

Abstract
Objective:
Metachromatic leukodystrophy (MLD) is a rare metabolic disorder leading to demyelination and rapid neurologic deterioration. As therapeutic options evolve, it seems essential to understand and quantify progression of the natural disease. The aim of this study was to assess cerebral volumetric changes in children with MLD in comparison to normal controls and in relation to disease course.
Method:
Eighteen patients with late-infantile MLD and 42 typically developing children in the same age range (20–59 months) were analyzed in a cross-sectional study. Patients underwent detailed genetic, biochemical, electrophysiologic, and clinical characterization. Cerebral gray matter (GM) and white matter (WM) volumes were assessed by multispectral segmentation of T1- and T2-weighted MRI. In addition, the demyelinated WM (demyelination load) was automatically quantified in T2-weighted images of the patients, and analyzed in relation to the clinical course.
Results:
WM volumes of patients did not differ from controls, although their growth curves were slightly different. GM volumes of patients, however, were on average 10.7% (confidence interval 6.0%–14.9%, p < 0.001) below those of normally developing children. The demyelination load (corrected for total WM volume) increased with disease duration (p < 0.003) and motor deterioration (p < 0.001).
Conclusion:
GM volume in patients with MLD is reduced when compared with healthy controls, already at young age. This supports the notion that, beside demyelination, neuronal dysfunction caused by neuronal storage plays an additional role in the disease process. The demyelination load may be a useful noninvasive imaging marker for disease progression and may serve as reference for therapeutic intervention.
Metachromatic leukodystrophy (MLD), a rare lysosomal storage disorder,1 is characterized in its most common late-infantile form by a disease onset during the first years of life and a rapid neurologic deterioration.2 Death occurs within the first decade as no causal therapy is yet available.2–4 New therapeutic approaches are currently emerging, including gene therapy, hematopoietic stem cell transplantation, and enzyme replacement therapy.1 However, due to its status as an “orphan disease” (1 per 100,000 live births3), systematic investigations on the natural disease course and standardized tools are scarce.
Leukodystrophy can sensitively be detected as signal hyperintensities on brain MRI.5 Recently, we applied a visual rating scale in a nationwide cohort to describe the natural course of brain changes in MLD.6,7 Advantages are the applicability to images acquired on different scanners using different acquisition modes and sequences, making it a robust tool for multicenter studies. However, this visual assessment is not sensitive enough to measure gradual white matter (WM) changes in order to evaluate disease progression or therapeutic effects in an individual patient. Moreover, gray matter (GM) changes are not captured by the scoring system.
The aim of this study was to quantify cerebral GM and WM in patients with MLD compared to typically developing children, and to quantify the volume of demyelinated WM in a group of clinically, biochemically, and genetically well-characterized children with late-infantile MLD. We related these imaging features to the disease course and deterioration of gross motor function, the clinical key feature.8
METHODS
Standard protocol approvals, registrations, and patient consents.
Patient data for this cross-sectional study were collected as part of a natural history study of the German leukodystrophy network LEUKONET,2 (n = 8) and from a therapeutic phase I/II trial investigating enzyme replacement9 in Copenhagen, Denmark (n = 13); 3 patients belonged to both groups. The trial registration number from the Danish study is EudraCT 2006–005341–11, and the ClinicalTrials.gov identifier is NCT00418561. For the purpose of this study, only baseline data were used from the latter trial, i.e., before the initiation of treatment. The study was approved by the ethical committees of the University of Tübingen, Germany, and Copenhagen, Denmark, respectively. Written informed consent was given by the parents.
Patients.
Demographic, clinical, biochemical, and genetic details are listed in the table. Patients showed very low arylsulfatase A (ASA) activity in leukocytes and abnormally high sulfatide excretion in urine. In addition, detailed sequencing analyses of the ARSA gene (NM_000487) were performed in all patients (table).
Table.
