

Abstract
Kaposi’s sarcoma-associated herpesvirus (KSHV) capsids can be produced in insect cells using recombinant baculoviruses for protein expression. All six capsid proteins are required for this process to occur and, unlike for alphaherpesviruses, the small capsid protein (SCP) ORF65 is essential for this process. This protein decorates the capsid shell by virtue of its interaction with the capsomeres. In this study, we have explored the SCP interaction with the major capsid protein (MCP) using GFP fusions. The assembly site within the nucleus of infected cells was visualized by light microscopy using fluorescence produced by the SCP–GFP polypeptide, and the relocalization of the SCP to these sites was evident only when the MCP and the scaffold protein were also present – indicative of an interaction between these proteins that ensures delivery of the SCP to assembly sites. Biochemical assays demonstrated a physical interaction between the SCP and MCP, and also between this complex and the scaffold protein. Self-assembly of capsids with the SCP–GFP polypeptide was evident. Potentially, this result can be used to engineer fluorescent KSHV particles. A similar SCP–His6 polypeptide was used to purify capsids from infected cell lysates using immobilized affinity chromatography and to directly label this protein in capsids using chemically derivatized gold particles. Additional studies with SCP–GFP polypeptide truncation mutants identified a domain residing between aa 50 and 60 of ORF65 that was required for the relocalization of SCP–GFP to nuclear assembly sites. Substitution of residues in this region and specifically at residue 54 with a polar amino acid (lysine) disrupted or abolished this localization as well as capsid assembly, whereas substitution with non-polar residues did not affect the interaction. Thus, this study identified a small conserved hydrophobic domain that is important for the SCP–MCP interaction.
Introduction
Herpesviruses can self-assemble capsids that possess icosahedral symmetry (Wildy et al., 1960) and are composed of six proteins. The four capsid shell proteins are the major capsid protein (MCP), triplex proteins 1 and 2, and small capsid protein (SCP), whereas the scaffold protein and maturational protease occupy the internal space of the capsid (reviewed by Brown & Newcomb, 2011; Cardone et al., 2012; Homa & Brown, 1997; Rixon, 1993; Steven & Spear, 1996). The MCP is the building block of the individual capsomeres, in both the hexameric (hexons) and pentameric (pentons) configurations. The SCP decorates the capsid shell by virtue of its location on the hexameric capsomeres (Lo et al., 2003; Trus et al., 2001; Wingfield et al., 1997; Zhou et al., 1995). The triplex proteins form a trimer that acts to stabilize capsid shell assembly by essentially linking the capsomeres together. The two internal proteins are present only in the precursor capsid structures; the scaffold protein drives the assembly of a closed structure and the maturational protease is essential during the transformation of the scaffold-containing structure to a DNA-containing capsid (reviewed by Brown & Newcomb, 2011; Cardone et al., 2012; Homa & Brown, 1997; Rixon, 1993; Steven & Spear, 1996). Capsid assembly for herpesviruses begins in the cytoplasm. Cellular localization studies show that MCP is transported into the nucleus by the scaffold protein and one of the triplex proteins ‘carries’ the other triplex protein into the nucleus (Adamson et al., 2006; Nicholson et al., 1994; Plafker & Gibson, 1998; Rixon et al., 1996). The SCP ‘piggy-backs’ onto the major capsid–scaffold protein complex and becomes concentrated at the assembly site (Desai & Person, 1998; Rixon et al., 1996). These small complexes come together (Newcomb et al., 1996; Spencer et al., 1998) in the nucleus to form a spherical closed capsid structure.
Self-assembly of herpes simplex virus type 1 (HSV-1), Kaposi’s sarcoma-associated herpesvirus (KSHV) and Epstein–Barr virus (EBV) capsids using recombinant baculoviruses for protein expression has been demonstrated (Henson et al., 2009; Newcomb et al., 1999; Perkins et al., 2008; Tatman et al., 1994; Thomsen et al., 1994). An important difference between the assembly pathways of these human herpesviruses was uncovered during these types of analyses and led to the discovery of the requirement of the gammaherpesvirus SCP for capsid self-assembly (Henson et al., 2009; Perkins et al., 2008). The SCP is not required for HSV-1 capsid assembly (Desai et al., 1998; Tatman et al., 1994; Thomsen et al., 1994) nor for other alphaherpesviruses (Antinone et al., 2006; Chaudhuri et al., 2008; Krautwald et al., 2008). Thus, the SCP specifies an essential function for assembly which was also confirmed in KSHV-infected cells (Sathish & Yuan, 2010).
The six gene products that compose KSHV capsids are ORF25 (MCP), ORF62 (triplex 1), ORF26 (triplex 2), ORF17 (protease), ORF17.5 (scaffold) and ORF65 (SCP) (Nealon et al., 2001). The capsid assembly domain of ORF65 resides in the N terminus (86 aa) and six residues (R14, D18, V25, R46, G66 and R70) within this domain are important for capsid assembly (Kreitler et al., 2012). The goal of this study was to use SCP fusions to GFP to discover how the KSHV SCP interacts with the MCP, the localization of this protein to nuclear sites of assembly and the role of a hydrophobic domain in this small molecule for assembly of capsids.
Results
Localization of ORF65–GFP fusion protein to nuclear assembly sites and self-assembly of capsids with SCP–GFP polypeptides
Previously, we fused GFP to the N terminus of HSV-1 SCP (VP26) and observed that the fusion protein localized to nuclear puncta in infected cells and could still decorate capsids (Desai & Person, 1998). We took a similar approach with KSHV ORF65 and, because the assembly domain resides at the N terminus (Kreitler et al., 2012), we initially made a C-terminal in-frame fusion with the EGFP ORF. The ORF65 gene, which codes for 170 aa (18 kDa) was amplified using KSHV BAC36 DNA (Zhou et al., 2002) as template and cloned in-frame with the EGFP gene. The baculovirus expressing ORF65CGFP was used in both single and co-infections to examine fluorescence distribution in Sf21 cells (Fig. 1a). When expressed by itself, the fluorescence observed was diffusely distributed throughout the cell with greater accumulations in the nucleus. This distribution changed significantly when both ORF25 (MCP) and ORF17.5 (scaffold protein) were present. The fluorescence accumulated in large nuclear puncta, which are potential nuclear assembly sites. There was no change in the distribution of the fluorescence in the presence of the triplex proteins. This was also evident in phase images of similar infected cells (Fig. 1b); the circular structures that displayed the fluorescence were internal to the nuclear membrane margins. We also examined the distribution of fluorescence that arose from the polypeptide when the GFP was fused to the N terminus of ORF65. Although the fluorescence was more or less similar in infected cells compared with ORF65CGFP, there was no change in the distribution in the presence of ORF25/17.5. In fact, the distribution was similar to the pattern observed in cells infected with a baculovirus expressing only EGFP (Fig. 1c). We used fusions of GFP to ORF25 and ORF17.5 to determine if these proteins gave rise to a similar phenotype in co-infected cells. Fluorescence observed with ORF17.5NGFP was diffusely distributed in the nucleus whether or not ORF25 or ORF65 was present, whereas fluorescence observed with ORF25GFP relocalized to the nucleus in a diffuse pattern in the presence of both ORF65 and ORF17.5 (data not shown). These fusion proteins did not localize to discrete nuclear puncta; thus, similar to the HSV-1 SCP (Desai et al., 2003), the phenotype observed for the KSHV SCP is a unique property displayed by these small proteins.
Fig. 1.

