313. [DOI] [PubMed] [Google Scholar]
- 29.You YJ, Kim J, Cobb M, Avery L. Starvation activates MAP kinase through the muscarinic acetylcholine pathway in Caenorhabditis elegans pharynx. Cell Metab. 2006;3:237–45. doi: 10.1016/j.cmet.2006.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, Ogawa M, Chou CJ, Xia B, Crawley JN, et al. Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature. 2001;410:207–12. doi: 10.1038/35065604. [DOI] [PubMed] [Google Scholar]