Demographic data of patients with MLDa
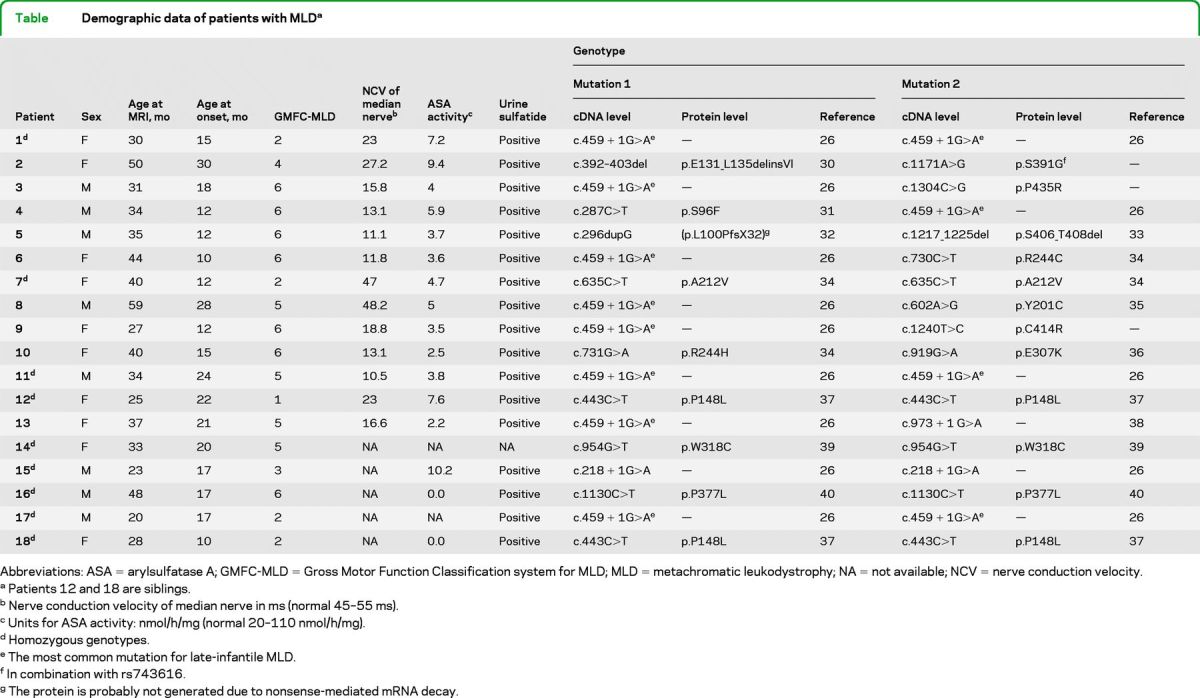
Abbreviations: ASA = arylsulfatase A; GMFC-MLD = Gross Motor Function Classification system for MLD; MLD = metachromatic leukodystrophy; NA = not available; NCV = nerve conduction velocity.
Patients 12 and 18 are siblings.
Nerve conduction velocity of median nerve in ms (normal 45–55 ms).
Units for ASA activity: nmol/h/mg (normal 20–110 nmol/h/mg).
Homozygous genotypes.
The most common mutation for late-infantile MLD.
In combination with rs743616.
The protein is probably not generated due to nonsense-mediated mRNA decay.
MLD was defined as deficiency of ASA together with an increase of urinary sulfatide level or pathogenic mutations in the MLD gene. Late-infantile MLD was defined as onset of first symptoms at age 30 months and younger.2 In our cohort, age at onset was between 10 and 30 months (mean 16.95 ± 5.986 months). Gross motor function was assessed using the Gross Motor Function Classification system for MLD (GMFC-MLD), which consists of 7 levels characterizing gross motor function, 0 meaning normal for age and 6 characterizing loss of all gross motor function.8 Patients' age at MRI was between 20 and 59 months (mean 35.9 ± 9.92 months). At the time of MRI, all patients were symptomatic. Nerve conduction velocity (NCV) was determined in 13 patients at the time of the MRI examination by stimulating motor fibers of the right median nerve, revealing clearly abnormal values (indicative of a peripheral neuropathy) in all but 2 patients (patients 7 and 8).
Controls.
Control MRI data of typically developing children used in this study were obtained from the Pediatric MRI Data Repository created by the NIH MRI Study of Normal Brain Development.10,11 Anatomic MRI from 42 children were selected who had the same age range and gender distribution as the patients (mean 38.95 ± 11.16 months, minimum 20 months, maximum 59 months, 23 female).
MRI data.
MRI sequences of patients were acquired on 1.5 T Siemens scanners and consisted of conventional clinical routine images with a high-resolution T1-weighted (magnetization-prepared rapid gradient echo sequence with echo time/repetition time = 11.4/4.4 msec and voxel size typically 1 × 1 × 1 mm, in 5 patients the voxel size was higher and varied between 0.39–0.86 × 0.39–0.86 × 1–1.4 mm) and a T2-weighted axial sequence (typically a spin echo sequence with echo time/repetition time = 99/5,940 msec, voxel size 0.78 × 0.78 × 4 mm, in 5 patients the in-plane resolution was higher with 0.39–0.78 × 0.39–0.78 mm and the slice thickness varied between 3.3 and 7.2 mm).
MRI of controls were acquired using 1.5 T General Electric or Siemens scanners. Sequences consisted of T1-weighted images (spatial resolution of 1.3 × 1 × 1 mm, 1 × 1 × 1 mm, or 1 × 1 × 3 mm), T2-weighted images (1 × 1 × 2 or 1 × 1 × 3 mm), and proton density–weighted images (1 × 1 × 2 or 1 × 1 × 3 mm). All images used in this study passed the quality control test for image artifacts (for details of sequences see Brain Development Cooperative Group10).