KSHV MCP interaction with the SCP–GFP fusion polypeptide. (a) ORF65GFP localizes to nuclear puncta only when both the MCP (ORF25) and scaffold proteins (ORF17.5) are present. Sf21 cells were infected with the baculovirus expressing ORF65CGFP or co-infected with viruses expressing different KSHV capsid proteins. The cells were imaged by confocal microscopy 48 h after infection. (b) Fluorescence and phase imaging of nuclear puncta. Sf21 cells co-infected with ORF65CGFP and the virus expressing both ORF25 and ORF17.5 were imaged by confocal microscopy 24 h after infection. The images show the fluorescence (left panel), the phase image (middle panel) and the merged image (right panel). The nucleus is marked in some cells with a black circle. The intranuclear structures, which overlap with the ORF65CGFP fluorescent puncta, are indicated with arrowheads. (c) ORF65 tagged at the N terminus with GFP (ORF65NGFP) or GFP expressed without fusion to ORF65 does not relocalize to nuclear assembly sites in the presence of ORF25/17.5. Cells were infected and imaged as described in (b). Bar, 20 μm in all panels. (d) Co-immunoprecipitation of ORF25 and ORF17.5 polypeptides in the presence of SCP–GFP. Sf21 cells were co-infected with viruses expressing ORF65CGFP and either ORF25NV5 (lane 3) or ORF17.5NV5 (lane 2), or both (lane 1). Both the MCP and scaffold proteins were co-precipitated by ORF65CGFP (lane 1, top panel). ORF25 was also co-precipitated by ORF65CGFP in relatively similar amounts in lane 3, but a much lower amount of ORF17.5 was precipitated when ORF25 was not present in the cells (lane 2, top panel). Input proteins were detected using anti-V5 (αV5) and anti-GFP (αGFP) antibodies (three bottom panels). Protein standards (kDa) are indicated. IP, Immunoprecipitation; WB, Western blot.
In order to demonstrate the interaction between ORF65 and ORF25 (MCP) biochemically, we performed a co-immunoprecipitation assay using lysates from similar co-infected cells. Sf21 cells were co-infected with ORF65CGFP (45 kDa)-, ORF25NV5 (153.4 kDa)- and ORF17.5NV5 (31 kDa)-expressing viruses. Both the ORF25NV5 and ORF17.5NV5 (scaffold) polypeptides were functional in that they supported self-assembly of capsids. The herpesvirus scaffold proteins have been shown to be extensively modified by phosphorylation (Casaday et al., 2004). Lysates prepared 48 h post-infection were immunoprecipitated with anti-GFP antibodies. The immunoprecipitates as well as the input lysates were immunoblotted using anti-V5 antibodies (Fig. 1d). In the presence of antibody to ORF65CGFP, ORF25 was co-precipitated regardless of the presence of ORF17.5 (compare Fig. 1d, lanes 1 and 3). Relatively similar levels (as judged by the signal from the same epitope) of ORF17.5 were precipitated when ORF25 was also present, but a much reduced amount of the scaffold protein was observed to co-precipitate when expressed without the MCP as judged by the signal in the immunoblot (compare Fig. 1d, lanes 1 and 2).
Previously, we demonstrated the incorporation of HSV-1 VP26–GFP in capsids purified after sedimentation through sucrose gradients (Desai & Person, 1998). As we could generate a particle decorated with a live fluorescent tag we also sought to determine if this could potentially be done with KSHV SCP; in this case, the protein is essential for assembly. Sf21 cells were co-infected with baculoviruses expressing all six KSHV capsid proteins and the infected cells were examined by ultrastructural methods using conventional thin-section, transmission electron microscopy (TEM) (Fig. 2a, b). Extensive areas within the nucleus of cells infected with viruses expressing either ORF65CHA (Kreitler et al., 2012) or ORF65CGFP containing dense material were visualized (Fig. 2a shows cells infected with ORF65CGFP-expressing virus); the same areas in each specific infected cell were imaged at higher magnification, revealing assembled capsids (Fig. 2b). The inset of Fig. 2(a) is a light microscopy image of different cells, which shows the intranuclear fluorescent puncta. We also purified these GFP-tagged capsids using sucrose gradient sedimentation (Fig. 2c). Light-scattering bands indicative of assembled capsids were observed in gradients regardless of whether the ORF65 was tagged with haemagglutinin (HA) or GFP. The intensity of the bands was similar for similar numbers of infected cells, which indicated normal levels of assembly of the GFP-decorated capsids (Fig. 3). Capsids were harvested by side-puncture aspiration after visualizing the light-scattering band using incident light. Negative-stained capsids were prepared and the images revealed closed icosahedral capsids in both gradients (Fig. 2c). The ORF65 proteins in the capsids purified by sucrose gradient sedimentation were detected using immunoblot methods with antibodies to ORF65 (Gao et al., 2003) (Fig. 2d), and were also confirmed by anti-HA and anti-GFP antibodies (data not shown). These data showed the incorporation of the GFP-tagged ORF65. We also tested whether the ORF65NGFP polypeptide could support self-assembly in this system. When sucrose gradient sedimentation of infected cell lysates was performed, there was no evidence of capsid assembly (Fig. 3).
Fig. 2.
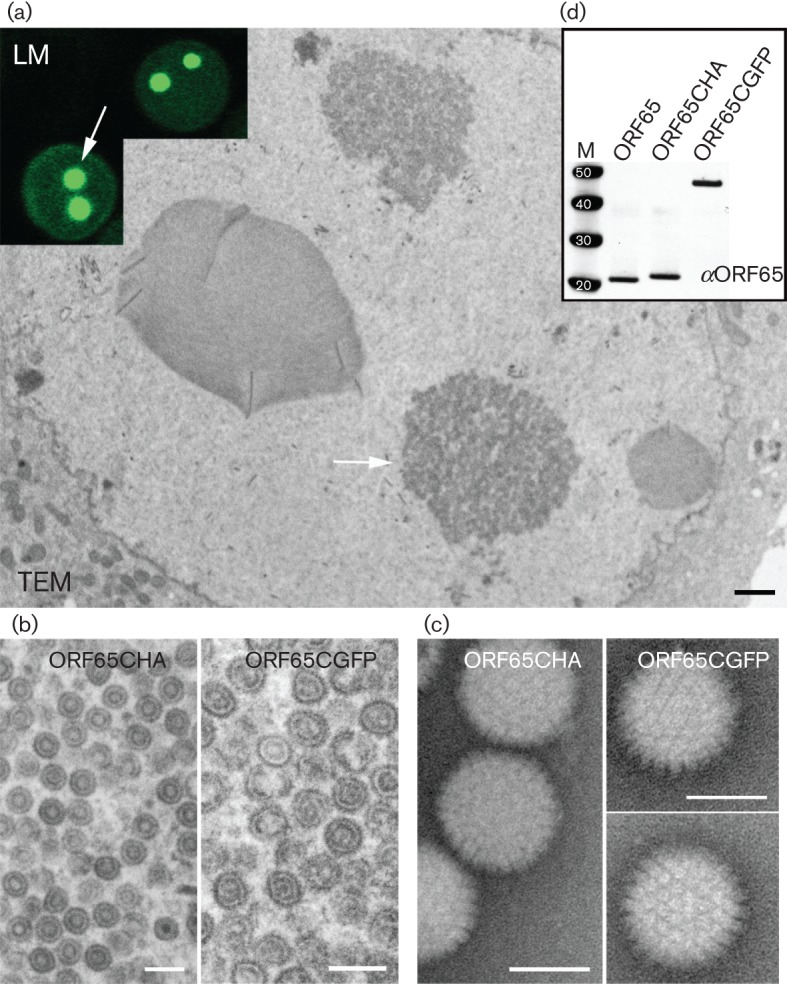
KSHV capsid self-assembly with the SCP–GFP fusion polypeptide. (a) Images derived from both light microscopy (LM) and TEM of different Sf21 cells co-infected with baculoviruses expressing all KSHV capsid proteins including ORF65CGFP (Bac-All). The assembly site in the nucleus of the cells is indicated by arrows. Bar in TEM image, 400 nm. (b) Similar assembly sites in the nucleus were examined at higher magnification, revealing assembled closed structures. Left panel, virus expressing ORF65CHA; right panel, virus expressing ORF65CGFP. Bars, 200 nm. (c) Negative-stained images of capsids extracted from sucrose gradients of lysates made from Bac-All ORF65CGFP or Bac-All ORF65CHA co-infected cells. Bars, 100 nm. (d) Immunoblot, using anti-ORF65 (αORF65) antibody (Gao et al., 2003), of capsids purified from sucrose gradients showed the presence of ORF65 proteins in the capsid fraction. Protein standards (kDa) are shown in lane M.
Fig. 3.

Fusion of GFP at the C terminus of the KSHV SCP supports self-assembly. Sf21 cells were co-infected with baculoviruses expressing all six capsid proteins. The ORF65-expressing virus was tagged at the C terminus with HA or EGFP and at the N terminus with EGFP. The capsid band that was observed by incident light following sucrose gradient sedimentation of infected cell lysates was photographed. Capsid assembly was evident only with the CHA- and CGFP-tagged SCP fusions. The arrow indicates the direction of sedimentation.
