

Abstract
Retinoid X receptor-alpha (RXRα) is implicated in the regulation of many biological processes and also represents a unique intracellular target for pharmacologic interventions. Efforts on discovery of small molecules targeting RXRα have been primarily focused on the molecules that bind to its classical ligand-binding pocket (LBP). Here, we report the identification and characterization of a new RXRα transcriptional antagonist by using structure-based virtual screening. The new antagonist binds with submicromolar affinity to RXRα (Kd = 4.88 × 10–7 M) and selectively inhibits RXRα transactivation. The compound does not bind to the LBP but to a hydrophobic groove on the surface of RXRα. The new compound also effectively suppresses AKT activation and promotes apoptosis of cancer cells in a RXRα-dependent manner by inhibiting tRXRα interaction with the p85α subunit of PI3K. Thus, the compound represents a new RXRα modulator that regulates the nongenomic actions of RXRα by surface binding.
Keywords: RXRα, coregulator-binding site, RXRα antagonist, nongenomic actions, virtual screening
Retinoid X receptor-α (RXRα), a unique member of the nuclear receptor (NR) superfamily, plays an important role in many biological processes ranging from apoptosis, cell differentiation, and growth to lipid metabolism.1−4 Altered expression and function of RXRα is implicated in the development of a number of diseases and cancer.1−4 Thus, RXRα has been an attractive and important target for pharmacologic interventions and therapeutic applications.1−4 The first identified natural RXRα ligand was the vitamin A derivative retinoid 9-cis retinoic acid (9-cis-RA).1−4 Some fatty acids such as docosahexaenoic acid, oleic acid, and phytanic acid also serve as ligands for RXRα. 9-cis-RA and synthetic ligands (rexinoids) are effective in preventing tumorigenesis and treating inflammatory diseases. Targretin (bexarotene), a rexinoid, was approved for treating human cutaneous T-cell lymphoma.1,5 RXRα acts primarily as a ligand-dependent transcription factor through forming homodimer with itself or heterodimer with other members of the NR family.
Structurally, RXRα is composed of three main functional domains: an N-terminal transcriptional activation function (AF-1) region, a DNA-binding domain and a ligand-binding domain (LBD).3,4 The LBD possesses a canonical ligand-binding pocket (LBP), a transactivation function domain 2 (AF-2) composed of Helix 12 of the LBD, a coregulator binding surface, and a dimerization surface.3,4 The ligand-dependent transcription regulation is predominately mediated through H12 that is highly mobile. The coregulator binding surface is a region where the binding sites of corepressor and the coactivator overlap. Canonical ligands bind to the LBP to mediate directly the transcriptional activity and so identifying and optimizing molecules that bind to its classical LBP has been the focus of drug discovery efforts targeting RXRα. A large pool of RXRα ligands that bind to the LBP have been designed and reported.1,6 However, there are key limitations of treatment with rexinoids including unwanted side effects such as rising of plasma triglyceride levels, suppression of the thyroid hormone axis, and induction of hepatomegaly.1,4−6 The current challenge is to discover selective RXRα modulators with the desired pharmacological activities but lacking undesired side effects.1,4,6 Therefore, targeting potential binding sites other than LBP could become a new paradigm for RXRα-based drug discovery. One of these potential binding sites is the coregulator-binding site.
Compounds that bind to the coregulator-binding site have been successfully demonstrated for other NRs, including estrogen receptor (ER),7 androgen receptor,8,9 vitamin D receptor, and thyroid receptor.10−12 However, compounds that bind to the coregulator-binding site of RXRα have not been reported. Inspired by the successes reported for other NRs, we employed docking-based virtual screening (VS) to identify RXRα modulators targeting the coregulator-binding site. Here we report the identification and characterization of a small molecule that binds to the coregulator-binding site of RXRα to regulate its nongenomic actions.