MRI data analysis.
Images were first denoised using a spatially adaptive nonlocal means filter as implemented in the voxel-based morphometry 8 toolbox (http://dbm.neuro.uni-jena.de/vbm). T2 images (and proton density–weighted images for tissue prior creation) were then spatially coregistered and resliced to the accompanying high-resolution T1 image using the routines implemented in the statistical parametric mapping (SPM8) software package (Wellcome Trust Centre for Neuroimaging, London, UK; www.fil.ion.ucl.ac.uk/spm), using a seventh degree b-splines interpolation algorithm.
In order to create representative (age-appropriate) tissue priors for the segmentation of this data, 156 anatomic MRI from 108 healthy children between 1 and 6 years of age were selected from the above mentioned NIH database (mean age 4.1 years, SD 1.62, 1.7–6.6 years, 76 female, 80 male). To this effect, T1-, T2-, and proton density–weighted images were segmented using a multispectral segmentation algorithm implemented in SPM8. The segmented images (in the individual native space) were affinely registered to template (Montreal Neurological Institute) space. From the registered images a common (new) set of 6 custom image priors was created using the template-O-matic toolbox as implemented in SPM8.12 As this approach estimates regression parameters, older and younger children were included in order to stabilize these calculations.
Preprocessed T1- and T2-weighted images of patients and controls were segmented using the multispectral image segmentation algorithm, now using the customized priors (figure 1). This segmentation algorithm is implemented as “New Segmentation” in SPM8 and is an extension of the “Unified Segmentation” model.13 This segmentation uses both prior tissue information and tissue intensities in a modified mixture model. The intensity distribution of the different tissue types is modeled by a mixture of Gaussians to account for partial volume effects within one tissue type. It thus allows 2 different tissue classes within the WM, which is important for the classification of both demyelinated and normal-appearing WM into one tissue type, i.e., the total WM. The volume of the resulting GM and WM tissue maps was calculated by summing over all voxels' probability values, taking into account voxel sizes. GM and WM volumes of patients and controls were statistically compared using Student t tests. As this comprised a cross-sectional analysis, only one MRI per subject was analyzed (most recent scan).
Figure 1. Summary of the methodologic approach for segmentation.

In a first step, multispectral image segmentation results in gray matter (GM) and white matter (WM) image volumes. In a second step, the WM segment is applied to the T2-weighted image and an automated intensity threshold within the WM of the T2-weighted image is calculated in order to separate normal-appearing and demyelinated WM (demyelination load).
In additional analyses, total GM was subdivided into subcortical (basal ganglia and thalamus), infratentorial, and the remaining (supratentorial cortical) GM volume. As there is no standard anatomic atlas available for this age range, cerebellar and subcortical GM was defined manually on the customized GM prior map from this cohort (see above). These masks were smoothed with a 3-mm isotropic kernel and binarized using a relative intensity threshold of 0.3 in order to account for the interindividual variability. These anatomic masks were then applied to the individual modulated GM image segments in stereotactic space14 in order to calculate the GM volumes in these regions; these were compared between patients and controls as described above.
Demyelination load.
As the T2-weighted sequence shows improved contrast of WM hyperintensities (compared to T1), only the T2-weighted image was used for determining demyelinated WM, termed demyelination load. Demyelination load was defined as the volume of hyperintense areas within cerebral WM on T2-weighted images (including cerebellar WM).
Within the WM segment of the T2-weighted image (obtained by multiplying the total WM segment with the T2-weighted image) it was assumed that 2 tissue classes were present in the WM, demyelinated and normal-appearing WM. Therefore, a mixture of 2 Gaussians was applied to the image histogram. In order to improve the fit of the Gaussians and to sensitize the model to higher image intensities, the histogram was smoothed and scaled between 0 and 1,000 using a quadratic function. The threshold distinguishing normal from hyperintense white matter was defined as the intercept of the 2 Gaussians. As demyelination was expected to be in larger clusters rather than single voxels, the resulting thresholded image was homogenized using a Markov Random Field approach to integrate neighborhood intensities.
To account for total WM volume of a patient (which changes substantially as a function of normal brain development15), demyelination load is provided as a fraction of the individual total WM volume. It was correlated with disease duration in order to assess disease progression, and nerve conduction velocity in order to relate central to peripheral demyelination (Pearson correlation coefficient), and with gross motor function as described by the GMFC-MLD (Spearman rank correlation coefficient).
