

Abstract
High throughput methods such as next generation sequencing are increasingly used in molecular diagnosis. The aim of this study was to develop a workflow for the detection of BRCA1 and BRCA2 mutations using massive parallel sequencing in a 454 GS Junior bench top sequencer. Our approach was first validated in a panel of 23 patients containing 62 unique variants that had been previously Sanger sequenced. Subsequently, 101 patients with familial breast and ovarian cancer were studied. BRCA1 and BRCA2 exon enrichment has been performed by PCR amplification using the BRCA MASTR kit (Multiplicom). Bioinformatic analysis of reads is performed with the AVA software v2.7 (Roche). In total, all 62 variants were detected resulting in a sensitivity of 100%. 71 false positives were called resulting in a specificity of 97.35%. All of them correspond to deletions located in homopolymeric stretches. The analysis of the homopolymers stretches of 6 bp or longer using the BRCA HP kit (Multiplicom) increased the specificity of the detection of BRCA1 and BRCA2 mutations to 99.99%. We show here that massive parallel pyrosequencing can be used as a diagnostic strategy to test for BRCA1 and BRCA2 mutations meeting very stringent sensitivity and specificity parameters replacing traditional Sanger sequencing with a lower cost.
1. Introduction
Germline mutations that inactivate BRCA1 and BRCA2 are responsible for breast and ovarian cancer susceptibility [1, 2]. The prevalence of BRCA1 and BRCA2 mutations where family history shows more than one occurrence of breast cancer under the age of 50 ranges from 8 to 21.2%. Mutation carriers are at an increased cumulative risk to the age of 70 of 36–70% and 10–65% for breast cancer and ovarian cancer, respectively [3, 4]. Moreover, BRCA1 and BRCA2 mutation carriers are also at increased risk of pancreatic, prostate, and endometrial cancer. Molecular diagnosis is an important factor in clinical decisions that include increased surveillance, chemoprevention, or prophylactic surgery [5, 6]. Predictive testing in family members allows the identification of other individuals at risk.
BRCA1 and BRCA2 mutation screening is offered to patients from high risk families. Direct Sanger sequencing allows the identification of the sequence alteration and is considered the gold standard. Sequencing of BRCA1 and BRCA2 genes is time consuming and costly due to the large size of the genes and the equal distribution of mutations along the whole BRCA1 and BRCA2 sequence (5589 and 10254 nucleotides, resp.). A high level of allelic heterogeneity has been described including single nucleotide variants (SNVs), short insertions and deletions (InDels), and large structural variants (see Breast Cancer Information Core database: http://www.research.nhgri.nih.gov/bic/). Currently, many laboratories include a scanning method that allows the detection of all different types of mutations with a sensitivity and specificity of 100% [7].
High throughput methods such as next generation sequencing are increasingly used in molecular diagnosis [8]. Massive parallel sequencing allows the generation of millions of DNA sequences in a single run with low cost per base [9]. The development of technologies to capture and enrich specific regions of the genome improves performance and reduces the cost allowing joint sample analysis of numerous individuals [10]. Several studies have demonstrated the potential of massive sequencing both in the field of research and in genetic diagnosis [11, 12]. Recently, next generation sequencing methods for the mutation analysis of the BRCA1 and BRCA2 genes in patients with breast and ovarian cancer have been described using both high capacity and bench top platforms [13–18]. Bench top sequencers are addressed to individual labs to suit the demand of midsize diagnostic laboratories.
Here, we developed a workflow using massive parallel pyrosequencing in a bench top 454 GS Junior sequencer together with homopolymer scanning to screen for mutations in the BRCA1 and BRCA2 genes. Our workflow was first validated in a panel of 23 patients previously Sanger sequenced. Subsequently, 101 patients with familial breast and ovarian cancer were studied. We found 18 pathogenic mutations and 10 variants with unknown clinical significant effect (VUS). We show here that our workflow performs as Sanger sequencing in terms of sensitivity and specificity with the advantage of taking less time and cost consuming being suitable for genetic diagnosis.