We then examined whether the alanine substitution mutants of ORF65 in the assembly domain (Kreitler et al., 2012) affected the interaction with the MCP. As the GFP relocalization assay is a measure of this interaction we transferred the alanine-scanning mutants in ORF65 from the pFB1-ORF65CHA-ΔEcoRI plasmid into pFB1-ORF65CGFP. The genes encoding substitutions R14A, D18A, V25A, R46A, G66A and R70A, which produced proteins that were defective for assembly (Kreitler et al., 2012), were moved into this transfer vector. The recombinant baculoviruses expressing these mutant proteins were then tested in co-infections with vFBD-ORF25/17.5. Under light microscopy, all of the six assembly-defective mutants displayed relocalization of GFP fluorescence to assembly sites (Fig. 4) indicative of normal ORF25/17.5 interaction.
Fig. 4.
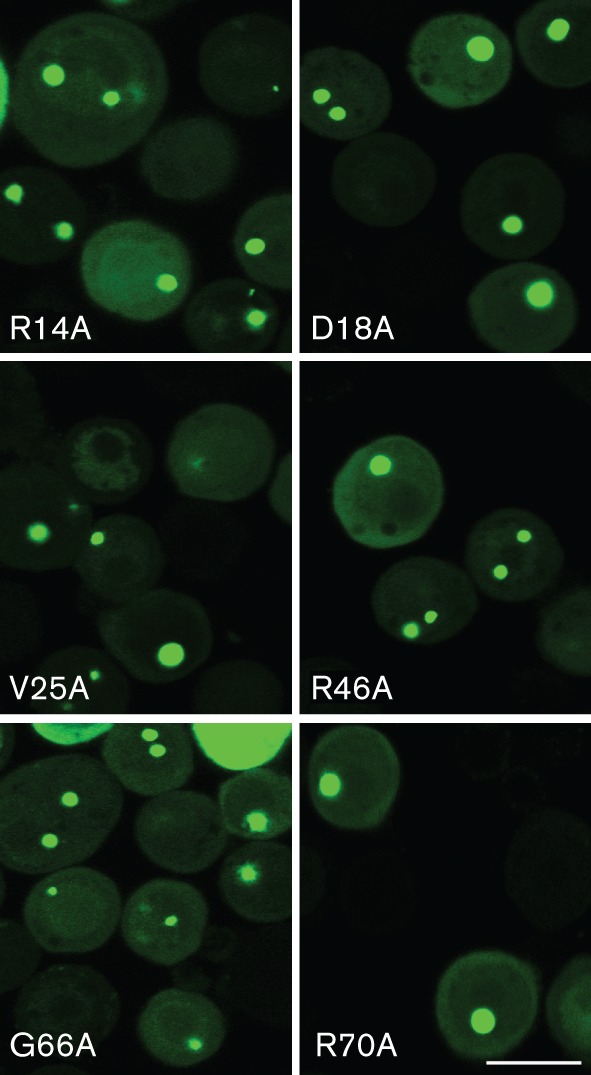
Assembly-defective alanine substitution mutants of ORF65 can still interact with the MCP–scaffold complex. Sf21 cells were co-infected with viruses expressing the ORF65 alanine substitution mutants fused to GFP and the ORF25/17.5 dual-expressing virus. The cells were imaged by confocal microscopy 24 h post-infection. Bar, 20 μm.
Use of a His6-affinity tag to purify capsids and image SCP decoration of capsids using gold label
The utility of the His6 tag for protein purification is well documented. We have tagged HSV-1 VP26 in the past and have now tagged KSHV SCP with a His66 sequence. For ORF65, the fusion was made at the C terminus of the protein. Our aim was to use this tag to see if we could purify capsids using this approach and to use it as an imaging tool. There are some advantages of this tag over the GFP tag for both purification and imaging purposes. The expression of the His6- tagged ORF65 in recombinant baculovirus-infected Sf21 cells was confirmed by Western blots using anti-His6 antibodies (Fig. 5a, top panel) as well as in assembled capsids (Fig. 5a, bottom panel). Purification of assembled capsids was carried out first on sucrose gradient-purified capsids and subsequently on whole infected cell extracts, both total soluble lysate and lysate that had been clarified by using a 20 % sucrose cushion. For each preparation, binding with Ni-nitrilotriacetic acid (NTA) agarose beads was followed by several washes and elution with 250 mM imidazole. The starting material and the eluted proteins were examined by PAGE (Fig. 5b) and the eluted samples were also prepared for imaging after negative staining (Fig. 5c). In each case, assembled capsids were eluted from the beads, indicating purification of the particles using this method. Preparations made from sucrose cushion-clarified lysates were generally purer, i.e. there were fewer co-purified proteins, but the ability to attain significant purification of capsids from the total lysate was remarkable considering the protein content in the starting lysate as judged by staining the proteins in the gel.
Fig. 5.

Purification of KSHV capsids using immobilized metal affinity chromatography (IMAC) methods and decoration of capsids with Ni-NTA-derivatized gold label. (a) Top panel, Western blot analysis of Sf21 cells infected with a vFB-ORF65C6XHIS or vFBD virus expressing ORF65C6XHIS and ORF17. MI, mock-infected cells. Bottom panel, Western blot analysis of infected cell lysates (Inf. cell) and capsids (Capsid) purified from sucrose gradients. The assembly infections were done with three capsid dual baculoviruses, in one case vFBD-ORF17/ORF65CHA was used and in the other vFBD-ORF17/ORF65C6XHIS was used. Both membranes were probed with anti-His tag (6XHIS) antibody. Protein standards are shown in the first lane. (b) IMAC purification of capsid proteins using Ni-NTA agarose. The starting material (SM) used in the purification is shown as well as the eluted (E) proteins. SG, sucrose gradient-purified capsids; SC, sucrose cushion enriched material; TOT, total infected cell lysate. Samples were prepared from Sf21-infected cells harvested 72 h post-infection. The gel was stained with SYPRO Ruby (Invitrogen). (c) Negative-stained images of IMAC-purified capsids in the elution buffer. Bars, 40 nm. (d) Capsids were purified using sedimentation methods and then incubated with Ni-NTA-derivatized 5 nm gold. Capsids that were assembled with ORF65CHA do not significant numbers of gold particles, whereas capsids containing ORF65C6XHIS are decorated with numerous gold particles. Bar, 100 nm.
Capsids purified using sucrose gradients were also incubated directly with Ni-NTA-derivatized gold particles (5 nm size). These conjugated gold particles permitted direct labelling of His6-tagged proteins without having to go through a two-step antibody-binding reaction, and because it is a direct label there was greater accuracy in the labelling of the protein and hence its location in a three-dimensional structure. The capsids containing ORF65CHA bound to a few gold particles or none at all, whereas the capsids containing ORF65CHIS were decorated with numerous gold particles as judged by the density of gold labelling (Fig. 5d). These data also suggested the C terminus of ORF65 was readily accessible on the capsid surface.
A domain resides in ORF65 that is required for interaction with the MCP
GFP relocalization to assembly sites in the nucleus was indicative of an interaction between the SCP and MCP. To map this interaction region we generated additional GFP fusion polypeptides of ORF65 that produced N-terminal and C-terminal truncations of the protein. Viruses expressing these polypeptides were used to infect Sf21 cells in the presence of ORF25/17.5-expressing virus and examined by confocal microscopy for localization to nuclear assembly sites (Fig. 6a). The N-terminal half of the polypeptide (86), which encodes the assembly domain, was able to relocalize GFP to assembly sites within the nucleus; the C-terminal half (87–170) did not. This was further delineated to an N-terminal 60 aa polypeptide, although further truncation to 50 aa resulted in loss of this interaction as judged by the appearance of diffuse fluorescence. An N-terminal 55 aa polypeptide displayed nuclear puncta, but there was also an increase in cytoplasmic fluorescence, as summarized in Fig. 6(e). All the truncation mutant viruses produced a stable polypeptide of the correct size in infected cells as judged by immunoblot examination of lysates using GFP antibodies (Fig. 6b).
Fig. 6.