Validation was done by manually segmenting the demyelination load, considered the gold standard in MRI volumetry, by 2 experienced raters (S.G. and P.C.), using ITK-Snap.16 Agreement was tested by using the dice coefficient for spatial overlap17 and the intraclass correlation coefficient (ICC) for volume measurements.18
RESULTS
GM and WM volume.
Regression analysis of the cross-sectional data showed an increase of GM and WM volumes over the observed age range for the healthy controls (figure 2). Patients had lower GM volumes (mean 0.601 ± 0.041 L) than controls (mean 0.673 ± 0.057 L, p < 0.001). This amounts to an average decrease in GM volume of 10.7% (95% confidence interval [CI] 6.0%–14.9%) in patients with MLD compared to controls. WM volume, comprising normal and affected WM, was not different between patients and controls (0.419 ± 0.034 L vs 0.416 ± 0.041 L, p = 0.746).
Figure 2. Gray matter (GM) and white matter (WM) volumes.
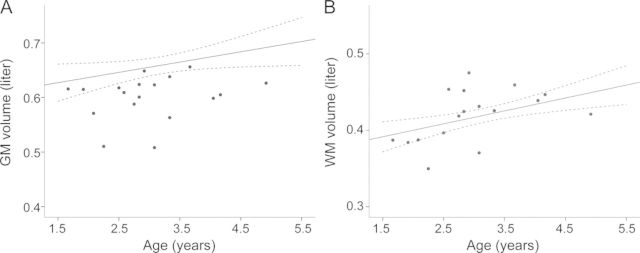
GM (A) and WM (B) volumes of controls (mean of linear regression line and its 95% confidence interval) and patients (single dots) showing less GM volume in patients, whereas WM volume was not different between patients and controls.
When analyzing regional volumes, we found that subcortical GM volume did not differ between patients and controls (mean 0.0152 ± 0.0017 L vs 0.0155 ± 0.0017 L, p = 0.537). However, both the cerebellar (0.0759 ± 0.0105 L vs 0.1016 ± 0.0095 L) as well as the supratentorial (0.5384 ± 0.0420 L vs 0.6061 ± 0.0522 L) GM volume differed between patients and controls (p < 0.001). Results are also shown in figure e-1 on the Neurology® Web site at www.neurology.org.
Demyelination load.
Demyelination load showed a rapid increase during the first months after disease onset (figure 3). There was a clear correlation with disease duration (correlation coefficient 0.65, p = 0.003). Uncorrected demyelination load (not as fraction per WM volume) also increased with disease duration (correlation coefficient 0.62, p = 0.04, data not shown). Increasing demyelination load was associated with gross motor deterioration as indicated by the GMFC-MLD (ρ = 0.66, p < 0.006) (figure 3). Demyelination load, however, did not correlate with NCV (correlation coefficient −0.165, p = 0.589). It is noteworthy that 2 patients did have normal NCV at the time of MRI examination (see table), when there was already clear cerebral demyelination and severely reduced gross motor function (GMFC-MLD level 2 and 5).
Figure 3. Demyelination load.
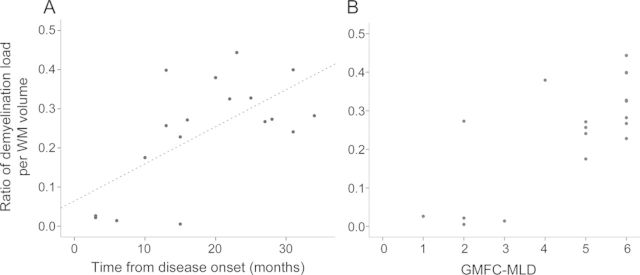
Demyelination load as a fraction of the total white matter (WM) volume in relation to time from disease onset (A) and to motor deterioration assessed with the Gross Motor Function Classification system for metachromatic leukodystrophy (GMFC-MLD) (B).
Validating the automatically determined demyelination load with the manually determined values revealed good agreement as given by an ICC of 0.987 (95% CI 0.967–0.995). The spatial overlap of the demyelination load measured by the 2 approaches showed a median dice coefficient of 0.78 and a mean of 0.67 (±0.266, minimum 0.09, maximum 0.88). The unequal distribution of the data was explained by outliers within the extremely low range of the demyelination load. When only comparing subjects with a demyelination volume >15 mL, the mean dice coefficient was 0.80 (±0.042, minimum 0.74, maximum 0.88).
The inter-rater test of the manual measurements revealed similarly good agreement between the 2 raters. The agreement of the volume values was demonstrated as an ICC of 0.997 (95% CI 0.991–0.999), the dice coefficient of spatial overlap was 0.81 (±0.101, minimum 0.59, maximum 0.89).
