
Figure 1.
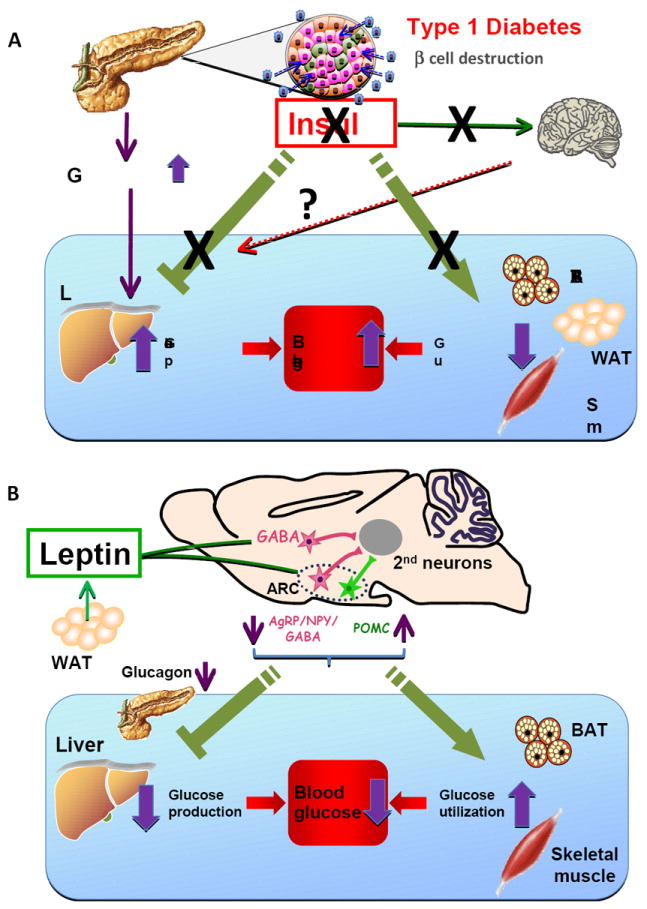
Metabolic effects of insulin deficiency and CNS leptin in Type 1 diabetes A. Alterations of glucose metabolism in T1DM. Autoimmune destruction of pancreatic ß cells markedly reduces insulin secretion, which increases glucagon secretion from pancreatic α cells. Hyperglucagonemia drives HGP resulting in hyperglycemia. Insulin deficiency reduces glucose utilization in peripheral tissues such as BAT, white adipose tissue (WAT) and skeletal muscle, further contributing to hyperglycemia. Insulin also regulates peripheral metabolism via the CNS, including inhibition of HGP and lipid metabolism in WAT. Whether impairment of these central effects of insulin contributes to the metabolic abnormalities in T1DM is unknown.
B. Anti-diabetic actions of leptin. Leptin is released mainly from white adipose tissue and acts through LEPR on GABAergic as well as POMC neurons, to induce anti-diabetic actions in the insulin-deficient state. Leptin inhibits the activity of AgRP/NPY/GABA neurons, while it activates that of POMC neurons in the ARC. The 2nd order neurons for leptin’s anti-diabetic actions may be located in the paraventricular hypothalamus (PVH), lateral hypothalamus (LH) VMH or brainstem. LEPR in GABAergic neurons in other hypothalamic regions such as the dorsomedial hypothalamus (DMH) may also mediate leptin’s actions on glycemia. Through its effects on GABAergic and POMC neurons, leptin suppresses HGP and increases glucose utilization in peripheral tissues such as BAT and skeletal muscle. Suppression of the hyperglucagonemia that is characteristic of insulin deficiency appears to be critical for the suppression of HGP by leptin, whereas the mechanism for the leptin-induced increase in glucose utilization is unknown.