Identification of a discrete domain in ORF65 required for interaction with MCP; the amino acid position at A54 is important for this interaction. (a) The region between aa 50 and 60 of ORF65 is required for efficient relocalization of GFP to the nuclear assembly sites. Sf21 cells were co-infected with the vFBD-ORF25/17.5 virus and viruses expressing WT ORF65 (ORF65-170/FL) or truncation polypeptide mutations fused to GFP. The cells were imaged 48 h after infection. Bar, 20 μm. (b) Immunoblot analysis of infected cell lysates with anti-GFP (αGFP) antibody demonstrated stable accumulation of all truncation mutant polypeptides. (c) Substitution of A54 with lysine or deletion of this region abolished interaction with the MCP as judged by the absence of fluorescent puncta in the nucleus. Cells were imaged 24 h after co-infection with viruses expressing ORF65CGFP and vFBD-ORF25/17.5. Bar, 20 μm. (d) Western blot analysis (anti-GFP) of lysates of infected cells demonstrated the presence of stable polypeptide accumulation of each ORF65 mutant polypeptide. Molecular mass standards (kDa) are in lane M. (e) Summary of truncation and in-frame deletion mutant ORF65 polypeptide structures and phenotypes in nuclear (NUC) site relocalization.
The alignment of ORF65 amino acid sequences from different gammaherpesviruses (Kreitler et al., 2012) revealed a string of conserved hydrophobic residues located in the region between aa 48 and 55. To map the MCP interaction site more precisely in the full-length polypeptide we created in-frame deletions and site-directed substitutions using QuikChange (Stratagene) mutagenesis. Two deletion mutants, Δ50–55 and Δ50–52, were made and the mutant polypeptide fused to GFP was examined in co-infected cells for assembly site localization (Fig. 6c). The Δ50–52 mutant displayed the ability to do so, whereas the Δ50–55 mutant did not (data summarized in Fig. 6e). We also tested whether the alanine substitution mutants of ORF65 in this region (Kreitler et al., 2012) affected the interaction with the MCP protein. The alanine-scanning mutants in each individual residue of ORF65 between aa 48–55 were moved from the pFB1-ORF65CHA-ΔEcoRI plasmid into the pFB1-ORF65CGFP vector. The recombinant baculoviruses expressing these mutant proteins were then tested in co-infections with vFBD-ORF25/17.5. Under light microscopy, none of the alanine substitution mutants affected the relocalization of GFP to assembly sites (data not shown).
ORF65 residue A54 is sensitive to substitution with a polar residue
As single alanine substitutions at the residues located in this sequence did not affect relocalization of the GFP fluorescence, we decided to create substitutions that would introduce polar residues in this region. Precise identification of residues or positions in the molecule sensitive to the presence of charge was achieved by lysine substitution at each amino acid that resided between positions 49 and 55. All of these changes were made in the ORF65CGFP gene so that the mutants could be tested for localization to assembly sites and in the ORF65CHA gene to examine capsid assembly. Confocal examination of infected cells showed that substitution of lysine at position A54 abolished assembly site localization (Fig. 6c). Lysine substitutions at the other positions did not affect ORF65 localization. All the ORF65 mutant proteins expressed in infected cells were stably expressed as judged by immunoblot assays with anti-GFP antibodies (Fig. 6d).
Biochemical assays were also used to confirm the abrogation of the SCP–MCP interaction by substitution at A54. Sf21 cells were co-infected as described above and the lysates were immunoprecipitated with anti-GFP antibody. The precipitated proteins as well as the input proteins were detected by immunoblot (Fig. 7). ORF25 (tagged with V5) was precipitated by WT ORF65CGFP (Fig. 7, lane 1), but the amounts of precipitated protein were much lower when the ORF65 contained the A54K substitution (Fig. 7, lane 2) or the Δ50–55 deletion (Fig. 7, lane 3). The amount of ORF25 and ORF65CGFP in the lysate prior to antibody precipitation was more or less similar between the different infected cells (Fig. 7).
Fig. 7.
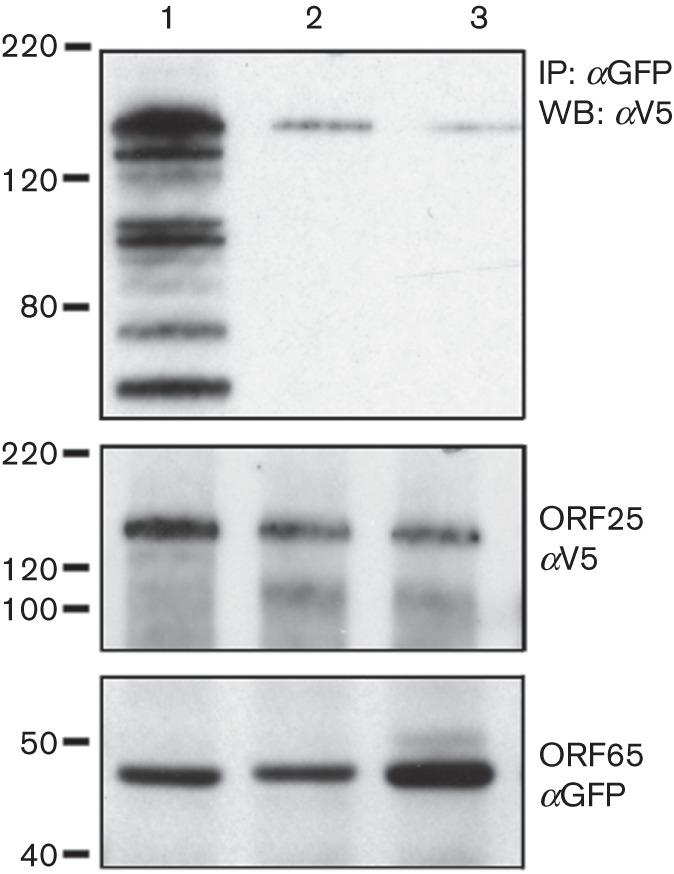
ORF65 polypeptides that contain mutations of the A54 residue do not bind to the MCP efficiently. Sf21 cells were infected as described in the legend to Fig. 1(d). The lysates were immunoprecipitated with anti-GFP antibodies, and the co-precipitated (top panel) and input proteins (two bottom panels) were revealed by immunoblot using anti-V5 (αV5) and anti-GFP (αGFP) antibodies. Lane 1, WT ORF65CGFP; lane 2, ORF65-A54KCGFP; lane 3, ORF65-Δ50–55CGFP. Molecular mass standards (kDa) are indicated on the left.
We then tested all the lysine mutants for their ability to support capsid self-assembly using proteins expressed in the ORF65CHA genetic background. This analysis revealed the importance of the hydrophobicity at additional sites, positions L49 and I53; self-assembly of capsids was affected as judged by sedimentation analysis of infected cell lysates (data not shown). When similar infected cells were examined by conventional TEM, assembled structures were not seen in cells infected with the virus expressing ORF65CHA A54K; however, some defective assemblies were evident in cells infected with viruses expressing ORF65CHA L49K or I53K (Fig. 8a). To further explore the effects of substitutions at position 54, changes that retained the hydrophobic nature of this site (M, V and L) were made. A proline was also introduced at this position. The ORF65CHA protein expressed by these mutants supported capsid self-assembly as judged by sucrose gradient sedimentation assays (Fig. 8b, data not shown for A54M). All mutant polypeptides were observed to accumulate in stable amounts in infected cells as judged by Western blotting (Fig. 8c). Two bands corresponding to the ORF65 proteins were sometimes evident in the gel, which could be due to post-translational modifications such as phosphorylation.
Fig. 8.

Substitution with polar residues at position 54 of ORF65 abolished capsid self-assembly. (a) Examination of infected cells using conventional thin-section TEM revealed the presence of aberrant structures (arrows) in cells infected with viruses expressing ORF65 L49K or I53K plus all the viruses expressing the five other capsid proteins. No capsid structures or shells were evident in cells infected with virus expressing ORF65 A54K, whereas closed capsid structures (arrowhead) were observed in cells infected with the virus expressing ORF65 WT protein. Bars, 500 nm. (b) ORF65 polypeptides containing substitution of residue 54 with L, V, P and K were tested for self-assembly as judged by sucrose gradient sedimentation of infected cell lysates. The arrow indicates direction of sedimentation. (c) Stable accumulation of ORF65 polypeptides tagged with HA was observed in infected cell lysates using immunoblot methods and anti-HA (αHA) antisera. Proteins standards (kDa) are shown in lane M.