DISCUSSION
As therapeutic options for MLD are currently being investigated, it seems essential to investigate the normal disease course and implement tools to describe it. This study successfully quantified volumes of GM, WM, and demyelinated WM (demyelination load) in patients with late-infantile MLD. The demyelination load increased with disease duration and deterioration of gross motor function. Thus, the demyelination load seems to be a clinically useful and pathologically relevant parameter to quantify MRI changes in MLD, qualifying it as a potential noninvasive surrogate parameter for monitoring therapeutic intervention.
It is interesting to note that the WM volume of patients with MLD was not different from the control group. This is in line with histopathology where the prominent aspect of demyelination and sulfatide accumulation in the WM leads to a “firmer”19 or “hard in consistency” aspect,20 probably due to astrocytosis and gliosis. This might be the reason for its preserved volumes in the earlier stages of the disease, followed by atrophy of the WM only very late in the disease course.
In contrast, this study provides compelling evidence that GM development is altered in children with late-infantile MLD. Compared with typically developing children of the same age, children with MLD had less global GM volume. When investigating this further, the effect was present in cortical and cerebellar, but not in deep GM structures (although it must be borne in mind that the thalamus, for example, also contains white matter, potentially weakening the effect in this region). This new observation is surprising as MLD is classically considered a WM disorder. There may be several explanations for this GM involvement. Considering the close neuron-oligodendrocyte interaction during development,21 disruption of myelin might also disturb neuronal growth in the developing brain. In fact, it has been demonstrated that formation of a normal myelin sheath is required for normal neuronal maturation and growth.22,23 This explanation seems to not be sufficient, however, as reduced GM volumes could already be seen early during the disease, when demyelination, as judged from MRI, was not yet as far progressed. Therefore, it is interesting to note that sulfatide storage in MLD is not restricted to glial cells but neurons are also affected.24 Neuronal accumulation of metachromatic material is well-described already in early cases of MLD.19,20,25 It was speculated that at least initial symptoms in humans could be a consequence of neuronal dysfunction caused by neuronal storage, while demyelination may dominate the phenotype only later in the disease.1 A recent magnetic resonance spectroscopy study further underlines the neuronal involvement by showing decreased N-acetylaspartate levels,9 generally interpreted as a marker of neuroaxonal integrity. Our findings suggest that the role of GM damage in MLD should be investigated more closely.
MLD of late-infantile onset has been shown to follow a very uniform and relentlessly downhill course.2 It has been suggested that patients with this early-onset form must be expected to show no residual ASA activity. The mutation most commonly reported in late-infantile MLD, 459 + 1 G>A,26 is predicted to be a null allele. It was also the most common mutation in our series, found homozygous in 17% of patients and as compound heterozygous with another mutation in 33%. Our patients showed several different genotypes; all of them resulted in almost no residual ASA activity, consistent with this prediction and likely the reason for the uniform clinical course that characterizes this subtype. Our data suggest that while different genotypes cause the late-infantile form of MLD, they all lead to very similar phenotypes. Moreover, there was no correlation between nerve conduction velocity and demyelination load, indicating that central and peripheral nervous system affection do not necessarily develop in parallel. Indeed, the positive correlation between gross motor deterioration and demyelination load indicates that it is the central, rather than the peripheral, demyelination which causes gross motor deterioration in late-infantile MLD.
In this study we applied and adapted a widely established segmentation-normalization framework,13 recently enhanced to use T1- and T2-weighted images, to the specific pathology of patients with MLD. High-resolution T1-weighted images have a better image-contrast between GM and WM and a higher spatial resolution compared to routine T2-weighted sequences, which makes fully automated segmentation approaches feasible.27 Conversely, T2-weighted images provide a better image contrast between normal and abnormal WM and therefore aid in tissue classification in patients with MLD. Combining both advantages and using routine clinical sequences (high-resolution T1, low-resolution T2) makes this method feasible for multicenter studies while not requiring longer acquisition times, which is especially helpful in children. Patients with the late-infantile form of MLD were expected to present with different brain morphology due to their young age and possible atrophy. Therefore, appropriate prior probability images for this study were created from a normal cohort of children with the same age range. This correctly identifies GM and WM, which, using an additional step of modeling 2 Gaussians, can then be subdivided into unaffected and affected WM. Validation yielded good agreement of this method with manual segmentation. However, although volume agreement was very high, the spatial overlap of small volumes was lower. This suggests that the automated method, although with sufficient sensitivity, might not be of high specificity in demyelination volumes below 15 mL. One of the reasons might be that early signs of demyelination are only very “faint”6 in signal intensity, without a clear border to the normal-appearing WM. Overlap measures around 0.8 are in general referred to as “good agreement” in MRI volumetry.28 Still, agreement between 2 manual raters was higher than between manual and automated segmentation. While a human expert inherently uses additional information about texture and location of the supposedly abnormal tissue within the WM, the automated thresholding algorithm is guided only by the information about the signal intensity. Therefore partial volume effects of GM or CSF might influence the variability when measuring the demyelination load. While a semiautomated approach might introduce expert knowledge into automated measurements,29 a fully automated approach is preferable for studies with larger datasets and in therapeutic trials, where maximum objectivity is required.
