

Abstract
Recent studies have shown that the meiosis-specific kinase, Mek1, plays a key role in promoting recombination between homologous chromosomes during meiosis in budding yeast by suppressing recombination between sister chromatids, as well as playing a role in the meiotic recombination checkpoint. Understanding how Mek1 regulates recombination requires the identification of direct substrates of the kinase. We have applied the semi-synthetic epitope method developed by Shokat and colleagues to Mek1. This method uses an analog-sensitive version of Mek1, Gst-mek1-as, in conjunction with an ATPγS analog, for kinase assays that detect only those proteins that are directly phosphorylated by Mek1. This method may be applicable to any kinase for which an analog-sensitive version is available. In addition, it provides a non-radioactive alternative for kinase assays with wild-type kinases.
Keywords: meiosis, Mek1, kinase assays, Rad54, semi-synthetic epitope, yeast
1. Introduction
In the past several years, protein kinases have been found to play key roles in a variety of meiotic processes, including the initiation of premeiotic DNA synthesis (Ime2, Cdc28) and recombination (Cdc28 and Cdc7), the promotion of recombination between homologous chromosomes instead of sister chromatids (Mek1/Mre4), resolution of recombination intermediates (Cdc5), mono-orientation of sister kinetochores at the first meiotic division (Cdc5 and Cdc7) and onset of the first meiotic division (Cdc28, Cdc7 and Ime2) (1-9). Understanding the molecular mechanisms by which these kinases work requires the identification of the direct substrates of these kinases and the phenotypic analysis of phosphorylation site mutants. A major problem inherent in kinase studies is specificity, since all protein kinases do essentially the same reaction— i.e., transfer the gamma phosphate from ATP onto serine, threonine or tyrosine residues in their targets. This specificity issue has been addressed by the development of analog-sensitive (as) kinases, in which the ATP binding pocket of a kinase of interest is enlarged by mutation (10, 11). Addition of inhibitors that are too bulky to fit into the ATP binding pockets of unmodified kinases results in the specific inactivation of the as kinase. In addition, use of ATP analogs in conjunction with an as kinase results in specific phosphorylation of target proteins (e.g., 12). These target proteins can be detected using the semi-synthetic epitope method (13) (Figure 1). Use of an ATPγS analog in the kinase reaction results in thiophosphorylation of substrate proteins specifically by the as kinase. This thiophosphorylation is then converted into an affinity tag by an alkylation reaction using p-nitrobenzyl mesylate (PNBM). The resulting thiophosphate ester is then detected on immunoblots using a commercially available rabbit monoclonal antibody referred to as the α-hapten antibody.
Figure 1.

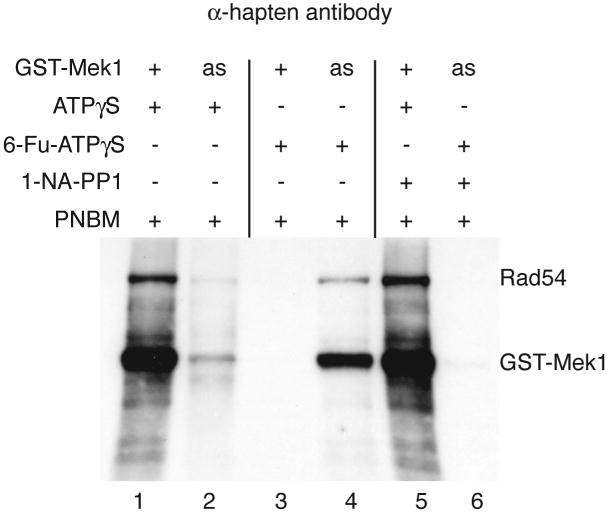
Mek1 is a meiosis-specific kinase that regulates meiotic recombination in a variety of ways (6, 12). Using the semi-synthetic epitope system we have shown that Mek1 itself, as well as the recombination protein, Rad54, are direct targets of the kinase (14) (Figure 2). Although Mek1 is involved suppressing Rad51-mediated strand invasion of sister chromatids as well as the meiotic recombination checkpoint, the substrates involved in these processes have not yet been identified (14, 15). The semi-synthetic epitope approach provides a relatively easy assay for testing whether candidate proteins are direct targets of Mek1. In addition, this protocol may be adapted for use with other as kinases such as Cdc5-as, Ime2-as, Cdc7-as and Cdc28-as, to name a few (1, 5, 16). Finally, the semi-synthetic epitope system can be used with wild-type kinases for non-radioactive kinase assays (Figure 2, lane 1).
