
Figure 6.
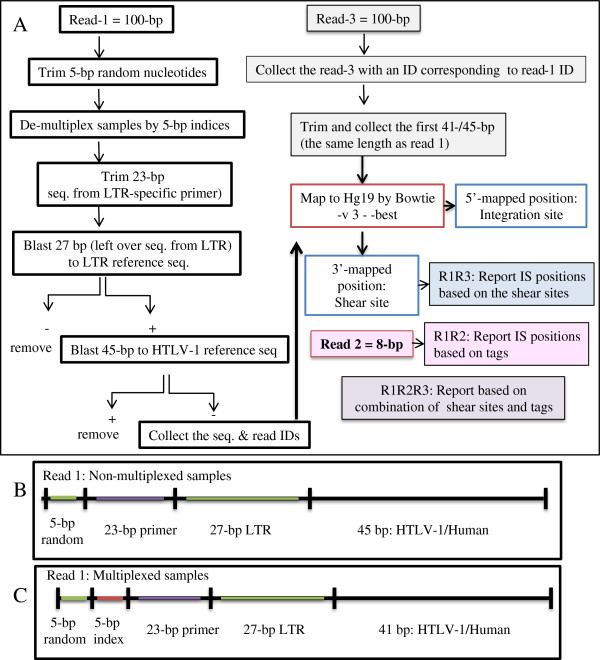
In-silico analysis work flow. (A) Illumina HiSeq 2000 platform outputs raw data of (Read-1 = 100 bp), (Read-3 = 100 bp), and (Read-2 = 8 bp). Data were analyzed according to this work flow after checking quality with the FastQC tool. In the case of Read-1, the first 5 bp were trimmed, and the next 5 bp were used to de-multiplex indexed samples. The downstream 23 bp, which correspond to the LTR primer (F2), were then removed. The next 27 bp were subjected to a blast search against the LTR reference sequence. For the blast search reads, the remaining 41/45 bp were subjected to a blast search against an HTLV-1 reference sequence. Reads were confirmed to be from HTLV-1 was removed, and the sequences and IDs from the remaining reads which considered as human, were collected. Subsequently, Read-3 with IDs corresponding to Read-1’s IDs were collected. The first 41/45 bp of Read-3 were trimmed and collected to have the same length as Read-1. The paired sequences of Read-1 and Read-3 (same lengths) were mapped against hg19 by Bowtie with -v 3 - -best parameters. The 5′-mapped positions were considered to be integration sites and the 3′-mapped positions as shear sites. Read-2 information was used to retrieve the clone size based on tags. Finally, the clone size was computed by combining tag and shear site information. All the analyses were done by our own Perl scripts, which resulted in the following reports. Report R1R3: the distribution of unique shear sites per integration site. Report R1R2: the distribution of unique tags per integration site. Report R1R2R3: the distribution of unique tags and shear sites per integration site. (B, C) The structure of Read-1 for the non-multiplexed and multiplexed samples.