Docking-based VS is a popular approach used in drug discovery where the structure of the target or target homologue is available.13 Many crystal structures of RXRα LBD have been determined either in apo form or in complex with ligands or with both ligand and coregulator peptide,4,6 offering an excellent opportunity to identify new RXRα binding compounds using docking-based VS. A chemical library of 200,000 compounds, commercially available from Specs (www.specs.net), was subjected to a Pipeline Pilot protocol14 to filter out compounds that failed the Lipinski rules15 and that are potentially reactive and contain undesired groups.16 About 102,000 compounds left were then docked using Glide17 to the coactivator binding site on RXRα using the structure of RXRα LBD in complex with CD3254 and a coactivator peptide (PDB code 3FUG).18 Fourteen compounds (Figure S1A, Supporting Information) were selected for purchase and biological testing after visual evaluation of the first 300 compounds with the best docking score. Compound 7 (Figure 1A) showed the strongest antagonist activity (Figure S1B, Supporting Information) among these candidate compounds. Interestingly, part of 7 is similar to a recently reported androgen receptor inhibitor that is a diarylhydrazide and functions also via binding to the coactivator-binding site.8 Similar to the classical RXRα antagonist BI1003,197 inhibited 9-cis-RA-induced RXRα transactivation in a dose-dependent manner (Figure 1A). So analogues of 7 were searched and selected for preliminary SAR studies. Nine analogues (Figure 1B) were available commercially and ordered and tested for their RXRα antagonist effect and their selectivity toward other nuclear receptors including ER, retinoic acid receptor-γ (RARγ), and Nur77 (Figure 1C), as well as glucocorticoid receptor (GR), PPARγ, and LXRα (Figure S2, Supporting Information). Among these compounds, 23 (ordered from www.specs.net under catalog number AE-848/34436002) showed an antagonist activity similar to 7, but demonstrated the best selectivity for RXRα. It significantly inhibited the activity of RXRα, but not ER, GR, and Nur77, and showed slight inhibition of transactivation of RARα, RARγ, PPARγ, and LXRα (Figures 1C and S2, Supporting Information), which are known to heterodimerize with RXRα.20 Further, compound 23 showed very little inhibitory effect on transactivation of RXRα/RARα and RXRα/LXRα heterodimers (Figure S3, Supporting Information). We also tested the antagonist effect of 23 toward the RXR subtypes RXRγ and RXRβ. The results showed that overall 23 is more selective toward RXRα though it demonstrated some inhibition of transactivation of RXRγ and RXRβ (Figure S4, Supporting Information). Thus, 23 is a new RXRα-selective antagonist.
Figure 1.

Identification of RXRα-selective antagonist 23. (A) Compound 7 and its antagonist effect on RXRα transactivation. MCF-7 cells cotransfected with the reporter plasmids pG5-Luc and pBind-RXRα-LBD were treated with 9-cis-RA (10–7 M) alone or together with 7 or BI1003 for 18 h. Luciferase reporter activities were measured by using the Dual-Luciferase Reporter Assay System. Transfection efficiency was normalized to Renilla luciferase activity. (B) Compounds related to 7 identified by computational approach. (C) Antagonist effect of compound 7 and analogues. MCF-7 cells cotransfected with pG5-Luc and pBind-RXRα-LBD, pBind-ER-LBD, pBind-RARγ-LBD, or pBind-Nur77-LBD were treated, respectively, with 9-cis-RA (10–7 M), propyl pyrazole triol (PPT) (10 μM), and all-trans-RA (10–7 M), in the presence or absence of compound (10 μM) for 18 h. Reporter activities were measured as described above. Data shown are mean ± SD.
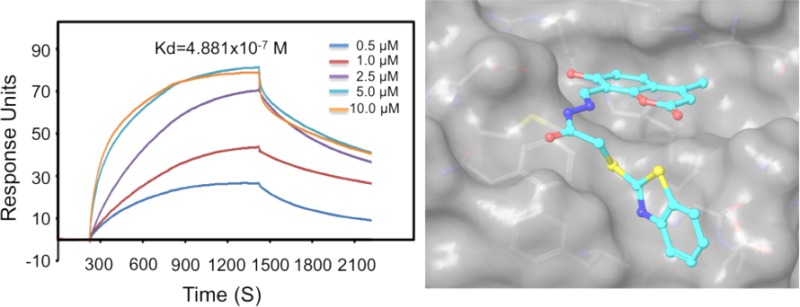
A dose-dependent study showed that 23 could inhibit 9-cis-RA-induced RXRα transactivation with an IC50 of 2.45 μM (Figure 2A). By using Biacore’s Surface plasmon resonance (SPR) technology, we found that 23 could bind to RXRα with a Kd of 4.88 × 10–7 M (Figure 2B). The binding of 23 to RXRα is unlikely due to its binding to the RXRα LBP, as it failed to compete with the binding of [3H]9-cis-RA to the RXRα LBP. By contrast, 9-cis-RA competed well with [3H]9-cis-RA for binding to RXRα (Figure 2C).