2. Methodology
2.1. Patients
A total of 23 samples containing 62 unique variants were used to evaluate the methodology. 49 variants corresponded to single nucleotide variants (SNV) while 13 corresponded to deletions (8), insertions (3), and combined insertions and deletions (2). Among the 62 variants tested 14 were pathogenic mutations (11 insertions/deletions, 1 missense mutation, 1 nonsense mutation, and 1 splice site mutation). DNA samples were obtained from the Hereditary Cancer Program at the Catalan Institute of Oncology (ICO-IDIBELL) and the Genetic Counselling Unit at the Hospital of Sabadell (Barcelona, Spain).
Then, 101 patients with breast and ovarian cancer were screened for mutations using our validated workflow. DNA samples were collected from patients referred to the Genetic Counselling Unit at the Hospital of Sabadell (Barcelona, Spain). Informed consent was obtained from all the patients included in our study. Genomic DNA was extracted from peripheral blood following standard procedures and using Gentra Puregene DNA reagents (Qiagen, Valencia, CA, USA).
2.2. Multiplex PCR Target Amplification, NGS Library Preparation, and Sequencing
BRCA1 and BRCA2 coding regions and exon intron boundaries were amplified using the BRCA MASTR kit (Multiplicom, Niel, Belgium). Samples used to evaluate the methodology performance were amplified using the BRCA MASTR kit v1.2 (7 samples) and v2.0 (16 samples) following manufacturer instructions. The samples screened for BRCA1 and BRCA2 mutations were amplified using the BRCA MASTR kit v2.1. The BRCA MASTR kit v1.2 amplifies BRCA1 and BRCA2 coding regions and exon intron boundaries in 169 amplicons while versions 2.0 and 2.1 amplify both genes in 94 and 93 amplicons, respectively. Briefly, 50 ng of genomic DNA was used in a two-step multiplex reaction to firstly amplify BRCA1 and BRCA2 coding regions followed by the incorporation of molecular barcodes (multiple identifiers, MIDs) and 454 adapters to each amplicon. A BRCA1 and BRCA2 amplicon library of each patient was generated and quantified using Quant-iT PicoGreen (Invitrogen, Life Technologies, San Diego, CA, USA). Equivalent amounts of the patient libraries were pooled to generate a unique sequencing library that is twice purified using Agencourt AMPure XP (Beckman Coulter, Beverly, MA, USA) and PicoGreen quantified. Emulsion PCR was performed using the GS Junior Titanium emPCR kit (Lib-A) and pyrosequenced in the sense and antisense strands with the GS Junior following manufacturer's instructions (Roche Applied Science, Mannheim, Germany).
2.3. Bioinformatic Analysis
Data analysis was performed using the GS Amplicon Variant Analyzer version 2.7 (AVAv2.7) software (Roche). After sequence quality filtering, specific primers, MIDs, and adapter sequences are trimmed. Reads are then mapped to BRCA1 and BRCA2 genomic reference sequences NG_005905 and NG_012772, respectively. Coverage was obtained for all amplicons and analysed to detect low coverage amplicons. Variants are filtered using the AVAv2.7 software according to two parameters, the presence of the variant in both strands and the percentage of reads with the variant. Finally, variants are annotated according to the Human Genome Variation Society guidelines (http://www.hgvs.org/). Functional significance of variants is assigned by the authors following established criteria [19].
2.4. Homopolymer Analysis
BRCA1 and BRCA2 coding homopolymers of 6 bp or longer were analyzed using the BRCA HP v.2.0 (Multiplicom). Briefly, 50 ng of genomic DNA is amplified in two multiplex reactions resulting in 39 fragments that comprise all coding homopolymers. Fragment length is analysed on the ABI 3130 sequencer using the Gene Mapper software (Applied Biosystems, Foster City, CA, USA).
2.5. Multiple Amplicon Quantification (MAQ) Analysis
BRCA1 and BRCA2 large rearrangements were analysed using the BRCA MAQ kit (Multiplicom). It consists in the simultaneous amplification of several fluorescently labelled target amplicons (BRCA1 and BRCA2 exons) and reference sequences. Fragments are then size separated on an ABI 3130 sequencer (Applied Biosystems). Comparison of the relative intensities of the target amplicons in the test individual and a control individual results in a dosage quotient, indicating the copy number of the CNV in the test sample.