Figure 2.
For Gst-mek1-as, the semi-synthetic epitope method can be broken down into four parts: (1) generating a culture of meiotic cells containing activated Gst-mek1-as kinase, (2) pulling down Gst-mek1-as from soluble extracts using glutathione-sepharose, (3) using the beads in kinase assays containing a substrate of interest and the ATP analog, 6-Fu-ATPγS, followed by alkylation of the thio-phosphorylated proteins and (4) probing the phosphorylated proteins on an immunoblot using the α-hapten antibodies.
2. Materials
2.1 Yeast strains and sporulation
SK1 diploid strain NH520 transformed with high copy number GST-mek1-as plasmid, pLW3 (6) MATaleu2∷hisG his4-X dmc1Δ hoΔ lys2 ura3 mek1Δ/2μ GST-mek1-as URA3 MATα leu2∷hisG his4-B dmc1Δ hoΔ lys2 ura3 mek1Δ (See Note 1)
SD-ura plates: 2% agar, 0.7% yeast nitrogen base without amino acids, 2% glucose, 1 g –uracil dropout powder (See Note 2)
YEP + glycerol plates: 2% agar, 2% bactopeptone, 1% yeast extract, 3% glycerol
Liquid YEPD medium: 2% bactopeptone, 1% yeast extract, 2% glucose
Liquid YPA medium: 2% bactopeptone, 1% yeast extract, 2% potassium acetate
Sporulation (Spo) medium: 2% potassium acetate
2.2 Yeast extracts and glutathione precipitation
Lysis buffer (LB) Make fresh the same day using ice-cold water and keep on ice. 50 mM Tris-HCl, pH 7.5, 10 mM EDTA, pH 8.0, 300 mM NaCl, 1 mM dithiothreitol, 10 mM NaF (Sigma, diluted from 1M stock made up in water and stored at 4°C), 10 mM Na4P2O4 (Sigma, diluted from 100 mM stock made up in water and stored at 4°C), 1 mM PMSF (Sigma, diluted from 100 mM stock made up in ethanol and stored at room temperature), 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin [USB Corporation, all diluted from 1 mg/ml stocks made up in either water (leupeptin and aprotinin) or ethanol (pepstatin) and frozen at -20°C in 1 ml aliquots]. Add PMSF immediately before use as it is unstable in aqueous solutions.
Glutathione-sepharose (Amersham Biosciences) The day of the experiment, glutathione-sepharose is equilibrated with LB. For each yeast cell pellet from 100 ml sporulating culture, use 40 μl of a 1:1 slurry of beads: liquid. Transfer slurry to a 1.7 ml microfuge tube and mark the volume on the tube. Add 1 ml LB, spin for 10 sec at 2000 x g, remove supernatant by aspiration, leaving ∼100 μl behind. Repeat wash two times with 1 ml LB. After the final wash, remove LB to the mark so that the 1:1 slurry is regenerated.
Glass beads, 0.5 mm (#11079105), Biospec Products, Inc. Use directly from bottle. Place on ice at the beginning of the procedure to cool beads down.
BioRad Protein Assay reagent (#500-0001). Store at 4°C.
30 1/2 gauge needle and 1 ml syringe
2.3 Kinase assays and alklyation
ATP (Sigma) Make 100 μM stock in water, aliquot and store at -80°C.
6-Fu-ATPγS (BioLog #F008) Comes as a 10 mM solution. Aliquot 6 μl/microfuge tube and store at -80°C. The nucleotide should not be reused after thawing.
Kinase buffer: 50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 10 mM MgCl2.
p-nitrobenzyl mesylate (PNBM) (Epitomics #3700-1) Add 420 μl DMSO to 5 mg powder in bottle to generate a 50 mM stock. Make 30 μl aliquots and store at -20°C. The PNBM can be refrozen and reused.
1-(1,1-Dimethylethyl)-3-(1-naphthalenyl)-1H-pyrazolo[3, 4-d]pyrimidin-4-amine (1-NA-PP1) (Tocris Bioscience #3063) Resuspend 10 mg in 3.15 ml dimethylsulfoxide (DMSO) to generate 10 mM stock. Store at -20°C. This stock can be refrozen and reused. Dilute to 375 μM in DMSO on the day of the experiment.