Discussion
Using mutants of ORF65 that result in polypeptide truncation, we had previously shown that the assembly domain of ORF65 resides in the N-terminal 86 resides. Within this domain, 6 aa (R14, D18, V25, R46, G66 and R70) abolish assembly when changed to alanine, as judged by sucrose gradient sedimentation of infected cell lysates, electron micrographs of fractions and radioactive quantification of the peak capsid fractions (Kreitler et al., 2012). In this study, we have expanded on our previous findings. We have demonstrated the ability to decorate the fluorescent GFP reporter to the capsids of HSV-1 (Desai & Person, 1998), EBV (Henson et al., 2009) and now KSHV. This flexibility was surprising because the fusion of a large tag to an essential protein did not disrupt its activity; HSV-1 VP26 is not required for assembly (Chen et al., 2001; Desai et al., 1998). ORF65 tagged at the C terminus with an autofluorescent protein is functional in that it can be used to follow the protein’s relocalization to assembly sites in the nucleus and supports self-assembly of capsids in insect cells. These data support the notion that the SCP could be used to follow both the capsid and mature virion in KSHV-infected cells, similar to what we have done with the HSV-1 SCP using a fluorescent tag that supports lytic virus replication (Desai & Person, 1998). It is still possible that the large GFP tag may affect the ability of the protein to function in the infected cell (A. Gallo & W. Brune, personal communication), but other smaller fluorescent chemical moieties or different engineered GFP variants/insertions could be tested for functionality (Bosse et al., 2012; Nagel et al., 2012). The insertion of the large sequence (GFP) at the N terminus of ORF65 affects the function of the SCP in self-assembly; however, as shown by Kreitler et al. (2012), a smaller HA tag does not.
The HSV-1 SCP, by virtue of its interaction with the MCP, becomes concentrated in the nucleus (Desai et al., 2003; Rixon et al., 1996). This was apparent from the two cited works, the first of which demonstrated VP26 nuclear localization only occured when both VP5 and pre-VP22a were present in transfected cells, the second from our studies in which we genetically introduced the VP26–GFP marker into the virus with a null mutation in VP5. VP26GFP remained diffusely distributed in the cell in the absence of VP5. As shown here, this is also true for KSHV ORF65 and we have now biochemically demonstrated this interaction using binding assays which reveal a complex between the SCP–GFP, MCP and scaffold proteins. KSHV ORF65 has been shown to interact with ORF25, as well as the triplex proteins (Sathish & Yuan, 2010). Our biochemical data confirm that the SCP likely binds directly to ORF25 independently of ORF17.5 and to the scaffold proteins indirectly only in the presence of the MCP. However, the relocalization of ORF65GFP to nuclear assembly sites requires both the MCP and scaffold proteins. In HSV-1 VP26–GFP-infected cells, the nuclear puncta are indicative of assembled capsids or capsid shells; in the absence of assembly the fluorescence always remained diffuse. However, in this system although the nuclear puncta are the sites of capsid assembly, their presence cannot be used as a phenotype to test for assembly because the puncta can form with only three proteins. Rather, this phenotype is a valid indicator for the interaction between the SCP and MCP. The nuclear localization signal in the scaffold protein drives the import of the MCP–scaffold complex into the nucleus (Nicholson et al., 1994; Plafker & Gibson, 1998). There is a predicted monopartite nuclear localization signal in ORF17.5 between residues 90 and 99, but none in ORF25 or ORF65 (Kosugi et al., 2009).
Using mutagenesis experiments, a region responsible for the MCP interaction spanning aa 50–60 was mapped in ORF65. A hydrophobic patch of residues resides in the MCP interaction region – an conclusion that we reach because introduction of charge at certain sites in this region disrupts the MCP interaction and capsid self-assembly. The amino acid at position 54 of ORF65 was particularly sensitive to introduction of the polar residue. This residue could readily accommodate other hydrophobic side chains or even a proline; thus, it is not the amino acid specifically that is important, but the location of this residue, potentially in the way it makes contact with the MCP. Using protein structure software (Rost et al., 2004), an α-helix is predicted to span from aa 38 to 65, which overlaps the region that may bind with the MCP. If these predictions correlate with the actual high-resolution structural data on ORF65 then it could indicate that one of the key functional regions of this protein is a structured domain. Based on the observation that the previously identified residues of ORF65 important for assembly (Kreitler et al., 2012) do not overlap with this region, this small protein displays a multi-faceted mechanism for functional activity – an observation noted in structure and functional analyses of the human herpesvirus SCPs (Apcarian et al., 2010; Borst et al., 2001; Chaudhuri et al., 2008; Dai et al., 2013; Desai et al., 2003; Lai & Britt, 2003; Lee et al., 2008). Thus, the alphaherpesvirus SCPs still impart important functional information. HSV-1 VP26 is required for efficient production of infectious virus in the mouse neuron; this was also observed for the pseudorabies SCP in both cell culture and a neuronal system (Krautwald et al., 2008). The varicella zoster virus SCP acts as a transporter of the MCP into the nucleus and thus plays an important role in initiating capsid assembly (Chaudhuri et al., 2008). When fused with GFP, it too displays fluorescence that localizes to dense nuclear structures (Lebrun et al., 2014). Genetic deletion/manipulation of the betaherpesvirus SCP gene results in elimination of virus production and high-resolution studies suggest it may act at a step beyond capsid assembly (Borst et al., 2001; Dai et al., 2013).
The KSHV SCP differs from the alphaherpesvirus SCPs by potentially specifying an additional stabilizing function, similar to phage capsid decoration proteins, which acts as a ‘glue’ or clamp to reinforce the capsid. Two studies on lambda phage gpD and T4 phage Soc protein show that these decoration proteins stabilize the capsid shell during DNA encapsidation (lambda) or at extremes of pH/temperature (T4) (Lander et al., 2008; Qin et al., 2010). Biophysical and structural studies reveal these proteins form trimers that ‘glue’ or clamp the capsid shell, specifically capsomeres, at the threefold axis of symmetry, which is considered one of the weakest points of the capsid shell (Singh et al., 2013). Herpesviruses utilize the triplex to do this, but KSHV may also use the SCP to act as an external ‘hinge’ or protein cross-link to create a stable structure. The KSHV SCP must act at a stage earlier than the phage decoration proteins; because there was no evidence of capsid assemblies for the alanine substitution mutants that abolished assembly.
It appears that this small protein has a complex mechanism for localization to the nucleus (binding to the MCP) and subsequent stabilization of the capsid shell. The initial interaction with the MCP–scaffold complex is probably important for targeting this small protein to the assembly site and is mediated by a discrete hydrophobic domain. Subsequent to this, other residues that are dispersed throughout the assembly domain are important for facilitating the formation of a stable shell structure that can mature into a closed angular capsid. The gammaherpesvirus SCPs clearly specify separate functional sites for these activities. This is also what was observed with the HSV-1 SCP (Desai et al., 2003). Although HSV-1 VP26 is not required for capsid assembly, we used two methods to identify functional sites: (i) a similar GFP relocalization assay to nuclear assembly sites and (ii) an in vitro capsid-binding assay. In the GFP localization assay, two residues were discovered that influenced VP26–GFP localization to assembly sites, whereas in the capsid-binding assay, an expanded set of amino acids was found to be important, which included a separate C-terminal conserved domain.
Based on this and our other recent studies that show the KSHV SCP is required for assembly of the capsid shell, we conclude that the gammaherpesvirus SCP is an important mediator of stable capsid shell assembly and thus a valid antiviral target. Therefore, a potential practical outcome of this study is the identification of a new antiviral target for gammaherpesvirus lytic replication. Another potentially useful outcome of this study has been the discovery that we can fuse a large polypeptide (GFP) to ORF65, and get an assembled structure and the ability to purify the capsid from a crude lysate using immobilized metal affinity chromatography (IMAC) methods. The latter observation will be particularly useful to purify capsids and subassemblies containing ORF65 as we proceed in our investigation of ORF65 function. The former could be useful to display complex peptides or polypeptides that are potential vaccine candidates, as has been done with phage capsids (Chackerian, 2007; Li et al., 2006). The high occupancy of ORF65 on the capsid surface and thus high density of a candidate vaccine molecule could be useful in generating immunological responses that target many viral and microbial diseases.