3. Results
3.1. Validation of Next Generation Sequencing Performance for BRCA1 and BRCA2 Mutation Screening
In order to evaluate our massive parallel sequencing approach 23 patients previously Sanger sequenced were pyrosequenced in a 454 GS Junior platform.
In total three runs were performed. In the first run 7 samples were sequenced using the BRCA MASTR kit v1.2 while in the last two runs 8 samples were simultaneously sequenced using the BRCA MASTR kit v2.0. The number of reads was variable between the runs. The average coverage per amplicon was higher in the third run. The use of the BRCA MASTR kit v2.0 that amplifies the two BRCA genes in 94 amplicons instead of 169 increases the average number of reads per amplicon as well as decreasing the number of amplicons with less than 38 reads even though in the first run we sequenced seven samples instead of eight (Table 1).
Table 1.
Summary of sequencing runs and coverage results of the validation set.
| Run 1 | Run 2 | Run 3 | |
|---|---|---|---|
| BRCAMASTRv1.2 | BRCAMASTRv2.0 | BRCAMASTRv2.0 | |
| Samples | 7 | 8 | 8 |
| Amplicons | 169 | 94 | 94 |
| Passed filter reads | 118006 | 78777 | 100771 |
| Mapped reads | 113374 | 78698 | 100344 |
|
| |||
| Coverage (number of reads/amplicon) | |||
| Minimum | 19 | 13 | 17 |
| Mean | 96,98 | 102,7 | 132,27 |
| Maximum | 277 | 445 | 414 |
| Standard deviation coverage | 35,8 | 46,92 | 70,51 |
| Amplicons <38 reads (%) | 32 (2.7) | 32 (4.2) | 13 (1.72) |
Bioinformatic analysis of reads is performed with the AVA software v2.7 (Roche). First, adapter and MIDs are trimmed from the obtained reads. Then, reads are mapped to the references sequences and variants are called and reported. We considered true variants those found in both strands and present in at least 25% of reads. The list of variants reported by the AVA software was further filtered excluding those variants present in amplicons with less than 38x coverage. It has been described that a minimum coverage of 38x is required to obtain a Phred score of 30 (or P = 99.9%) when using a variant detection filter of 25% [20]. The number of amplicons with less than 38x coverage ranged from 13 to 32 which represent less than 5% of the total number of amplicons sequenced. All 49 distinct substitutions were detected both in heterozygosity and homozygosity. Heterozygous substitutions were detected between 25% and 76.47% of the reads while homozygous substitutions were detected between 97.44% and 100% of the reads (Supplementary Table 1 availabe online at http://dx.doi.org/10.1155/2014/542541). As expected the variant detection is closer to 50% with high coverage. All the deletions and insertions, except from c.548-58delTT located in a homopolymeric stretch of 7 T in intron 7 of the BRCA1 gene, were detected in both the forward and the reverse strands and between 26% and 82.14% of the reads. We detected the variant c.548-58delTT in all samples at high frequency even though it was not present in all samples resulting in a false positive. Deletion of c.6841+79delTTAA in intron 11 of the BRCA2 gene was detected both in heterozygosity and homozygosity. The pathogenic variants were all detected in heterozygosity (Table 2) except for the c.8946delA in the BRCA2 gene which was detected in homozygosity in the forward reads. This is due to the location of the c.8946delA in a homopolymer stretch. In total, all 62 variants were detected resulting in a sensitivity of 100%.
Table 2.
Pathogenic mutations in the validation set tested for the evaluation of the AVA 2.7 software.