5 X Protein Sample Buffer: 310 mM Tris-HCl, pH 6.8, 15% SDS, 25% 2-mercaptoethanol, 25% glycerol (weight/volume) and 0.25% bromophenol blue.
2.4 Protein gels and immunblots
8% SDS-polyacrylamide gel a. Resolving gel For one mini-gel make 5 ml solution using 2.3 ml water, 1.3 ml 30% Acrylamide/Bis (29:1) (BioRad), 1.3 ml 1.5 M Tris-HCl, pH 8.8, 50 μl 10% SDS, 50 μl 10% ammonium persulfate (APS) (BioRad). APS can be stored at 4°C for up to a week. Polymerization is initiated by addition of 3 μl N,N,N,N′-Tetramethyl-ethylenediamine (TEMED) (BioRad) and should only be added immediately prior to pouring the gel. b. Stacking gel For a single gel, make 2 ml using 1.4 ml water, 330 μl 30% acrylamide/bis (29:1), 250 μl 1 M Tris-HCl, pH 6.8, 20 μl 10% SDS, 20 μl 10% APS and 2 μl TEMED.
Benchmark Prestained Protein Ladder (Invitrogen)
Whatman 0.45 mM Opitran BA-S85 Reinforced nitrocellulose (VWR)
Whatman 3MM filter paper (Fisher Scientific)
Running buffer: 0.3% Tris Base, 0.1% SDS, 1.44% glycine. Can be stored at room temperature indefinitely either as a 10 X or 1 X solution.
Transfer buffer: 1.47% glycine, 0.3% Tris base, 20% methanol. Can be stored indefinitely at room temperature as a 1 X solution.
TBST: 20 mM Tris-HCl, pH 7.5, 250 mM NaCl, 0.1% Tween-20 This solution can be made in a large volume (e.g. 2 l) as a stock that is stored at room temperature.
Blocking buffer TBST plus 5% non-fat milk. Dissolve 2.5 g milk powder in 50 ml TBST (See Note 3).
Primary antibody Thiophosphate ester rabbit monoclonal antibody (the α-hapten antibody) (Epitomics, #2686-1)
Secondary antibody Goat anti-rabbit IgG (H+L) HRP antibody (Epitomics #3053-1)
Amersham ECL Plus Western Blotting Detection System (GE Healthcare RPN2132)
Amersham Hyperfilm ECL (GE Healthcare 28-9068-39)
3. Methods
3.1 Sporulation
Using a sterile toothpick, streak out NH520/pLW3 onto SD-ura plates to select for single colonies that contain the plasmid. Invert plate and incubate at 30°C for 2-3 days.
Inoculate a single colony into 5 ml YEPD in a 15 ml test tube. Incubate on a roller at 30°C overnight. Patch cells from the same colony onto YEP + glycerol plates to check for petite colonies (see Note 4).
At 5:00 pm the next day, dilute 1.2 ml and 2.0 mls of overnight culture into 600 ml YPA in two 2.8 L flasks, respectively. Incubate at 30°C shaking at 250 rpm for 16 hrs (See Notes 5 and 6).
At 9:00 am the next day, blank the spectrophotometer with 1 ml YPA and read the absorbance at a wavelength 660 nm of 1 ml of undiluted culture. The OD660 reading should be between 1.2 and 1.4. Using the conversion chart in Table 1, convert the OD660 to cell density and multiply times the total volume of YPA to determine the total number of cells in the culture. Calculate the volume of Spo medium necessary to give a cell density of 3 × 107 cells/ml. Aliquot this volume minus 5 ml into a 2.8 L flask (See Note 7).
Divide cells between two 500 ml bottles and pellet in a centrifuge at 3000 x g for 5 min. Resuspend each pellet in 5 ml water, combine, transfer to a 15 ml test tube and pellet again in a tabletop centrifuge. Resuspend pellet in 5 ml Spo medium and add to flask with Spo medium. Place on shaker (250 rpm) at 30°C for five hours (See Note 8).
Divide the sporulating culture into 100 ml aliquots in 250 ml bottles and pellet in a centrifuge. Resuspend each pellet in 10 ml water, transfer to a 15 ml test tube and spin in the tabletop centrifuge. Pour off supernatant and resuspend the cells in 1 ml 25% glycerol. Transfer to 1.7 ml graduated microfuge tubes (Posi-Click from Denville Scientific) and store at -80°C.