Figure 2.
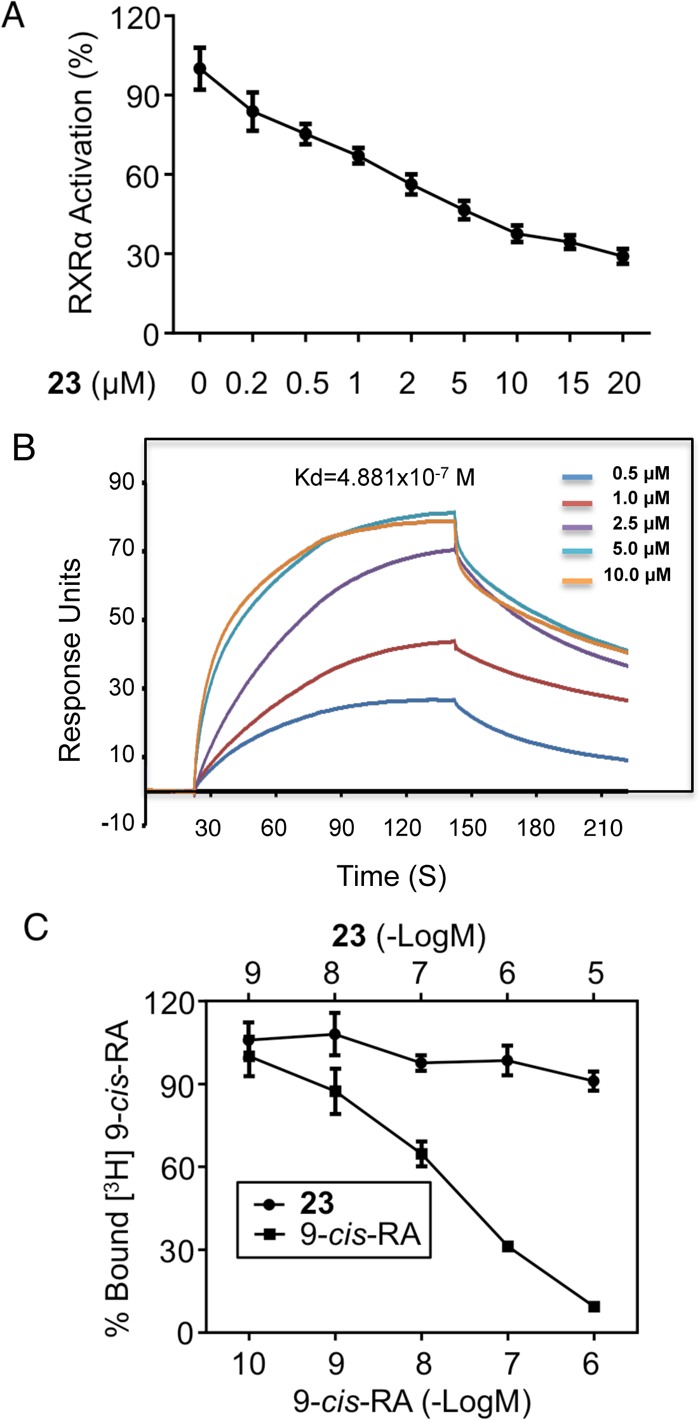
RXRα antagonist 23 binds RXRα at a site other than LBP. (A) Dose-dependent effect of 23 on inhibiting RXRα transactivation. HEK293T cells cotransfected with pG5-Luc and pBind-RXRα-LBD were treated with 9-cis-RA (10–-7 M) alone or together with the indicated concentration of 23 for 18 h. (B) SPR assay. Gradient concentrations of 23 were injected through flow cells immobilized with RXRα-LBD. The kinetic profiles are shown. The dissociation constant (Kd) of the 23/RXRα-LBD complex was calculated to be 4.881 × 10–7 M. (C) Compound 23 fails to compete with 9-cis-RA for binding to RXRα LBP. The bacterially expressed His-tagged RXRα-LBD was incubated with 7.5 nM [3H]-9-cis-RA in the presence or absence of the indicated concentrations of 9-cis-RA or 23. The RXRα LBD was captured by nickel-coated beads. Bound [3H]-9-cis-RA was quantitated by liquid scintillation counting. Data shown are mean ± SD.