| Variant HGVS | Gene | Variant freq. % (number of reads) | |
|---|---|---|---|
| forward | reverse | ||
| c.70_71insTGTC | BRCA1 | 55.88 (68) | 59.32 (59) |
| c.1121-1123delCACinsT | BRCA1 | 35 (60) | 49.25 (67) |
| c.1961delA | BRCA1 | 38.68 (106) | 31.07 (103) |
| c.2921T>A (p.L974X) | BRCA1 | 51.16 (43) | 36.21 (58) |
| c.3767_3768delCA | BRCA1 | 26 (50) | 54.69 (64) |
| c.3770-3771delAG | BRCA1 | 50 (50) | 47.46 (59) |
| c.4107-4110dupATCT | BRCA1 | 54.24 (59) | 51.85 (54) |
| c.5123C>A | BRCA1 | 52.38 (42) | 56.25 (48) |
| c.1842dupT | BRCA2 | 56.00 (25) | 55.17 (29) |
| c.5350-5351delAAinsT | BRCA2 | 48.92 (139) | 54.08 (98) |
| c.6275-6276delTT | BRCA2 | 47.41 (116) | 38.13 (139) |
| c.7617+1G>A | BRCA2 | 34.78 (23) | 48.28 (29) |
| c.8946delAa | BRCA2 | 100 (52) | 44.44 (36) |
| c.9026_9030delATCAT | BRCA2 | 50.94 (53) | 40.54 (37) |
aMutation located in a homopolymeric region.
We detected 37 different false positives with the AVA software 2.7 (Supplementary Table 2). All of them correspond to deletions located in homopolymeric stretches and are generated as a result of the use of the pyrosequencing technology as has been described previously [21]. 35 out from 37 correspond to deletions in homopolymeric stretches of 6 bp or longer. The remaining 2 false positives correspond to two deletions at homopolymers of 4 nucleotides. In the total of three runs 71 false positives were called resulting in a specificity of 97.35%. The analysis of the homopolymers stretches of 6 bp or longer using the BRCA HP v2.0 kit (Multiplicom) allows the exclusion of all variants detected in homopolymers ≥ 6 bp from the variant list reported by the AVA 2.7 software, increasing the specificity of the detection of BRCA1 and BRCA2 mutations to 99.99%.
3.2. Detection of BRCA1 and BRCA2 Mutations in a Cohort of 101 Patients with Inherited Breast and Ovarian Cancer
We next decided to implement our parallel pyrosequencing protocol and sequence analysis approach to screen for mutations in the BRCA1 and BRCA2 genes in a series of 101 patients with breast and ovarian cancer. Our objective was to further analyze the performance of massive parallel pyrosequencing in terms of number of sequences obtained per run, coverage uniformity, and number of variants detected as well as in the identification of pathogenic mutations responsible for the disease.
All samples were first analysed for mutations in the homopolymer stretches of >6 bp using the BRCA HP v2.0 kit. Three frameshift mutations were detected. Although they are not strictly located in the homopolymer stretch, they are within the fragments amplified by the BRCA HP v2.0 kit (Table 4). Sanger sequencing identified one deletion, one insertion, and a combined InDel (c.4030del6insC, c.5189dupA, and c.5722_5723delCT in the BRCA2 gene).
Table 4.
Summary of BRCA1 and BRCA2 pathogenic mutations and variants of unknown significance (VUS) detected using our proposed workflow.
| Variant HGVS | Gene | Detected with assay | Clinical significance | |
|---|---|---|---|---|
| c.68_69delAG (p.Glu23Valfs∗16) | BRCA1 | NGS | Pathogenic | Spanish recurrent mutation |
| c.211A>G (p.Arg71Gly) | BRCA1 | NGS | Pathogenic | Spanish recurrent mutation |
| c.2410C>T (p.Gln804∗) | BRCA1 | NGS | Pathogenic | Reported |
| c.2900_2901dupCT (p.Pro968Leufs∗32) | BRCA1 | NGS | Pathogenic | Novel |
| c.3406C>A p.(Pro1136Thr) | BRCA1 | NGS | VUS | Novel |
| c.3708T>G (p.Asn1236Lys) | BRCA1 | NGS | VUS | Reported |
| c.4935G>C (p.Arg1645Ser) | BRCA1 | NGS | VUS | Reported |
| c.5078_5080delCTG (p.1692del26) | BRCA1 | NGS | Pathogenic | Reported |
| c.5123C>A (p.Ala1708Glu) | BRCA1 | NGS | Pathogenic | Spanish recurrent mutation |
| Δ Exons 16/17 | BRCA1 | MAQ | Pathogenic | Reported |
|
| ||||
| c.68-7T>A | BRCA2 | NGS | VUS | Reported |
| c.754G>A (p.Asp252Asn) | BRCA2 | NGS | VUS | Novel |
| c.3264dupT (p.Gln1089Serfs∗8) | BRCA2 | NGS | Pathogenic | Spanish recurrent mutation |
| c.4030del6insC (p.Asn1344Hisfs∗5) | BRCA2 | HP | Pathogenic | Novel |