Table 1.
Conversion of optical density660 (OD660) values to cell density.
| OD660 | Cells/ml (×107) |
|---|---|
| 0.80 | 0.63 |
| 0.85 | 0.69 |
| 0.90 | 0.76 |
| 0.95 | 0.83 |
| 1.00 | 0.92 |
| 1.05 | 1.02 |
| 1.10 | 1.12 |
| 1.15 | 1.24 |
| 1.20 | 1.35 |
| 1.25 | 1.48 |
| 1.30 | 1.61 |
| 1.35 | 1.76 |
| 1.40 | 1.91 |
| 1.45 | 2.06 |
| 1.50 | 2.24 |
| 1.55 | 2.42 |
| 1.60 | 2.61 |
3.2 Yeast extracts and glutathione precipitation
This protocol uses a frozen cell pellet from 100 ml sporulating culture.
Thaw pellet on ice for approximately 10 minutes. If necessary, melting can be accelerated by gentle flicking of the tube.
Everything should be kept as cold as possible to reduce the chance of proteolysis. Pellet cells by spinning in a 4°C microfuge for 1 min at 3300 × g.
To wash the cells, resuspend pellet in 0.5 ml LB and transfer to a 14 ml round bottom graduated Falcon tube (2059, Fisher Scientific) containing 4.5 ml LB (See Note 9).
Pellet 1 min in a tabletop centrifuge. Discard supernatant and resuspend in 1 ml LB. Measure 1 ml glass beads using a graduated microfuge tube and add to cells (the total volume should be ∼2 ml).
Vortex 10 × 1 min at top speed with 1 min rests on ice in between.
Spin 1 min in the tabletop centrifuge to remove bubbles. Using a pipetman, transfer the supernatant to a new 1.7 ml microfuge tube. Measure the volume using the graduations on the side of the tube and add 25% Triton X-100 to a final concentration of 1%. Mix gently by flicking. Incubate on ice for 10 min. During this time equilibrate the glutathione-sepharose with lysis buffer (See Note 10).
Pellet insoluble material by spinning the extract in a 4°C microfuge for 10 min at 16,200 × g.
To measure the protein concentration, make a 1:10 dilution of the soluble extract in LB. For each extract, add 800 μl water and 200 μl BioRad Protein Assay reagent to a microfuge tube and add 1 μl of the diluted extract. Blank the spectrophotometer with 1 X BioRad Protein Assay reagent at OD595 and compare the absorbance value for each extract to a standard curve previously generated using known quantities of a standard protein such as bovine serum albumin. Multiply by 10 to calculate the protein concentration of the extract. The concentration should be at least 20 mg/ml (See Note 11).
Transfer the entire soluble extract (∼800-900 μl) to a microfuge tube containing 40 μl 1:1 glutathione-sepharose slurry equilibrated in LB, being careful not to transfer any insoluble material.
Rock tube for 1.5 hr at 4°C to allow the Gst-mek1-as to bind the glutathione-sepharose.
Pellet beads by spinning in a microfuge for 30 sec at 3300 × g. It is difficult to see the beads at this point so leave ∼200 μl extract behind when removing the supernatant by either aspiration or with a pipetman.
Wash beads by resuspending them in 1 ml lysis buffer and spinning as in step 11. Remove supernatant and repeat for a total of three times, then wash twice with 1 ml ice-cold kinase buffer. After the final wash, remove any residual liquid using a 1ml syringe attached to a 30 1/2 gauge needle.
3.3 Kinase assays and alkylation
Kinase reactions contain between 24-28 μl and include 22 μl Gst-mek1-as-bound bead slurry, 1 μl 100 μM ATP, 1 μl 10 mM 6-Fu-ATPγS, 1-4 μl substrate and 1 μl 375 μM 1-NA-PP1 (if appropriate). Determine the number of desired reactions and multiply by 22 μl to determine the volume of kinase buffer required to resuspend the kinase bound beads (See Note 12). For four reactions, use 88 μl kinase buffer. Transfer 22 μl bead:buffer slurry to three 1.7 ml microfuge tubes, leaving 22 μl in the original microfuge tube. Before pipetting the slurry, mix well with the pipette tip as the beads tend to settle to the bottom of the tube. Add 0.2-1.0 μg of putative substrate to each reaction.