To confirm that 23 binds to the coregulator-binding site on the surface of RXRα, mutagenesis in the site was carried out to validate its importance in the antagonist effect of 23. Mutations in the coactivator-binding site might impact not only the binding of 23 but also the binding of coactivator, which would preclude our evaluation by the reporter assay that depends on the binding of the coactivator. However, comparison of the interactions of 23 and coactivator to RXRα identified that Val298 in the Helix 4 of RXRα is critical for the binding of 23 but not the coactivator (Figure 3A). Thus, Val298 was mutated to Ser, and the resulting mutant, RXRα-V298S, was subjected to evaluation by the reporter assay. 9-cis-RA was able to activate RXRα-V298S, similar to its effect on RXRα (Figures 3B and S5, Supporting Information), suggesting that mutation of Val298 did not impair the ability of RXRα to bind to 9-cis-RA and to recruit the coactivator. Classical antagonists, such as BI100319 and UVI3003,21 also potently inhibited the 9-cis-RA-induced RXRα-V298S activity. By contrast, the antagonist effect of 23 was largely reduced as compared to its inhibitory effect on RXRα. Thus, Val298 is crucial for the binding of 23 but not classical RXRα ligands and coregulators.
Figure 3.
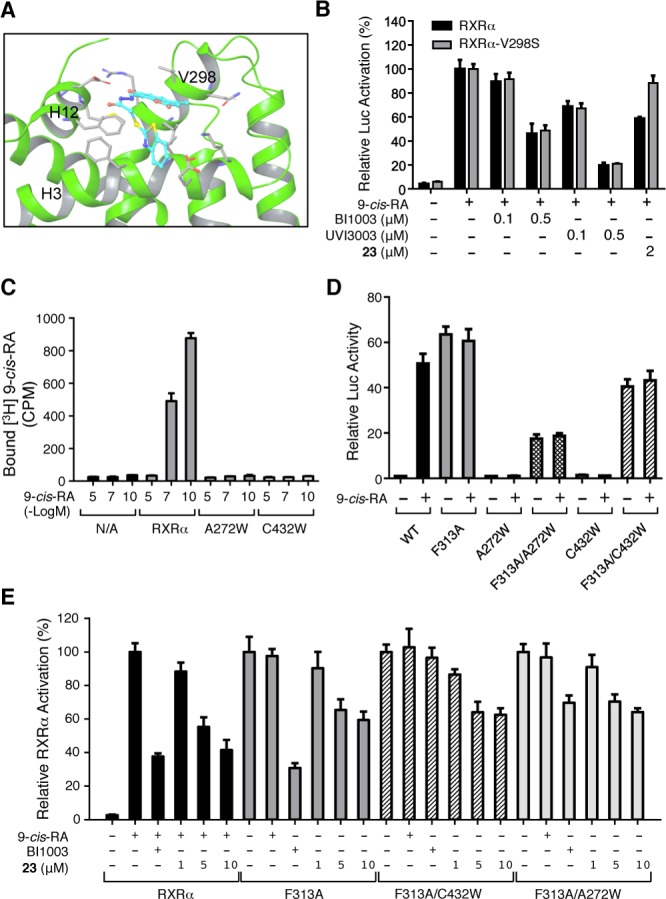
Coregulator-binding site of RXRα but not its LBP is critical for the antagonist effect of 23. (A) Val298 is critical for 23 binding revealed by modeling. (B) Mutation of Val298 impairs the antagonist effect of 23. HEK-293T cells cotransfected with pG5-Luc and pBind-RXRα/V298S were treated with the indicated compounds for 18 h. (C) Substitution of Cys432 and Ala272 in RXRα with Trp impairs 9-cis-RA binding. Lysates of HEK-293T cells transfected with RXRα, RXRα-C432W, or RXRα/A272W were incubated with 7.5 nM [3H]-9-cis-RA in the presence or absence of unlabeled 9-cis-RA. The Myc-RXRα was captured by hydroxylapatite. Bound [3H]-9-cis-RA was quantitated. (D,E) Transactivation of RXRα LBP mutants. HEK-293T cells cotransfected with pG5-Luc and the indicated RXRα or mutant expression vector were treated with 9-cis-RA (10–7 M), BI1003 (1 μM), or the indicated concentration of 23 for 18 h. Data shown are mean ± SD.