| c.4316C>A (p.Ala1439Asp) | BRCA2 | NGS | VUS | Reported |
| c.4681C>A (p.His1561Asn) | BRCA2 | NGS | VUS | Reported |
| c.4965C>A (p.Tyr1655∗) | BRCA2 | NGS | Pathogenic | Reported |
| c.5189dupA (p.Asn1730Lysfs∗12) | BRCA2 | HP | Pathogenic | Novel |
| c.5722_5723delCT (p.Leu1908Argfs∗1) | BRCA2 | HP | Pathogenic | Reported |
| c.6215C>G (p.Ser2072Cys) | BRCA2 | NGS | VUS | Reported |
| c.6613G>A (p.Val2205Met) | BRCA2 | NGS | VUS | Reported |
| c.7180A>T (p.Arg2394∗) | BRCA2 | NGS | Pathogenic | Reported |
| c.7480C>T (p.Arg2494∗) | BRCA2 | NGS | Pathogenic | Reported |
| c.8009delC (p.Ser2670Trpfs∗2) | BRCA2 | NGS | Pathogenic | Novel |
| c.9274delT (p.Tyr3092Ilefs∗11) | BRCA2 | NGS | Pathogenic | Novel |
NGS: next generation sequencing; HP: homopolymer; MAQ: multiple amplicon quantification.
The remaining 98 samples were distributed in 14 Junior runs in groups of seven samples. We decided to sequence seven samples per run instead of eight in order to increase the coverage per amplicon and to decrease the number of amplicons with low coverage (<38 reads).
The number of reads obtained per run was very variable. The reads that passed the quality filters ranged between 69455 reads and 150722 reads with an average of 99864 reads (±28215) (Table 3). As a result of the differences between the reads obtained per run, the average coverage per amplicon and most importantly the number of amplicons with less than 38 reads were also variable (Table 3). Depending on the run the number of amplicons with less than 38 reads ranged between 1 and 26.
Table 3.
Summary of reads obtained and coverage results in 14 GS Junior runs.
| Run | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Passed filter reads | 113809 | 69455 | 82287 | 73127 | 88687 | 138625 | 72728 | 110271 | 90416 | 136237 | 69811 | 121332 | 80589 | 138238 |
| Mapped reads | 113445 | 69184 | 81904 | 72835 | 87529 | 137794 | 72522 | 109691 | 89982 | 135982 | 69613 | 12107 | 77080 | 138190 |
|
| ||||||||||||||
| Coverage (number of reads/amplicon) | ||||||||||||||
| Mean | 213,28 | 106,88 | 127,26 | 113,81 | 134,03 | 202,36 | 110,44 | 167,54 | 137,11 | 207,24 | 106,07 | 184,66 | 117 | 196.9 |
| Minimum | 5 | 29 | 22 | 34 | 19 | 24 | 26 | 27 | 31 | 7 | 23 | 21 | 11 | 8 |
| Maximum | 750 | 377 | 394 | 417 | 1560 | 1455 | 823 | 556 | 439 | 663 | 383 | 728 | 509 | 734 |
| Amplicons <38 reads (%) | 7 | 13 | 10 | 1 | 20 | 2 | 20 | 2 | 1 | 10 | 8 | 8 | 26 | 18 |
In the total 14 GS Junior runs, we identified 14 patients with deleterious mutations of which 7 are frameshift mutations (one mutation was found in three different patients), 4 are nonsense, 2 are missense, and 1 is an in frame deletion that affects splicing (Table 4). All mutations were confirmed by Sanger sequencing discarding the presence of false positives. One BRCA1 mutation, c.68_69delAG, was found in three different patients. This mutation accounts for the 30.4% BRCA1 mutations in the Mediterranean area [22]. Although only found once in our series, mutations c.211A>G, c.5123C>A in BRCA1, and c.3264dupT in BRCA2 are also considered recurrent in the Spanish population [22]. We have identified 5 novel mutations in our cohort. Mutation c.2900_2901dupCT in the BRCA1 gene and mutations c.4030del6insC, c.5189dupA, c.8009delC, and c.9274delT in the BRCA2 gene are mutations not described previously. In addition we detected 9 variants with unknown clinical significance (VUS). All of them are missense mutations except one located in an intronic sequence (c.68-7T>A in the BRCA2 gene). Finally, large genomic deletions and duplications were screened using the MAQ assay, which consists in a semiquantitative PCR that amplifies all exons in the BRCA1 and BRCA2 genes together with control regions. We detected an exonic deletion that comprises exons 16 and 17 of the BRCA1 gene. This mutation is predicted to produce an inframe deletion of 132 amino acids that disrupts the BRCT-N domain (p.Glu1559_Thr1691del) and it has been described to be deleterious by functional analysis [23].
