
Figure 1.
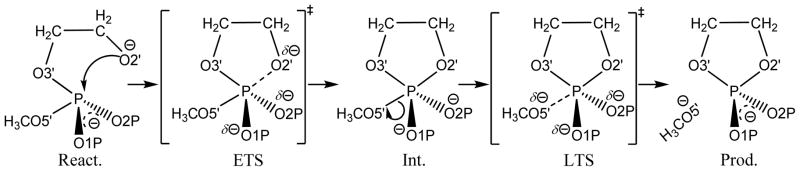
Adiabatic energy profiles for the native, S3′, and S5′ simplest models of RNA transphosphorylation in both (A) gas and (B) solution phases, as a function of the difference in bond length (Δbond) between the breaking P—X5′ bond (X = O for native and S3′; X = S for S5′) and the forming P—O2′ bond (Δbond = P—X5′ - P—O2′). The level of all the electronic structure calculations is at MP2/6-311+G(d,p). The profiles are mapped from the intrinsic reaction coordinate paths and have been shifted to match the respective rate-limiting free-energy barriers calculated in the decoupled rigid-rotor harmonic-oscillator approximation at 37°C. Other than this shift, however, there is no zero-point energy or thermal corrections included in the profiles.