To further exclude the possibility that 23 binds to the LBP of RXRα, we designed and constructed RXRα mutants with its LBP blocked. Computational modeling suggested that substitution of Cys432 and Ala272 in the LBP with a bulky amino acid such as Trp could block the passage of a ligand to the LBP. Indeed, RXRα/C432W and RXRα/A272W failed to show any binding to [3H]-9-cis-RA (Figure 3C), even though the mutant proteins were well expressed (Figure S6, Supporting Information). In contrast, RXRα bound well to [3H]-9-cis-RA, which was competed away by unlabeled 9-cis-RA. F313A mutation is known to shift RXRα from an apo-receptor to a constitutively active form.22 In order to utilize LBP-blocked mutants to evaluate the antagonist effect of 23, Phe313 in RXRα/C432W and RXRα/A272W was substituted with Ala. As reported,22 RXRα-F313A is constitutived as active in the absence of 9-cis-RA. Similar to RXRα-F313A, both RXRα-C432W/F313A and RXRα-A272W/F313A mutants showed strong constitutive transcriptional activity (Figure 3D), making them ideal mutants to evaluate the LBP-independent antagonist effect of 23. Treatment of cells with 23 could effectively inhibit their constitutive activity in a dose-dependent manner, similar to its effect on RXRα (Figure 3E). By contrast, BI1003, which showed potent inhibitory effect on the transactivation of RXRα and RXRα-F313A by binding to their LBP, had much reduced inhibitory effect on the constitutive transactivation of both mutants. Thus, the blockage of the LBP of RXRα, which affects the activity of 9-cis-RA and the classical RXRα antagonist BI1003, has no effect on the antagonist activity of 23, further confirming unique RXRα binding activity of 23.
We recently reported that modulation of RXRα activity by certain RXRα ligands, such as Sulindac and analogues, could inhibit AKT activation in cancer cells.23,24 We therefore asked whether the LBP-independent binding of 23 could suppress AKT activation. A549 lung cancer and HepG2 liver cancer cells were treated with 23, and the activation of AKT was examined. As shown in Figure 4A, AKT activation in these cells was inhibited by 23 dose-dependently, with apparent inhibition observed when 5–10 μM of 23 was used. Since TNFα could induce RXRα-dependent AKT activation,24 cells were also treated with TNFα, and its activation of AKT in the absence or presence of 23 was examined. Treatment of cells with TNFα enhanced AKT activation, which was also suppressed by 23 (Figure 4A). Similar results were obtained in PC-3 prostate cancer cells (Figure S7, Supporting Information) and other cancer cells including colon cancer and pancreatic cancer cells (data not shown). We also evaluated the apoptotic effect of 23 in cancer cells. A549 and HepG2 cells were treated with 23 in the absence or presence of TNFα, and the cleavage of PARP, an indication of apoptosis in cancer cells, was examined by immunoblotting (Figure 4B). Treatment of cancer cells with 23 induced PARP cleavage, which was further enhanced by TNFα treatment. Such apoptosis induction by 23 correlated well with its inhibition of AKT activation, suggesting that AKT inhibition might play a role in its induction of apoptosis.
Figure 4.