4. Discussion
Molecular genetic testing of mutations in the BRCA1 and BRCA2 genes is currently performed using highly sensitive but labour-intensive direct Sanger sequencing of individual exons. The advances in sequencing technologies have increased the speed and efficiency of DNA testing and next generation platforms are becoming the standard in molecular genetic diagnosis.
Here, we have tested and implemented a method for the molecular analysis of the BRCA1 and BRCA2 genes based on massive parallel pyrosequencing of pooled BRCA1 and BRCA2 gene enriched samples. BRCA1 and BRCA2 exon enrichment has been performed by PCR amplification using the Multiplicom BRCA MASTR kit, which amplifies all BRCA1 and BRCA2 coding exons in 97 amplicons. PCR enrichment was chosen over a hybridisation based method because PCR enrichment has been shown to cover all the amplicons of interest and to provide less variation in coverage between regions [10]. In addition, PCR enrichment has also a lower cost and requires less DNA compared to hybridisation based methods. Currently, PCR based enrichment is chosen for molecular diagnosis when analysing few genes simultaneously.
We validated our approach in a cohort of 23 patients with previously characterised BRCA1 and BRCA2 mutations and polymorphisms. We detected all mutations and polymorphisms in both heterozygosity and homozygosity achieving 100% sensitivity and 97.35% specificity. To increase the specificity of the method the variants called in homopolymeric regions should be excluded. Because both BRCA1 and BRCA2 genes comprise homopolymeric stretches in their coding regions a complementary assay is then needed to screen for changes in homopolymers. We used the BRCA HP assay developed by Multiplicom which screens for deletions and insertions in all exonic homopolymers of 6 bp or longer. This allowed the exclusion from our final variant list all changes detected in homopolymeric regions of ≥6 nucleotides resulting in a specificity of 99.99%.
Other works have analysed the performance of pyrosequencing in the detection of mutations in the BRCA1 and BRCA2 genes using different approaches to obtain the BRCA1 and BRCA2 DNA library and using the 454 GS FLX and GS Junior platforms [15–18]. Here, we used a multiplex amplicon based assay which amplifies all BRCA1 and BRCA2 coding regions and exon-intron boundaries and attaches the MIDs and sequencing adaptors in a second PCR (BRCA MASTR, Multiplicom). Multiplex PCR has been demonstrated to result in higher coverage per amplicon compared to singleplex [15] or long PCR fragments [17] and allows the joint sequencing of seven samples in each run. Recently, Feliubadaló et al. [18] have developed and validated a workflow using the BRCA MASTR kit amplicon followed by 454 GS Junior pyrosequencing. Data analysis combines the use of the three types of software VIP, R, and AVA and numerous filters followed by visual inspection of fragments. Their workflow achieves a specificity of 99.99% and a sensitivity of 100% when adding the BRCA HP assay to detect insertions and deletions in homopolymeric regions. In contrast to Feliubadaló et al. [18] our data analysis is based exclusively on the AVA 2.7 software making it simpler and completely automated. The AVA2.7 software in contrast to previous versions is able to call small InDels and achieve a sensitivity of 100% in variant calling ([16] and this report). Using our filtering parameters in the AVA 2.7 software together with BRCA HP assay we achieve a specificity of 99.99% and a sensitivity of 100%.