It should be noted that the present study is cross-sectional. Changes over time in a given individual can only be found in a longitudinal study; however, a group study such as this one, including patients at different timepoints after disease onset, allows us to assess typical patterns evolving as a function of time. Naturally, the here-reported volume increases or decreases should be replicated in such a longitudinal study, but it must be borne in mind that, in a rapidly progressing and ultimately fatal disorder, repeated investigations are difficult to justify.
The results provide evidence for a new aspect of the disease, not previously reported, namely lower cortical GM volume in patients compared to controls, already at young age. With a newly introduced morphometric measure, the demyelination load, we could confirm that WM changes steadily increase with disease duration and with deterioration of gross motor function. These data may contribute to a better understanding of disease pathomechanisms and may serve as reference for family counseling and especially for evaluating the effect of therapeutic interventions.
Supplementary Material
ACKNOWLEDGMENT
The authors thank V. Gieselmann, MD, PhD, coordinator of the German LEUKONET, for support; Dr. Norman Barton (Shire plc) for feedback and support of this study; Dr. Eduard Paschke and Dr. Paul Karl (Laboratory of Metabolic Diseases, Department of Pediatrics, Medical University of Graz, Austria) and Dr. Jürgen Kohlhase (Center for Human Genetics, Freiburg, Germany) for contributing genetic information; Dr. Birgit Kustermann-Kuhn (Neurometabolic Laboratory, University Children's Hospital Tübingen, Germany) for contributing biochemical details of the patients; and the Brain Development Cooperative Group for their work.
GLOSSARY
- ASA
arylsulfatase A
- CI
confidence interval
- GM
gray matter
- GMFC-MLD
Gross Motor Function Classification system for metachromatic leukodystrophy
- ICC
intraclass correlation coefficient
- MLD
metachromatic leukodystrophy
- NCV
nerve conduction velocity
- SPM
statistical parametric mapping
- WM
white matter
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Samuel Groeschel drafted the manuscript and undertook MRI analysis and interpretation. Christine í Dali contributed patient data and was involved in revision of the manuscript and interpretation of the data. Philipp Clas was involved in establishing MRI analysis procedure, in validating the data, and in revision of the manuscript. Judith Böhringer performed part of the genetic analysis and was involved in manuscript revision and interpretation of the data. Morten Dunoe performed part of the genetic analysis and was involved in manuscript revision and interpretation of the data. Christian Krarup performed the neurophysiologic analysis and was involved in manuscript revision and interpretation of the data. Christiane Kehrer contributed patient data and was involved in manuscript revision and interpretation of the data. Marko Wilke was involved in study concept, establishing methodology, manuscript revision, and interpretation of the data. Ingeborg Krägeloh-Mann was involved in study concept, manuscript revision, and interpretation of the data.
Study Funding
This study was funded by a grant of the German Federal Ministry of Education and Research (LEUKONET: A network for coordinated clinical and basic research on leukodystrophies) and an institutional research grant from Shire plc. Marko Wilke was partly funded by the German Research Council (DFG WI3630). The control datasets used in the preparation of this manuscript were obtained from the Pediatric MRI Data Repository created by the NIH MRI Study of Normal Brain Development. This multisite, longitudinal study of typically developing children was conducted by the Brain Development Cooperative Group supported by the National Institute for Child Health and Human Development, National Institute on Drug Abuse, National Institute of Mental Health, and National Institutes of Neurological Disorders and Stroke, contract nos. N01-HD023343, N01-MH90002, N01-NS92314, -2315, - 2316, -2317, -2319, -2320. This article reflects the views of the authors and may not reflect the opinions or views of the Brain Development Cooperative Group Investigators or the NIH.