For reactions containing inhibitor, add 1 μl 375 μM 1-NA-PP1 (final concentration is 15 μM) and wait five minutes at room temperature (see Note 13).
Add 6 μl 100 μM ATP to the 6 μl aliquot of 10 mM 6-Fu-ATPγS. To start the kinase reaction, add 2 μl of this mixture to the bead slurry + substrate and stir gently with pipette tip. Incubate at 30°C for 30 min.
To alkylate the proteins, add 1.3 μl 50 mM PNBM for a final concentration of 2.5 mM. Mix well with pipette tip and leave at room temperature for 2 hours (See Note 14).
Stop the alkylation reaction by adding 6 μl of 5 X Protein Sample Buffer and incubate the tubes at 95°C for five minutes. Before loading gel, spin the samples for 10 sec at 16,200 × g in a microfuge.
3.4 Protein gels and immunoblots
This protocol assumes the use of a Hoeffer Mini-gel apparatus but other apparatuses may also be used. Take a square glass plate and put a 1 mm spacer on either side. Place a notched plate on top. Holding the plates and spacers together, tap the bottom of the sandwich gently on the bench top to make sure the plates and spacers are flush and then clamp onto a Hoeffer gel casting apparatus. Placing a piece of parafilm underneath the bottom of the plates helps prevent leaks.
Acrylamide is a neurotoxin and therefore gloves should be worn while pouring the gel. To make the resolving gel, combine all of the solutions except the TEMED using disposable pipets in a 15 ml plastic tube and mix well. Add TEMED, mix and using a Pasteur pipet, immediately fill up ¾ of the volume between the glass plates. Using a squirt bottle, layer ethanol on top of the acrylamide. Leave at room temperature for at least 30 min to polymerize. Check the residual acrylamide in the plastic tube to confirm that the polymerization reaction has occurred. After polymerization, this tube can be discarded in the regular trash.
To pour the stacking gel, combine all the solutions except the TEMED in a 15 ml plastic tube. Pour ethanol off the resolving gel. Blot away residual ethanol using 3 MM filter paper. Add TEMED and fill to the top of the gel using a Pasteur pipette. Immediately insert comb and let polymerize for at least 15 min.
To run the gel, remove the plates from the gel casting apparatus and assemble onto gel tank with the notched plate facing inwards and the notch on top. Fill upper and lower reservoirs with running buffer, being sure to submerge the teeth of the comb. Place a stencil on the outer glass plate to indicate the positions of the wells and remove comb.
Load 10 μl Benchmark protein markers and 15-25 μl kinase reaction into separate wells using 1-200 μl Microcapillary Tips from Nortech (RSE-01R-204). Adding 15 μl of 1 X Protein Sample Buffer to empty lanes helps the gel run more evenly.
Run gel at 100 V (constant voltage) for 2 hr. The pink marker should not run off the gel (See Note 15).
To stop gel, turn off power supply and remove gel sandwich from the apparatus. To disassemble, remove the spacers and use one spacer to leverage the notched gel plate off the gel. The gel should remain stuck to the unnotched plate. Cut off the stacking gel with a scalpel.
Measure the dimensions of the gel and cut four pieces of 3MM filter paper and one piece of nitrocellulose to those dimensions.
Wet the nitrocellulose and pieces of filter paper in transfer buffer. Place one piece of wet filter paper on top of the gel and smooth out any bubbles by rolling a glass rod over the paper. Using a flat thin spatula, separate a corner of the gel from the glass plate and then, holding the gel and paper together, pull gently to peel the gel away from the plate.
With the gel side up, place the filter paper holding the gel onto a second piece of wet filter paper on a smooth surface. Layer the nitrocellulose and the remaining pieces of filter paper on top of the gel for a sandwich that is composed from bottom to top of the following: 2 pieces of filter paper, gel, nitrocellulose membrane, 2 pieces of filter paper. Remove bubbles using the glass rod each time a layer is added.
Proteins are transferred onto nitrocellulose using a Transblot SD Semi-Dry Transfer Cell apparatus from BioRad. Invert the sandwich so that the nitrocellulose membrane is under the gel, place on the transfer apparatus and connect the lid. Attach the transfer apparatus to a power supply such that current flows from top to bottom in a negative to positive direction so that the SDS-bound negatively charged proteins travel from the gel onto the membrane. For a mini-gel, use 70 mAmp (constant current) for 1 hour.