Methods
Cell lines and antibodies.
Spodoptera frugiperda (Sf9 and Sf21) cells were grown in Grace’s insect cell medium, supplemented with 10 % FCS (Gibco-Invitrogen) and passaged as described in Okoye et al. (2006). A rat mAb to influenza HA was purchased from Roche (clone 3F10), mouse V5 (R960) and rabbit GFP (A11122) antibodies from Invitrogen, and mouse histidine tag antibodies from Novagen (70796-3) and Invitrogen (P-21315). The Ni-NTA-derivatized gold was purchased from Nanoprobes. The mAb to ORF65 was provided generously by S. J. Gao (University of Southern California, CA, USA).
Plasmids.
ORF65 was cloned previously into the baculovirus transfer vector pFastBac1 (pFB1) as a SpeI–HindIII fragment (Perkins et al., 2008). We used the same cloning strategy to generate ORF65 genes with C-terminal fusions to the HA, V5 and GFP tags. For the HA and V5 fusions, the reverse primer specified the HindIII site and the HA/V5 epitope nucleotide sequences (Table 1). For the C-terminal GFP fusion, ORF65 was amplified as a SpeI–HindIII fragment without the amber stop codon and cloned into pFB1, and designated pFB1-ORF65amb−. The EGFP ORF was PCR-amplified from pEGFP-N2 (Clontech) as a HindIII fragment and cloned into pFB1-ORF65amb− in the correct orientation. The resulting plasmids were named pFB1-ORF65CHA, pFB1-ORF65CV5 and pFB1-ORF65CGFP. To clone the KSHV capsid ORFs into the baculovirus transfer vector pFastBac Dual (pFBDual; Invitrogen), we cloned ORF25/17.5, ORF26/62 and ORF17/65 as pairs. ORF17.5, ORF26 and ORF65 were PCR-amplified as XhoI–KpnI fragments (Table 1). The templates for these capsid ORF amplifications were sequence-confirmed plasmids containing the genes cloned into pFB1 (Perkins et al., 2008). ORF25, ORF62 and ORF17 were cloned as EcoRI–SpeI, EcoRI–HindIII and EcoRI–HindIII fragments, respectively, derived from pFB1 cloned copies (Perkins et al., 2008). To facilitate cloning among the various ORF65 plasmids, the EcoRI recognition site (at 200 bp) in ORF65 was removed, via a silent mutation, from plasmid pFB1-ORF65CHA (Table 1). This was done by QuikChange mutagenesis (see below) and the resulting plasmid was abbreviated pFB1-ORF65CHA-ΔEcoRI. PCR methods were also used to insert a His66 sequence in-frame with the C terminus of ORF65. Plasmid pFB1-ORF65CHA-ΔEcoRI was used as the template and reverse primer specified the His tag sequence (Table 1). PCR-amplified fragments were cloned as EcoRI–SpeI or XhoI–KpnI fragments into pFB1 or pFBDual, respectively. ORF25 and ORF17.5 were cloned into a modified pFastBac1 to generate a V5 epitope sequence at the N terminus of the cloned gene. The V5 sequence was cloned using annealed oligonucleotides into the BamHI and EcoRI sites of pFastBac1. The ORF25 and ORF17.5 genes were cloned as EcoRI–SpeI or EcoRI–HindIII fragments, respectively (Perkins et al., 2008). The PCR primers used to amplify the above genes are listed in Table 1 and the published sequence of KSHV strain BAC36 (Yakushko et al., 2011) was used to design each primer pair. All PCR assays used either PfuUltra (Stratagene) or Phusion polymerase (Finnzyme-NEB). The cloned genes were sequenced to check for authentic amplification. Confirmed plasmids were designated by the transfer plasmid abbreviation followed by the gene name, e.g. pFBD-ORF25/17.5.
Table 1. Primer list.
| Primer | Sequence |
| ORF65-SpeI-F | GGACTAGTACCATGTCCAACTTTAAGGTGAGAGAC |
| ORF65-Hind3amb-R | GGAAGCTTTTTCTTTTTGCCAGAGGGGGG |
| ORF65-CHA-Hind3-R | GGAAGCTTCTATGCGTAGTCCGGAACGTCGTATGGGTATTTCTTTTTGCCAGAGGGGGGTTTCCTCGC |
| ORF65-CV5-Hind3-R | GGAAGCTTCTACGTAGAATCGAGACCGAGGAGAGGGTTAGGGATAGGCTTACCTTTCTTTTTGCCAGAGGGGGGTTTCCTCGC |
| EGFP-Hind3-F | GGAAGCTTATGGTGAGCAAGGGCGAGGAGCTG |
| EGFP-Hind3-R | GGAAGCTTCTACAGCTCGTCCATGCCGAGAGTGATC |
| ORF65ΔEcoRI-F | CTGAGGAGGATGGGTGGGATTCAAAGGCGGGAT |
| OR65-EcoRI-F | GGAATTCACCATGTCCAACTTTAAGGTGAGAGAC |
| ORF65-SpeIamb-R | GGACTAGTTTTCTTTTTGCCAGAGGGGGG |
| ORF65C6XHIS-SpeI-R | CACTAGTCTAGTGGTGGTGGTGGTGGTGACTGCCTTTCTTTTTGCCAGAGGGGGGTTTC |
| pFastBac Dual primers | |
| ORF26-XhoI-F | GCTCGAGACCATGGCACTCGACAAGAGTATAGTG |
| ORF26-KpnI-R | GGGGTACCTTAGCGTGGGGAATACCAACAGGAG |
| ORF17.5-XhoI-F | GCTCGAGACCATGAACAGCTCTGGTCAAGAGG |
| ORF17.5-KpnI-R | GGGGTACCCTACTGCTTGTTCAGGAGCTCCTC |
| ORF65-XhoI-F | GGCTCGAGACCATGTCCAACTTTAAGGTGAGAGAC |
| ORF65-KpnI-R | GGGGTACCCTATTTCTTTTTGCCAGAGGGGGG |
| ORF65C6XHIS-KpnI-R | GGGGTACCCTAATGGTGATGGTGATGGTGTCCTCCGCCTTTCTTTTTGCCAGAGGGGGGTTTCC |
Truncations.
ORF65 truncations were cloned into pFB1-CEGFP (Desai et al., 2012) as EcoRI–SpeI fragments. These truncations were made with PCR assays using the pFB1-ORF65CHA-ΔEcoRI template. The DNA fragments were made with a forward primer that specified an EcoRI site (Table 1), and a reverse primer that specified the truncation and a SpeI site, but lacked the amber stop codon.
Mutagenesis.
Site-directed mutants were created using either QuikChange mutagenesis (Stratagene) or cassette mutagenesis. Both methods involved PCR assays using the pFB1-ORF65CHA-ΔEcoRI template. For the QuikChange mutagenesis procedure, complementary primers containing 15–20 nt on either side of the mutation were made. To aid with clone identification, a silent mutation was used to introduce a diagnostic, restriction enzyme recognition site into the mutant ORF65 gene. Following PCR assays, the DNA was digested with DpnI and transformed into Escherichia coli according to the manufacturer’s protocol. Positive clones were isolated, and proper introduction of the site-directed mutation was confirmed by diagnostic enzyme cleavage and ultimately by sequence analysis. ORF65 QuikChange mutants were moved from the pFB1-ORF65CHA-ΔEcoRI plasmid into the pFB1-ORF65CGFP vector via a SpeI–Bsu36I restriction digest and subsequent ligation.
Recombinant baculoviruses production.
We used the Bac-to-Bac system from Invitrogen and the E. coli strain DH10BAC, using both the manufacturer’s protocol (Invitrogen) and modifications described by Okoye et al. (2006) to generate recombinant baculoviruses. The Bacmid DNA was transfected into Sf9 cells and viruses were amplified in the same cell type (Okoye et al., 2006; Perkins et al., 2008).
Co-immunoprecipitation assays.