It is recommended that mutations detected by NGS technologies be validated by Sanger sequencing in the context of molecular diagnostics. Here, all deleterious mutations and VUS detected in our cohort of 101 patients have been confirmed by Sanger sequencing. These results together with the ones obtained in our validation set show that when using massive parallel pyrosequencing only deleterious mutations detected in homopolymeric tracts should be confirmed by Sanger sequencing [16].
Analysis of the coverage in our series of 14 runs showed that the number of amplicons with less than 38x ranged from 1 to 26 (0.14–3.8%) of a total number of 679 amplicons sequenced per run. This means that seven samples can be screened in a single GS Junior run with more than 95% of sequences covered sufficiently to provide a minimum power of 99.9% to detect heterozygous mutations in at least 25% of the reads. We detected that the number of reads obtained per run was very variable. After carefully reviewing our whole procedure, we realised that the addition of a lower number of molecules of DNA library per bead in the emulsion PCR resulted in the higher number of reads that passed quality filtering. Taking into account this observation we are now increasing the number of samples per run, which will result in a lower cost per sample analysed. We have checked that the cost and time consuming per sample of our sequencing approach improves the overall cost (approximately 50% less) and makes the process faster compared to direct Sanger sequencing alone.
Our proposed workflow to screen for mutations in the BRCA1 and BRCA2 genes consists first in the use of the BRCA1 and BRCA2 homopolymer assay (BRCA HP) followed by massive parallel sequencing with the 454 GS Junior sequencer and using the BRCA MASTR amplicon kit to generate the patient libraries. Coverage and variant calling is done using the AVA 2.7 software. Amplicons with low coverage should be Sanger sequenced. Finally, large rearrangements in the BRCA1 and BRCA2 genes are detected using the BRCA MAQ kit (Figure 1). Using our validated workflow, we have identified 18 deleterious mutations in 101 patients (17,8%) which is in accordance with the prevalence of BRCA1 and BRCA2 mutations reported in the Spanish hereditary breast and ovarian cancer population. In addition, we have detected 10 VUS, nine of which are unique and two of them have not been previously reported.
Figure 1.
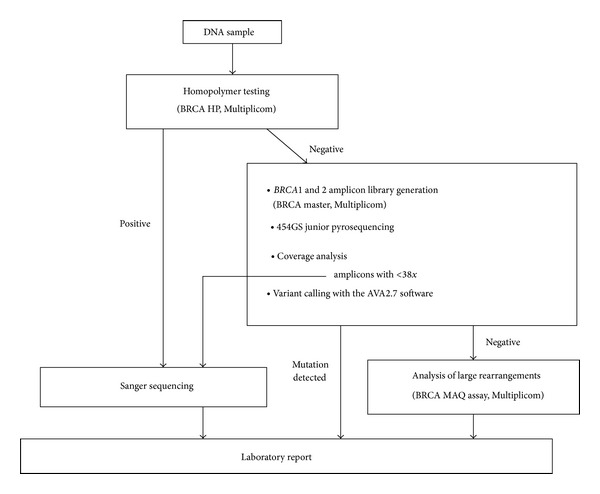
Proposed workflow using massive parallel pyrosequencing for analysing BRCA1 and BRCA2 genes.
5. Conclusions
We show here that massive parallel pyrosequencing can be used as a diagnostic strategy to test for BRCA1 and BRCA2 mutations meeting very stringent sensitivity and specificity parameters and could be used in diagnostic laboratories replacing traditional Sanger sequencing.
Supplementary Material
Supplementary Table 1. shows the frequency of detection of each variant both in the forward and reverse reads.
Supplementary Table 2. shows the false positives detected by the AVA2.7 software.