Remove the lid and disassemble the sandwich using forceps to peel the nitrocellulose membrane off the gel. If transfer has occurred properly, the prestained standards should be visible on the membrane. Place the membrane in a small plastic container that is slightly larger than the filter. Cover the membrane with blocking buffer (5-10 ml) and rotate gently on a platform shaker at room temperature for 1 hour. (See Note 16).
Pour off the blocking buffer. Rinse the membrane by adding ∼10 ml of TBST, briefly swirling the container by hand, and pouring the TBST into the sink. For the primary antibody incubation, add 5 ml of blocking buffer and a 1:10,000 dilution (0.5 μl) of α-hapten antibodies. Shake overnight (usually between 12-14 hrs) at 4°C.
Pour off blocking buffer with antibody and wash the membrane by adding ∼10 ml TBST and shaking for 10 min at room temperature. Repeat for a total of three washes.
For the secondary antibody, add 5 ml of blocking buffer with a 1:5000 dilution (1 μl) of goat anti-rabbit IgG HRP antibody and shake at room temperature for 1 hr.
Wash membrane three times with TBST for 10 min each, shaking at room temperature. After the final wash, drain the TBST from the membrane. Cut a piece of parafilm that is larger than the membrane and place the membrane face up on the parafilm so that the prestained molecular weight markers are visible.
In a test tube, mix two ml of ECL Plus kit Solution A with 50 μl Solution B. Using a pipette gently apply the mixture to the top of the membrane (See Note 17). Incubate at room temperature for 5 min.
Pick up the membrane with a forceps and remove any excess liquid by touching the edges of the membrane gently onto a paper towel.
For assaying the chemiluminescent signal, cover the membrane with plastic. Go to a dark room and put the membrane on film (See Note 18).
Develop film with an X-ray film developer (See Note 19).
Acknowledgments
We thank Jasmina Allen, Kevan Shokat and Beatrice Wang for help with ideas and reagents in the early development of this protocol. Patrick Sung generously provided bacterially purified Rad54 protein. Aaron Neiman provided helpful comments on the manuscript. This work was supported by an NIH grant to N. M. H. (R01 GM50717).
Footnotes
Mek1 is activated in response to meiotic DSBs by auto-phosphorylation and therefore it is necessary to isolate the kinase from meiotic cells (17). Mek1 is constitutively active in dmc1Δ-arrested cells (6). Therefore purifying Mek1 from dmc1Δ diploids increases the number of cells containing active kinase, as does using a high copy plasmid to over-express GST-mek1-as. In vitro autophosphorylation of Gst-mek1-as serves as a good internal positive control for the kinase reaction. Using strains derived from the SK1 background is useful for two reasons: (1) sporulation is highly efficient with wild-type cells typically exhibiting >90% asci and (2) the dmc1Δ arrest is very tight (18, 19). Yeast strains and plasmids are available from the Hollingsworth lab.
Although this method uses Gst-mek1-as, it may be applicable to any as-kinase for which an ATPγS analog can be identified and the kinase can be precipitated from an extract. An excellent description of the analog-senstive kinase approach can be found in (20), including which amino acids to mutate and how to analyze the resulting mutants. In addition to the 6-Fu-ATPγS analog, 6-phenethyl-ATPγS (6-PhEt-ATPγS) and 6-benzyl-ATPγS (6-Bn-ATPγS) are also commercially available from BioLog (http://www.biolog.de). Since different as-kinases exhibit preferences for different analogs, all of the analogs should be tried to see which one works best.
The semi-synthetic epitope system can also be used with wild-type kinases by substituting ATPγS for the analog. Although these reactions lack the specificity provided by as-kinases and therefore may have some additional background due to co-precipitating kinases that can also use ATPγS (Figure 2, compare lanes 1 and 4), they have the advantage over traditional kinase assays in being non-radioactive.
To make –ura dropout powder, combine in a blender: 5 g Adenine-HCl, 5 g tryptophan, 5 g histidine-HCl, 5 g Arginine-HCl, 5 g Methionine, 7.5 g Tyrosine, 7.5 g Leucine, 7.5 g Isoleucine, 7.5 g Lysine-HCl, 7.5 g valine, 7.5 g threonine, 7.5 g serine, 12 g phenylalanine. Blend for 5 × 1 min bursts using the “mix” setting. Pour into sterile bottle and use 2 g/L.