Biological evaluation of 23. (A,B) Inhibition of AKT activation (A) and induction of apoptosis (B). Cells were pretreated with 23 for 24 h before being exposed to TNFα (20 ng/mL) for an additional 30 min. Lysates prepared were analyzed by Western blotting for AKT activation (A) or PARP cleavage (B). (C) RXRα-dependent effects of 23. A549 cells transfected with RXRα siRNA or control siRNA for 48 h were treated with 23 for 24 h before being exposed to TNFα (20 ng/mL) for an additional 30 min. Lysates prepared were analyzed by Western blotting. (D) Inhibition of p85α interaction with tRXRα by 23. A549 cells transfected with myc-RXRα-Δ80 expression vector were analyzed for their interaction with endogenous p85α by coimmunoprecipitation assay using anti-Myc antibody. Immunoprecipitates were analyzed by Western blotting for the presence of p85α and Myc-RXRα-Δ80. One of three to five similar experiments is shown.
To determine whether the expression of RXRα plays a role in the inhibition of AKT activation and the induction of apoptosis by 23, A549 cells were transfected with RXRα siRNA and evaluated for its effect on the role of 23 in AKT activation and apoptosis induction. Transfection of RXRα siRNA reduced the levels of RXRα and its truncated version, tRXRα,24 and diminished the effect of 23 on inducing PARP cleavage and inhibiting AKT activation (Figure 4C). Furthermore, the inhibitory effect of compound 23 on the growth of cancer cells can be significantly enhanced by RXRα (Figure S8, Supporting Information). These results demonstrate that RXRα plays a crucial role in mediating the biological effect of 23 on cell death.
We next determined whether 23 could affect tRXRα interaction with the p85α regulatory subunit of PI3K, an event known to activate PI3K/AKT.24 A549 cells were transfected with Myc-tagged RXRα-Δ80, a mutant that mimics tRXRα,24 and treated with or without TNFα and/or 23. Co-immunoprecipitation assays using anti-Myc antibody showed that p85α was coimmunoprecipitated together with Myc-RXRα-Δ80 in cells treated with TNFα (Figure 4D), demonstrating their interaction. However, when cells were cotreated with 23, TNFα-induced interaction of Myc-RXRα-Δ80 with p85α was almost completely inhibited. Thus, 23 might induce apoptosis by suppressing AKT activation through its inhibition of tRXRα interaction with p85α.
We report here through virtual screening our identification of a unique RXRα antagonist, 23, which modulates RXRα activities through LBP-independent binding. Several lines of evidence showed that 23 acts through its binding to the surface of RXRα. First, despite its high affinity for binding to RXRα revealed by SPR study, 23 failed to compete with 9-cis-RA for binding to the LBP of RXRα revealed by the classical ligand competition assay (Figure 2C). Second, mutation of Val298, a critical amino acid residue in the hydrophobic groove on the surface of RXRα, impaired the antagonist effect of 23 (Figure 3A). Third, 23 could effectively inhibit the transcriptional activity of RXRα mutants with impaired LBP (Figure 3D). To our knowledge, compounds that bind to the surface site of RXRα have not been reported. Thus, 23 represents the first small molecule capable of functionally binding to the surface site of RXRα with submicromoloar affinity.
Recent studies demonstrated that RXRα could crosstalk extensively with signal transduction pathways through its interaction with various signaling proteins,24 which are likely mediated through its surface binding sites. When the effect of 23 on the PI3K/AKT signaling was examined, we found that it could suppress basal and TNFα-induced AKT activation (Figure 4A) and induce apoptosis (Figure 4B) in cancer cells in a RXRα-dependent manner (Figure 4C). Our coimmunoprecipitation assays demonstrated that 23 could block tRXRα interaction with p85α (Figure 4D). Thus, the unique surface binding of 23 could interfere with the binding of some RXRα-interacting proteins, providing an opportunity to regulate RXRα activities by targeting the coregulator-binding sites. The report of the structure of estrogen receptor-β with a second molecule of 4-hydroxytamoxifen bound in its coactivator-binding surface provides insight into the possible pharmacological effects of the drug through its binding to the surface site on NR.25 Thus, our demonstrations that 23 could bind to the coregulator-binding site of RXRα and regulate the PI3K/AKT signaling pathway and apoptosis in cancer cells provide a new approach to target a functionally important surface site on RXRα that could represent a new strategy to tackle the specificity issue.