Sf21 cells (2×106) were co-infected with baculoviruses expressing ORF65CGFP, ORF25NV5 and ORF17.5NV5. The infected cells were harvested 48 h after infection, lysed in 650 µl 1× capsid lysis buffer (CLB) buffer (Walters et al., 2003) plus 1 mM arginine and HALT protease inhibitor (Pierce), and incubated on ice for 15 min. The lysates were clarified in a refrigerated microcentrifuge at 18 000 g for 30 min. The soluble lysate was precleared using 50 µl Protein A/G beads (Santa Cruz) (30 min at room temperature) and then 200 µl cleared lysate added to 50 µl of Protein A Sepharose (Sigma) slurry. This was incubated for 1 h at room temperature on a rotator and the beads then washed with RIPA buffer (Desai et al., 2012) five times for 5 min each time. The final beads were resuspended in 2× Laemmli buffer and analysed by immunoblot methods.
Western blot analysis.
Sf21 cells were seeded in 12-well trays (1×106 cells per well) and, for each well, 100 µl baculovirus was added. Infected cells were harvested 48 h post-infection, pelleted and resuspended in RIPA buffer. The cell lysate was then clarified at 18 000 g for 30 min. The resulting supernatant was resolved by SDS-PAGE using the NuPAGE Bis-Tris gel system (Invitrogen) and transferred to a nitrocellulose membrane with an iBlot dry transfer apparatus. Transfer procedures were carried out according to the manufacturer’s protocol (Invitrogen). Western blots were carried out using primary antibody incubations for 60 min and anti-rat HRP secondary antibody incubations for 60 min combined with Amersham ECL Western blotting detection reagents (GE Healthcare) using the protocol provided by the manufacturer. Primary and secondary antibody dilutions were usually 1 : 5000 or 1 : 10 000.
Sedimentation analysis.
Sf21 cells (1.2×107 cells) in a 100 mm Petri dish were infected with 300 µl of each of vFB-ORF17, vFBD-ORF17.5/25 and vFBD-ORF26/62 baculovirus P3 stocks along with 300 µl of the mutant ORF65 baculovirus P2 stock. At 68–72 h after infection the cells were harvested and processed for sedimentation as described by Perkins et al. (2008). Capsids were harvested by side-puncture aspiration after visualizing the light-scattering band using incident light.
Confocal microscopy.
Sf21 cells seeded in a 35 mm FluoroDish (WPI) (1×106 cells) were infected with 50 µl of each baculovirus. The infected cells were analysed in a Zeiss LSM 510 confocal microscope 48 h after infection as described by Henson et al. (2009).
Electron microscopy.
Sf21 cells were used for all TEM experiments. Infected cells were processed for TEM at 68–72 h after infection. Infected cells were processed for conventional thin-section TEM as described by Huang et al. (2007) and Perkins & McCaffery (2007). Negative-stained capsids were prepared as described by Perkins et al. (2008) using 1 % phosphotungstic acid as the staining solution. Samples were examined using Phillips EM 420 and Tecnai 12 transmission electron microscopes (FEI); images were obtained with an SIS Megaview III camera (Olympus).
IMAC purification of capsids.
Infected cells (2.4×107 cells) were lysed in 1 ml 2× CLB buffer (minus EDTA) and sonicated for 30 s. Capsids were purified from: a sucrose gradient fraction containing capsids (see above); lysates clarified using a 20 % sucrose cushion and resuspended in 500 µl 2× CLB [500 µl lysate layered over 500 µl 20 % w/v sucrose (500 mM NaCl and 10 mM Tris/HCl, pH 7.5) and pelleted in a microcentrifuge for 60 min at 14 000 g] or total soluble lysate. Binding of 500 µl of each type of lysate (containing 20 mM imidazole) with 100 µl Ni-NTA agarose slurry (Qiagen, equilibrated in PBS) was done for 60 min at room temperature with rotation. The beads were washed with 500 µl native wash buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0) five times (5 min rotation during wash) and capsids were eluted by incubation with 100 µl elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, pH 8.0) for 30–60 min at room temperature in an Eppendorf shaker. All buffers and washes contained HALT protease inhibitor cocktail (EDTA free) (Pierce).
Gold labelling of capsids using Ni-NTA-derivatized gold.
Formvar and carbon-coated grids were freshly glow discharged, and 10 μl capsids in solution was adsorbed onto the grids for ~15 min. Grid loops were used to float grids on four drops of room temperature wash buffer (2.5 % FBS in PBS) and left for 5 min on the last drop. Grids were incubated in a humid chamber for 30 min in 10 μl 1 : 20 solution of 5 nm Ni-NTA-Nanogold (Nanoprobes) in wash buffer. Grid loops were used to float the grids through eight drops of wash buffer and then through eight drops of double-distilled H2O. The grids were then stained in 1 % phosphotungstic acid for 30 s, excess stain was wicked off and they were allowed to dry before imaging.
Data and figure preparation.
Scanned autoradiographs and digital electron micrographs were imported into Adobe Photoshop for figure compilation. Confocal images were converted to TIFFs using ImageJ software and imported into Photoshop for figure compilation. Files were adjusted for brightness and contrast, and trimmed for final figure versions.
Acknowledgements
We thank Brandon Henson for technical help with cloning and virus preparations. Funding for this research was provided by Public Health Service grants from the National Institutes of Health (R21 AI107530 and R21 AI097912) and ARRA funding in support of summer students AI061382-S4 (P. D.). Additional funding for shared equipment was provided by NIH-NCRR 1S10RR023454-01 (shared instrumentation grant for the Tecnai 12 G2 Spirit transmission electron microscope) and NIH-NCI 1U54CA143868-01 (Physical Sciences of Cancer Grant) (M. J. M.). KSHV BAC36 and ORF65 antibody were provided generously by S. J. Gao (University of Southern California, CA, USA).
References
- Adamson W. E., McNab D., Preston V. G., Rixon F. J. (2006). Mutational analysis of the herpes simplex virus triplex protein VP19C. J Virol 80, 1537–1548 10.1128/JVI.80.3.1537-1548.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinone S. E., Shubeita G. T., Coller K. E., Lee J. I., Haverlock-Moyns S., Gross S. P., Smith G. A. (2006). The herpesvirus capsid surface protein, VP26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J Virol 80, 5494–5498 10.1128/JVI.00026-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apcarian A., Cunningham A. L., Diefenbach R. J. (2010). Identification of binding domains in the herpes simplex virus type 1 small capsid protein pUL35 (VP26). J Gen Virol 91, 2659–2663 10.1099/vir.0.019984-0 [DOI] [PubMed] [Google Scholar]
- Borst E. M., Mathys S., Wagner M., Muranyi W., Messerle M. (2001). Genetic evidence of an essential role for cytomegalovirus small capsid protein in viral growth. J Virol 75, 1450–1458 10.1128/JVI.75.3.1450-1458.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse J. B., Bauerfeind R., Popilka L., Marcinowski L., Taeglich M., Jung C., Striebinger H., von Einem J., Gaul U. & other authors (2012). A beta-herpesvirus with fluorescent capsids to study transport in living cells. PLoS ONE 7, e40585 10.1371/journal.pone.0040585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. C., Newcomb W. W. (2011). Herpesvirus capsid assembly: insights from structural analysis. Curr Opin Virol 1, 142–149 10.1016/j.coviro.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone G., Heymann J. B., Cheng N., Trus B. L., Steven A. C. (2012). Procapsid assembly, maturation, nuclear exit: dynamic steps in the production of infectious herpesvirions. Adv Exp Med Biol 726, 423–439 10.1007/978-1-4614-0980-9_19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaday R. J., Bailey J. R., Kalb S. R., Brignole E. J., Loveland A. N., Cotter R. J., Gibson W. (2004). Assembly protein precursor (pUL80.5 homolog) of simian cytomegalovirus is phosphorylated at a glycogen synthase kinase 3 site and its downstream “priming” site: phosphorylation affects interactions of protein with itself and with major capsid protein. J Virol 78, 13501–13511 10.1128/JVI.78.24.13501-13511.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chackerian B. (2007). Virus-like particles: flexible platforms for vaccine development. Expert Rev Vaccines 6, 381–390 10.1586/14760584.6.3.381 [DOI] [PubMed] [Google Scholar]