DISCLOSURE
S. Groeschel received institutional research support from the German Federal Ministry of Education and Research and from Shire plc. C. í Dali serves on a scientific advisory board for Shire plc and has received institutional research support from Shire plc and Zymenex A/S. P. Clas was funded by the IZKF-Promotionskolleg PK2011–6, Medical Faculty, University of Tübingen. J. Böhringer received institutional research support from the German Federal Ministry of Education and Research. M. Dunoe and C. Krarup report no disclosures. C. Kehrer received institutional research support from the German Federal Ministry of Education and Research. M. Wilke was partly funded by the German Research Council (DFG WI3630). I. Krägeloh-Mann was a member of the scientific board of the Scandinavian biopharmaceutical company ZYMENEX for a worldwide clinical trial I study for enzyme replacement with the ZYMENEX's product “Metazym” in 2007. This did not imply any involvement or funding in the present study. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Gieselmann V.Metachromatic leukodystrophy: genetics, pathogenesis and therapeutic options.Acta Paediatr Suppl 2008;97:15–21 [DOI] [PubMed] [Google Scholar]
- 2.Kehrer C, Blumenstock G, Gieselmann V, Krageloh-Mann I.The natural course of gross motor deterioration in Metachromatic Leukodystrophy.Dev Med Child Neurol 2011;53:850–855 [DOI] [PubMed] [Google Scholar]
- 3.Gieselmann V, Krageloh-Mann I.Metachromatic leukodystrophy: an update.Neuropediatrics 2010;41:1–6 [DOI] [PubMed] [Google Scholar]
- 4.Kohler W.Leukodystrophies with late disease onset: an update.Curr Opin Neurol 2010;23:234–241 [DOI] [PubMed] [Google Scholar]
- 5.van der Voorn JP, Pouwels PJ, Kamphorst W, et al. Histopathologic correlates of radial stripes on MR images in lysosomal storage disorders.AJNR Am J Neuroradiol 2005;26:442–446 [PMC free article] [PubMed] [Google Scholar]
- 6.Eichler F, Grodd W, Grant E, et al. Metachromatic leukodystrophy: a scoring system for brain MR observations.AJNR Am J Neuroradiol 2009;30:1893–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groeschel S, Kehrer C, Engel C, et al. Metachromatic Leukodystrophy: natural course of cerebral MRI changes in relation to clinical course.J Inherit Metab Dis 2011;34:1095–1102 [DOI] [PubMed] [Google Scholar]
- 8.Kehrer C, Blumenstock G, Raabe C, Krageloh-Mann I.Development and reliability of a classification system for gross motor function in children with metachromatic leucodystrophy.Dev Med Child Neurol 2011;53:156–160 [DOI] [PubMed] [Google Scholar]
- 9.i Dali C, Hanson LG, Barton NW, Fogh J, Nair N, Lund AM.Brain N-acetylaspartate levels correlate with motor function in metachromatic leukodystrophy.Neurology 2010;75:1896–1903 [DOI] [PubMed] [Google Scholar]
- 10.Evans AC.The NIH MRI study of normal brain development.Neuroimage 2006;30:184–202 [DOI] [PubMed] [Google Scholar]
- 11.Almli CR, Rivkin MJ, McKinstry RC.The NIH MRI study of normal brain development (Objective-2): newborns, infants, toddlers, and preschoolers.Neuroimage 2007;35:308–325 [DOI] [PubMed] [Google Scholar]
- 12.Wilke M, Holland SK, Altaye M, Gaser C.Template-O-Matic: a toolbox for creating customized pediatric templates.Neuroimage 2008;41:903–913 [DOI] [PubMed] [Google Scholar]
- 13.Ashburner J, Friston KJ.Unified segmentation.Neuroimage 2005;26:839–851 [DOI] [PubMed] [Google Scholar]
- 14.Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS.A voxel-based morphometric study of ageing in 465 normal adult human brains.Neuroimage 2001;14:21–36 [DOI] [PubMed] [Google Scholar]
- 15.Groeschel S, Vollmer B, King MD, Connelly A.Developmental changes in cerebral grey and white matter volume from infancy to adulthood.Int J Dev Neurosci 2010;28:481–489 [DOI] [PubMed] [Google Scholar]
- 16.Yushkevich PA, Piven J, Hazlett HC, et al. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability.Neuroimage 2006;31:1116–1128 [DOI] [PubMed] [Google Scholar]
- 17.Dice LR.Measures of the amount of ecologic association between species.Ecology 1945;26:297–302 [Google Scholar]
- 18.Shrout PE, Fleiss JL.Intraclass correlations: uses in assessing rater reliability.Psychol Bull 1979;86:420–428 [DOI] [PubMed] [Google Scholar]
- 19.Black JW, Cumings JN.Infantile metachromatic leukodystrophy.J Neurol Neurosurg Psychiatry 1961;24:233–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jervis GA.Infantile metachromatic leukodystrophy. (Greenfield's disease).J Neuropathol Exp Neurol 1960;19:323–341 [DOI] [PubMed] [Google Scholar]