Glossary
ABBREVIATIONS
- RXRα
retinoid X receptor alpha:
- tRXRα
N-terminally truncated RXRα
- PI3K
phosphatidylinositol-3-OH kinase
- NR
nuclear receptor
- LBD
ligand-binding domain
- LBP
ligand-binding pocket
- PPT
propyl pyrazole triol
- TNFα
tumor necrosis factor-α
Biographies
Xiao-kun Zhang is the Thousand-Talent Professor and Dean of the School of Pharmaceutical Sciences, Xiamen University. He received his Ph.D. in biochemistry from the University of Vermont. After spending three years as a postdoctoral fellow at Sanford-Burnham Medical Research Institute, he joined the faculty at the same institute and was appointed as a professor in 2006. His work involves nuclear receptor signaling and drug development, with over 120 publications, 10 patents, and numerous awards. He is the coinventor of FDA-approved drug Targretin (bexarotene) for treating lymphoma patients.
Ying Su, Ph.D., is a computational chemist with over 15 years of experience with computer-aided drug design and Cheminformatics. Ying Su received her Ph.D. from the University of California, San Diego. After a postdoctoral appointment at the Scripps Research Institute, she worked for several local biotech companies. She joined the Sanford-Burnham Medical Research Institute in 2005 to build and lead a HTS informatics and CADD group. Dr. Su is coauthor of over 40 peer-reviewed scientific publications.
Supporting Information Available
Supplemental Figures 1 to 8 and detailed experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Army Medical Research and Material Command (W81XWH-11-1-0677), the National Institutes of Health (CA140980, GM089927, and CA179379), the 985 Project from Xiamen University, the National Natural Science Foundation of China (NSFC-91129302), and the fund from the Ministry of Education of China are acknowledged for financial support.
The authors declare no competing financial interest.
This paper posted ASAP on May 14, 2014. Figures 2 and 4 were corrected and the Supporting Information paragraph was ammended. The revised version was reposted on May 16, 2014.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dawson M. I.; Zhang X. K. Discovery and design of retinoic acid receptor and retinoid X receptor class- and subtype-selective synthetic analogs of all-trans-retinoic acid and 9-cis-retinoic acid. Curr. Med. Chem. 2002, 9, 623–637. [DOI] [PubMed] [Google Scholar]
- Liby K. T.; Yore M. M.; Sporn M. B. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat. Rev. Cancer 2007, 7, 357–369. [DOI] [PubMed] [Google Scholar]
- Szanto A.; Narkar V.; Shen Q.; Uray I. P.; Davies P. J.; Nagy L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004, 11Suppl 2S126–143. [DOI] [PubMed] [Google Scholar]
- de Lera A. R.; Bourguet W.; Altucci L.; Gronemeyer H. Design of selective nuclear receptor modulators: RAR and RXR as a case study. Nat. Rev. Drug Discovery 2007, 6, 811–820. [DOI] [PubMed] [Google Scholar]
- Querfeld C.; Nagelli L. V.; Rosen S. T.; Kuzel T. M.; Guitart J. Bexarotene in the treatment of cutaneous T-cell lymphoma. Expert Opin. Pharmacother. 2006, 7, 907–915. [DOI] [PubMed] [Google Scholar]
- Perez E.; Bourguet W.; Gronemeyer H.; de Lera A. R. Modulation of RXR function through ligand design. Biochim. Biophys. Acta 2012, 1821, 57–69. [DOI] [PubMed] [Google Scholar]