The blocking buffer should be made fresh for each experiment as the milk may go sour with time. Do not add sodium azide as this will inhibit the horseradish peroxidase reaction used to detect the antibody. Keep at 4°C during the overnight incubation.
One characteristic of the SK1 background is that it gives rise to petite (respiration-deficient) colonies. Because sporulation requires respiration, cells with defective mitochondria are unable to undergo meiosis. Every colony should therefore be checked for its ability to grow on a non-fermentable carbon source such as glycerol before sporulating the cells.
The synchrony and efficiency of sporulation is maximized if log phase cells at a density of 3 × 107 cells/ml are used. Because strains may exhibit different growth rates depending upon the starting inoculum, two different dilutions are used for a standard number of hours to increase the chances that the cells will be at the appropriate density at the start of the experiment.
The most time consuming part of this protocol is sporulating the cells. We therefore sporulate several hundred mls of cells at a time and store the meiotically arrested cells in 100 ml aliquots at -80°C. To achieve the correct cell density, at least two times as much YPA is needed as Spo medium. OD660 readings between 1.2 and 1.4 from 600 ml of YPA will produce from 270 to 380 ml sporulating culture (Table 1).
Good aeration is critical for sporulating yeast cells. Therefore the flask volume should always be at least five times the volume of Spo medium.
Cell are incubated in Spo medium for five hours to give the cells a chance to enter the meiotic program and arrest with unrepaired DSBs due to the meiotic recombination checkpoint. The time in Spo medium may be increased up to 8 hours if necessary.
Mek1 is activated by phosphorylation of its activation loop (17). Therefore it is important to include phosphatase inhibitors in the lysis buffer to prevent loss of this phosphorylation and therefore kinase activity.
Detergent is used to dissolve membranes and is added after vortexing to prevent foaming.
If making more than one extract, the amount of protein used for the Gst-mek1-as precipitation can be equalized by varying the volume used for the pulldown.
The amount of Gst-mek1-as pulled down from 100 ml sporulating cell pellet is more than enough for four reactions. If more reactions are necessary, the amount of kinase/reaction can be reduced by resuspending the beads in a larger volume of kinase buffer before distributing 22 μl into tubes for kinase reactions.
Addition of inhibitor should block the kinase reaction and serves as a good control for the specificity of the reaction (Figure 2, compare lanes 4 and 6).
The kinase buffer must have less than 0.5 mM reductant (e. g. DTT) because otherwise the PNBM will be consumed. If necessary, reactions can be quenched by the addition of EDTA to final concentration twice that of the magnesium concentration.
This protocol is designed to detect proteins greater than 50 kD. If smaller substrates are being tested, a higher percentage acrylamide gel may be necessary and the length the gel is run should be adjusted accordingly.
Incubating the membrane in milk prior to the addition of antibody decreases non-specific binding of the antibody to the membrane.
The HRP reaction occurs using a small volume (∼ 2 ml) of ECL Plus reagents. This volume can be placed directly on the membrane if the membrane is on a hydrophobic surface such as parafilm or saran wrap. The mixture must be added carefully so that the surface tension is not broken, or the mixture will spill off the membrane.
The membrane may be placed on a piece of plastic wrap that is then folded over to cover both sides. Alternatively, we slit the side of a hybridization bag and insert the membrane inside to eliminate creases that frequently arise when using plastic wrap. To conserve film, the membrane can be pressed down onto different areas of the same film for exposures ranging from 1 sec to 5 min before the film is developed.
The ECL Plus kit generates both a chemiluminescent signal that is transient (gone after ∼ 30 min) and a stable chemifluorescent signal. The chemiluminescent signal can be detected by exposing blots to X-ray film while the chemifluorescent signal can be detected and quantitated using a phosphoimager.