- 21.Piaton G, Gould RM, Lubetzki C.Axon-oligodendrocyte interactions during developmental myelination, demyelination and repair.J Neurochem 2010;114:1243–1260 [DOI] [PubMed] [Google Scholar]
- 22.Colello RJ, Pott U, Schwab ME.The role of oligodendrocytes and myelin on axon maturation in the developing rat retinofugal pathway.J Neurosci 1994;14:2594–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brady ST, Witt AS, Kirkpatrick LL, et al. Formation of compact myelin is required for maturation of the axonal cytoskeleton.J Neurosci 1999;19:7278–7288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng L, Suzuki K.Ultrastructural study of neurons in metachromatic leukodystrophy.Clin Neuropathol 1987;6:224–230 [PubMed] [Google Scholar]
- 25.Russel Brain W, Greenfield JG.Late infantile metachromatic leuko-encephalopathy, with primary degeneration of the interfascicular oligodentroglia.Brain 1950;73:291–317 [DOI] [PubMed] [Google Scholar]
- 26.Polten A, Fluharty AL, Fluharty CB, Kappler J, von Figura K, Gieselmann V.Molecular basis of different forms of metachromatic leukodystrophy.N Engl J Med 1991;324:18–22 [DOI] [PubMed] [Google Scholar]
- 27.Ashburner J, Csernansky JG, Davatzikos C, Fox NC, Frisoni GB, Thompson PM.Computer-assisted imaging to assess brain structure in healthy and diseased brains.Lancet Neurol 2003;2:79–88 [DOI] [PubMed] [Google Scholar]
- 28.Babalola KO, Patenaude B, Aljabar P, et al. An evaluation of four automatic methods of segmenting the subcortical structures in the brain.Neuroimage 2009;47:1435–1447 [DOI] [PubMed] [Google Scholar]
- 29.Clas P, Groeschel S, Wilke M.A semi-automatic algorithm for determining the demyelination load in metachromatic leukodystrophy.Acad Radiol 2012;19:26–34 [DOI] [PubMed] [Google Scholar]
- 30.Luyten JA, Wenink PW, Steenbergen-Spanjers GC, et al. Metachromatic leukodystrophy: a 12-bp deletion in exon 2 of the arylsulfatase A gene in a late infantile variant.Hum Genet 1995;96:357–360 [DOI] [PubMed] [Google Scholar]
- 31.Gieselmann V, Fluharty AL, Tonnesen T, von Figura K.Mutations in the arylsulfatase A pseudodeficiency allele causing metachromatic leukodystrophy.Am J Hum Genet 1991;49:407–413 [PMC free article] [PubMed] [Google Scholar]
- 32.Lugowska A, Wlodarski P, Ploski R, et al. Molecular and clinical consequences of novel mutations in the arylsulfatase A gene.Clin Genet 2009;75:57–64 [DOI] [PubMed] [Google Scholar]
- 33.Regis S, Filocamo M, Stroppiano M, Corsolini F, Caroli F, Gatti R.A 9-bp deletion (2320del9) on the background of the arylsulfatase A pseudodeficiency allele in a metachromatic leukodystrophy patient and in a patient with nonprogressive neurological symptoms.Hum Genet 1998;102:50–53 [DOI] [PubMed] [Google Scholar]
- 34.Draghia R, Letourneur F, Drugan C, et al. Metachromatic leukodystrophy: identification of the first deletion in exon 1 and of nine novel point mutations in the arylsulfatase A gene.Hum Mutat 1997;9:234–242 [DOI] [PubMed] [Google Scholar]
- 35.Hermann S, Schestag F, Polten A, et al. Characterization of four arylsulfatase A missense mutations G86D, Y201C, D255H, and E312D causing metachromatic leukodystrophy.Am J Med Genet 2000;91:68–73 [DOI] [PubMed] [Google Scholar]
- 36.Cesani M, Capotondo A, Plati T, et al. Characterization of new arylsulfatase A gene mutations reinforces genotype-phenotype correlation in metachromatic leukodystrophy.Hum Mutat 2009;30:E936–E945 [DOI] [PubMed] [Google Scholar]
- 37.Qu Y, Shapira E, Desnick RJ.Metachromatic leukodystrophy: subtype genotype/phenotype correlations and identification of novel missense mutations (P148L and P191T) causing the juvenile-onset disease.Mol Genet Metab 1999;67:206–212 [DOI] [PubMed] [Google Scholar]
- 38.Eng B, Nakamura LN, O'Reilly N, et al. Identification of nine novel arylsulfatase a (ARSA) gene mutations in patients with metachromatic leukodystrophy (MLD).Hum Mutat 2003;22:418–419 [DOI] [PubMed] [Google Scholar]
- 39.Onder E, Sinici I, Mujgan SF, Topcu M, Ozkara HA.Identification of two novel arylsulfatase A mutations with a polymorphism as a cause of metachromatic leukodystrophy.Neurol Res 2009;31:60–66 [DOI] [PubMed] [Google Scholar]
- 40.Zlotogora J, Bach G, Bosenberg C, Barak Y, von Figura K, Gieselmann V.Molecular basis of late infantile metachromatic leukodystrophy in the Habbanite Jews.Hum Mutat 1995;5:137–143 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.