Acknowledgments
The authors thank Conxi Lázaro and Lidia Feliubadaló for their collaboration in providing samples for the validation set and helpful advice. They thank Orland Diez for helping with the analysis of the validation set.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1 . Science. 1994;266(5182):66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 2.Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2 . Nature. 1995;378(6559):789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- 3.Antoniou A, Pharoah PDP, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. The American Journal of Human Genetics. 2003;72(5):1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King M, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2 . Science. 2003;302(5645):643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 5.Pruthi S, Gostout BS, Lindor NM. Identification and management of women with BRCA mutations or hereditary predisposition for breast and ovarian cancer. Mayo Clinic Proceedings. 2010;85(12):1111–1120. doi: 10.4065/mcp.2010.0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paradiso A, Formenti S. Hereditary breast cancer: clinical features and risk reduction strategies. Annals of Oncology. 2011;22(1) supplement 1:i31–i36. doi: 10.1093/annonc/mdq663. [DOI] [PubMed] [Google Scholar]
- 7.Gerhardus A, Schleberger H, Schlegelberger B, Gadzicki D. Diagnostic accuracy of methods for the detection of BRCA1 and BRCA2 mutations: a systematic review. European Journal of Human Genetics. 2007;15(6):619–627. doi: 10.1038/sj.ejhg.5201806. [DOI] [PubMed] [Google Scholar]
- 8.Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clinical Chemistry. 2009;55(4):641–658. doi: 10.1373/clinchem.2008.112789. [DOI] [PubMed] [Google Scholar]
- 9.Bentley DR. Whole-genome re-sequencing. Current Opinion in Genetics and Development. 2006;16(6):545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Mamanova L, Coffey AJ, Scott CE, et al. Target-enrichment strategies for next-generation sequencing. Nature Methods. 2010;7(2):111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- 11.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan JE, Carr IM, Sheridan E, et al. Genetic diagnosis of familial breast cancer using clonal sequencing. Human Mutation. 2010;31(4):484–491. doi: 10.1002/humu.21216. [DOI] [PubMed] [Google Scholar]
- 14.Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(44):18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Leeneer K, Hellemans J, de Schrijver J, et al. Massive parallel amplicon sequencing of the breast cancer genes BRCA1 and BRCA2: opportunities, challenges, and limitations. Human Mutation. 2011;32(3):335–344. doi: 10.1002/humu.21428. [DOI] [PubMed] [Google Scholar]
- 16.Michils G, Hollants S, Dehaspe L, et al. Molecular analysis of the breast cancer genes BRCA1 and BRCA2 using amplicon-based massive parallel pyrosequencing. Journal of Molecular Diagnostics. 2012;14(6):623–630. doi: 10.1016/j.jmoldx.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 17.Hernan I, Borràs E, de Sousa Dias M, et al. Detection of genomic variations in BRCA1 and BRCA2 genes by long-range PCR and next-generation sequencing. Journal of Molecular Diagnostics. 2012;14(3):286–293. doi: 10.1016/j.jmoldx.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 18.Feliubadaló L, Lopez-Doriga A, Castellsagué E, et al. Next-generation sequencing meets genetic diagnostics: Development of a comprehensive workflow for the analysis of BRCA1 and BRCA2 genes. European Journal of Human Genetics. 2013;21(8):864–870. doi: 10.1038/ejhg.2012.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tavtigian SV, Greenblatt MS, Goldgar DE, Boffetta P. Assessing pathogenicity: overview of results from the IARC unclassified genetic variants working group. Human Mutation. 2008;29(11):1261–1264. doi: 10.1002/humu.20903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Leeneer K, de Schrijver J, Clement L, et al. Practical tools to implement massive parallel pyrosequencing of PCR products in next generation molecular diagnostics. PLoS ONE. 2011;6(9) doi: 10.1371/journal.pone.0025531.e25531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biology. 2007;8(7, article R143) doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diez O, Gutiérrez-Enríquez S, Balmaña J. Heterogeneous prevalence of recurrent BRCA1 and BRCA2 mutations in Spain according to the geographical area: implications for genetic testing. Familial Cancer. 2010;9(2):187–191. doi: 10.1007/s10689-009-9301-5. [DOI] [PubMed] [Google Scholar]
- 23.Carvalho M, Pino MA, Karchin R, et al. Analysis of a set of missense, frameshift, and in-frame deletion variants of BRCA1 . Mutation Research. 2009;660(1-2):1–11. doi: 10.1016/j.mrfmmm.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. shows the frequency of detection of each variant both in the forward and reverse reads.
Supplementary Table 2. shows the false positives detected by the AVA2.7 software.