References
- 1.Benjamin KR, Zhang C, Shokat KM, Herskowitz I. Control of landmark events in meiosis by the CDK Cdc28 and the meiosis-specific kinase Ime2. Genes Dev. 2003;17:1524–39. doi: 10.1101/gad.1101503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee BH, Amon A. Role of Polo-like kinase CDC5 in programming meiosis I chromosome segregation. Science. 2003;300:482–6. doi: 10.1126/science.1081846. [DOI] [PubMed] [Google Scholar]
- 3.Matos J, Lipp JJ, Bogdanova A, Guillot S, Okaz E, Junqueira M, Shevchenko A, Zachariae W. Dbf4-dependent CDC7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell. 2008;135:662–678. doi: 10.1016/j.cell.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Pak J, Segall J. Regulation of the premiddle and middle phases of expression of the NDT80 gene during sporulation of Saccharomyces cerevisiae. Mol Cell Biol. 2002;22:6417–29. doi: 10.1128/MCB.22.18.6417-6429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sourirajan A, Lichten M. Polo-like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev. 2008;22:2627–2632. doi: 10.1101/gad.1711408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wan L, de los Santos T, Zhang C, Shokat K, Hollingsworth NM. Mek1 kinase activity functions downstream of RED1 in the regulation of meiotic DSB repair in budding yeast. Mol Biol Cell. 2004;15:11–23. doi: 10.1091/mbc.E03-07-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan L, Niu H, Futcher B, Zhang C, Shokat KM, Boulton SJ, Hollingsworth NM. Cdc28-Clb5 (CDK-S) and Cdc7-Dbf4 (DDK) collaborate to initiate meiotic recombination in yeast. Genes Dev. 2008;22:386–397. doi: 10.1101/gad.1626408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed NT, Bungard D, Shin ME, Moore M, Winter E. The Ime2 protein kinase enhances the disassociation of the Sum1 repressor from middle meiotic promoters. Mol Cell Biol. 2009;29:4352–4362. doi: 10.1128/MCB.00305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henderson KA, Kee K, Maleki S, Santini P, Keeney S. Cyclin-dependent kinase directly regulates initiation of meiotic recombination. Cell. 2006;125:1321–1332. doi: 10.1016/j.cell.2006.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bishop AC, Buzko O, Shokat KM. Magic bullets for protein kinases. Trends Cell Biol. 2001;11:167–172. doi: 10.1016/s0962-8924(01)01928-6. [DOI] [PubMed] [Google Scholar]
- 11.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 12.Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859–64. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- 13.Allen JA, Li M, Brinkworth CS, Paulson JL, Wang D, Hubner A, Chou WH, Davis RJ, Burlingame AL, Messing RO, Katayama CD, Hedrick SM, Shokat KM. A semisynthetic epitope for kinase substrates. Nature Methods. 2007;4:511–516. doi: 10.1038/nmeth1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niu H, Wan L, Busygina V, Kwon Y, Allen JA, Li X, Kunz RC, Kubota K, Wang B, Sung P, Shokat KM, Gygi SP, Hollingsworth NM. Regulation of meiotic recombination via Mek1-mediated Rad54 phosphorylation. Mol Cell. 2009;36:393–404. doi: 10.1016/j.molcel.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu L, Weiner BM, Kleckner N. Meiotic cells monitor the status of the interhomolog recombination complex. Genes Dev. 1997;11:106–118. doi: 10.1101/gad.11.1.106. [DOI] [PubMed] [Google Scholar]
- 16.Wan L, Zhang C, Shokat KM, Hollingsworth NM. Chemical inactivation of Cdc7 kinase in budding yeast results in a reversible arrest that allows efficient cell synchronization prior to meiotic recombination. Genetics. 2006;174:1667–1774. doi: 10.1534/genetics.106.064303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niu H, Li X, Job E, Park C, Moazed D, Gygi SP, Hollingsworth NM. Mek1 kinase is regulated to suppress double-strand break repair between sister chromatids during budding yeast meiosis. Mol Cell Biol. 2007;27:5456–67. doi: 10.1128/MCB.00416-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bishop DK, Park D, Xu L, Kleckner N. DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation and cell cycle progression. Cell. 1992;69:439–456. doi: 10.1016/0092-8674(92)90446-j. [DOI] [PubMed] [Google Scholar]
- 19.Padmore R, Cao L, Kleckner NR. Temporal comparison of recombination and synaptonemal complex formation during meiosis in Saccharomyces cerevisiae. Cell. 1991;66:1239–1256. doi: 10.1016/0092-8674(91)90046-2. [DOI] [PubMed] [Google Scholar]
- 20.Blethrow JD, Zhang C, Shokat KM, Weiss EL. Design and use of analog-sensitive kinases. Curr Protoc Mol Biol. 2004;Chapter 18 doi: 10.1002/0471142727.mb1811s66. Unt 18.11. [DOI] [PubMed] [Google Scholar]