- Gunther J. R.; Moore T. W.; Collins M. L.; Katzenellenbogen J. A. Amphipathic benzenes are designed inhibitors of the estrogen receptor α/steroid receptor coactivator interaction. ACS Chem. Biol. 2008, 3, 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caboni L.; Kinsella G. K.; Blanco F.; Fayne D.; Jagoe W. N.; Carr M.; Williams D. C.; Meegan M. J.; Lloyd D. G. True antiandrogens—selective non-ligand-binding pocket disruptors of androgen receptor–coactivator interactions: novel tools for prostate cancer. J. Med. Chem. 2012, 55, 1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther J. R.; Parent A. A.; Katzenellenbogen J. A. Alternative inhibition of androgen receptor signaling: peptidomimetic pyrimidines as direct androgen receptor/coactivator disruptors. ACS Chem. Biol. 2009, 4, 435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzon V.; Carbo L. R.; Estruch S. B.; Fletterick R. J.; Estebanez-Perpina E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric control. Mol. Cell. Endocrinol. 2012, 348, 394–402. [DOI] [PubMed] [Google Scholar]
- Moore T. W.; Mayne C. G.; Katzenellenbogen J. A. Minireview: Not picking pockets: nuclear receptor alternate-site modulators (NRAMs). Mol. Endocrinol. 2010, 24, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caboni L.; Lloyd D. G. Beyond the ligand-binding pocket: targeting alternate sites in nuclear receptors. Med. Res. Rev. 2013, 33, 1081–1118. [DOI] [PubMed] [Google Scholar]
- Cheng T.; Li Q.; Zhou Z.; Wang Y.; Bryant S. H. Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J. 2012, 14, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan M.; Brown R. D.; Varma-O’brien S.; Rogers D. Cheminformatics analysis and learning in a data pipelining environment. Mol. Diversity 2006, 10, 283–299. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [DOI] [PubMed] [Google Scholar]
- Rishton G. M. Reactive compounds and in vitro false positives in HTS. Drug Discovery Today 1997, 2, 382–384. [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Perez Santin E.; Germain P.; Quillard F.; Khanwalkar H.; Rodriguez-Barrios F.; Gronemeyer H.; de Lera A. R.; Bourguet W. Modulating retinoid X receptor with a series of (E)-3-[4-hydroxy-3-(3-alkoxy-5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-y l)phenyl]acrylic acids and their 4-alkoxy isomers. J. Med. Chem. 2009, 52, 3150–3158. [DOI] [PubMed] [Google Scholar]
- Lu J.; Dawson M. I.; Hu Q. Y.; Xia Z.; Dambacher J. D.; Ye M.; Zhang X. K.; Li E. The effect of antagonists on the conformational exchange of the retinoid X receptor alpha ligand-binding domain. Magn. Reson. Chem. 2009, 47, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre P.; Benomar Y.; Staels B. Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol. Metab. 2010, 21, 676–683. [DOI] [PubMed] [Google Scholar]
- Nahoum V.; Perez E.; Germain P.; Rodriguez-Barrios F.; Manzo F.; Kammerer S.; Lemaire G.; Hirsch O.; Royer C. A.; Gronemeyer H.; de Lera A. R.; Bourguet W. Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 17323–17328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivat V.; Zechel C.; Wurtz J. M.; Bourguet W.; Kagechika H.; Umemiya H.; Shudo K.; Moras D.; Gronemeyer H.; Chambon P. A mutation mimicking ligand-induced conformational change yields a constitutive RXR that senses allosteric effects in heterodimers. EMBO J. 1997, 16, 5697–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. G.; Chen L.; Chen J.; Zheng J. F.; Gao W.; Zeng Z.; Zhou H.; Zhang X. K.; Huang P. Q.; Su Y. Synthesis and SAR study of modulators inhibiting tRXRalpha-dependent AKT activation. Eur. J. Med. Chem. 2013, 62, 632–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Liu W.; Su Y.; Wei Z.; Liu J.; Kolluri S. K.; Wu H.; Cao Y.; Chen J.; Wu Y.; Yan T.; Cao X.; Gao W.; Molotkov A.; Jiang F.; Li W. G.; Lin B.; Zhang H. P.; Yu J.; Luo S. P.; Zeng J. Z.; Duester G.; Huang P. Q.; Zhang X. K. NSAID sulindac and its analog bind RXRalpha and inhibit RXRalpha-dependent AKT signaling. Cancer Cell 2010, 17, 560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Chirgadze N. Y.; Briggs S. L.; Khan S.; Jensen E. V.; Burris T. P. A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor beta. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9908–9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.