- Chaudhuri V., Sommer M., Rajamani J., Zerboni L., Arvin A. M. (2008). Functions of varicella-zoster virus ORF23 capsid protein in viral replication and the pathogenesis of skin infection. J Virol 82, 10231–10246 10.1128/JVI.01890-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. H., Jakana J., McNab D., Mitchell J., Zhou Z. H., Dougherty M., Chiu W., Rixon F. J. (2001). The pattern of tegument–capsid interaction in the herpes simplex virus type 1 virion is not influenced by the small hexon-associated protein VP26. J Virol 75, 11863–11867 10.1128/JVI.75.23.11863-11867.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X., Yu X., Gong H., Jiang X., Abenes G., Liu H., Shivakoti S., Britt W. J., Zhu H. & other authors (2013). The smallest capsid protein mediates binding of the essential tegument protein pp150 to stabilize DNA-containing capsids in human cytomegalovirus. PLoS Pathog 9, e1003525 10.1371/journal.ppat.1003525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P., Person S. (1998). Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J Virol 72, 7563–7568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P., DeLuca N. A., Person S. (1998). Herpes simplex virus type 1 VP26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology 247, 115–124 10.1006/viro.1998.9230 [DOI] [PubMed] [Google Scholar]
- Desai P., Akpa J. C., Person S. (2003). Residues of VP26 of herpes simplex virus type 1 that are required for its interaction with capsids. J Virol 77, 391–404 10.1128/JVI.77.1.391-404.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P. J., Pryce E. N., Henson B. W., Luitweiler E. M., Cothran J. (2012). Reconstitution of the Kaposi’s sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J Virol 86, 594–598 10.1128/JVI.05988-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S. J., Deng J. H., Zhou F. C. (2003). Productive lytic replication of a recombinant Kaposi’s sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J Virol 77, 9738–9749 10.1128/JVI.77.18.9738-9749.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson B. W., Perkins E. M., Cothran J. E., Desai P. (2009). Self-assembly of Epstein–Barr virus capsids. J Virol 83, 3877–3890 10.1128/JVI.01733-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homa F. L., Brown J. C. (1997). Capsid assembly and DNA packaging in herpes simplex virus. Rev Med Virol 7, 107–122 [DOI] [PubMed] [Google Scholar]
- Huang E., Perkins E. M., Desai P. (2007). Structural features of the scaffold interaction domain at the N terminus of the major capsid protein (VP5) of herpes simplex virus type 1. J Virol 81, 9396–9407 10.1128/JVI.00986-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S., Hasebe M., Tomita M., Yanagawa H. (2009). Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc Natl Acad Sci U S A 106, 10171–10176 10.1073/pnas.0900604106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krautwald M., Maresch C., Klupp B. G., Fuchs W., Mettenleiter T. C. (2008). Deletion or green fluorescent protein tagging of the pUL35 capsid component of pseudorabies virus impairs virus replication in cell culture and neuroinvasion in mice. J Gen Virol 89, 1346–1351 10.1099/vir.0.83652-0 [DOI] [PubMed] [Google Scholar]
- Kreitler D., Capuano C. M., Henson B. W., Pryce E. N., Anacker D., McCaffery J. M., Desai P. J. (2012). The assembly domain of the small capsid protein of Kaposi’s sarcoma-associated herpesvirus. J Virol 86, 11926–11930 10.1128/JVI.01430-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L., Britt W. J. (2003). The interaction between the major capsid protein and the smallest capsid protein of human cytomegalovirus is dependent on two linear sequences in the smallest capsid protein. J Virol 77, 2730–2735 10.1128/JVI.77.4.2730-2735.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander G. C., Evilevitch A., Jeembaeva M., Potter C. S., Carragher B., Johnson J. E. (2008). Bacteriophage lambda stabilization by auxiliary protein gpD: timing, location, and mechanism of attachment determined by cryo-EM. Structure 16, 1399–1406 10.1016/j.str.2008.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun M., Thelen N., Thiry M., Riva L., Ote I., Condé C., Vandevenne P., Di Valentin E., Bontems S., Sadzot-Delvaux C. (2014). Varicella-zoster virus induces the formation of dynamic nuclear capsid aggregates. Virology 454–455, 311–327 10.1016/j.virol.2014.02.023 [DOI] [PubMed] [Google Scholar]
- Lee J. H., Vittone V., Diefenbach E., Cunningham A. L., Diefenbach R. J. (2008). Identification of structural protein–protein interactions of herpes simplex virus type 1. Virology 378, 347–354 10.1016/j.virol.2008.05.035 [DOI] [PubMed] [Google Scholar]
- Li Q., Shivachandra S. B., Leppla S. H., Rao V. B. (2006). Bacteriophage T4 capsid: a unique platform for efficient surface assembly of macromolecular complexes. J Mol Biol 363, 577–588 10.1016/j.jmb.2006.08.049 [DOI] [PubMed] [Google Scholar]
- Lo P., Yu X., Atanasov I., Chandran B., Zhou Z. H. (2003). Three-dimensional localization of pORF65 in Kaposi’s sarcoma-associated herpesvirus capsid. J Virol 77, 4291–4297 10.1128/JVI.77.7.4291-4297.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel C. H., Döhner K., Binz A., Bauerfeind R., Sodeik B. (2012). Improper tagging of the non-essential small capsid protein VP26 impairs nuclear capsid egress of herpes simplex virus. PLoS ONE 7, e44177 10.1371/journal.pone.0044177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nealon K., Newcomb W. W., Pray T. R., Craik C. S., Brown J. C., Kedes D. H. (2001). Lytic replication of Kaposi’s sarcoma-associated herpesvirus results in the formation of multiple capsid species: isolation and molecular characterization of A, B, and C capsids from a gammaherpesvirus. J Virol 75, 2866–2878 10.1128/JVI.75.6.2866-2878.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W. W., Homa F. L., Thomsen D. R., Booy F. P., Trus B. L., Steven A. C., Spencer J. V., Brown J. C. (1996). Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid formation. J Mol Biol 263, 432–446 10.1006/jmbi.1996.0587 [DOI] [PubMed] [Google Scholar]
- Newcomb W. W., Homa F. L., Thomsen D. R., Trus B. L., Cheng N., Steven A., Booy F., Brown J. C. (1999). Assembly of the herpes simplex virus procapsid from purified components and identification of small complexes containing the major capsid and scaffolding proteins. J Virol 73, 4239–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson P., Addison C., Cross A. M., Kennard J., Preston V. G., Rixon F. J. (1994). Localization of the herpes simplex virus type 1 major capsid protein VP5 to the cell nucleus requires the abundant scaffolding protein VP22a. J Gen Virol 75, 1091–1099 10.1099/0022-1317-75-5-1091 [DOI] [PubMed] [Google Scholar]
- Okoye M. E., Sexton G. L., Huang E., McCaffery J. M., Desai P. (2006). Functional analysis of the triplex proteins (VP19C and VP23) of herpes simplex virus type 1. J Virol 80, 929–940 10.1128/JVI.80.2.929-940.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins E. M., McCaffery J. M. (2007). Conventional and immunoelectron microscopy of mitochondria. Methods Mol Biol 372, 467–483 10.1007/978-1-59745-365-3_33 [DOI] [PubMed] [Google Scholar]
- Perkins E. M., Anacker D., Davis A., Sankar V., Ambinder R. F., Desai P. (2008). Small capsid protein pORF65 is essential for assembly of Kaposi’s sarcoma-associated herpesvirus capsids. J Virol 82, 7201–7211 10.1128/JVI.00423-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plafker S. M., Gibson W. (1998). Cytomegalovirus assembly protein precursor and proteinase precursor contain two nuclear localization signals that mediate their own nuclear translocation and that of the major capsid protein. J Virol 72, 7722–7732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L., Fokine A., O’Donnell E., Rao V. B., Rossmann M. G. (2010). Structure of the small outer capsid protein, Soc: a clamp for stabilizing capsids of T4-like phages. J Mol Biol 395, 728–741 10.1016/j.jmb.2009.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rixon F. J. (1993). Structure and assembly of herpesviruses. Semin Virol 4, 135–144 10.1006/smvy.1993.1009 [DOI] [Google Scholar]
- Rixon F. J., Addison C., McGregor A., Macnab S. J., Nicholson P., Preston V. G., Tatman J. D. (1996). Multiple interactions control the intracellular localization of the herpes simplex virus type 1 capsid proteins. J Gen Virol 77, 2251–2260 10.1099/0022-1317-77-9-2251 [DOI] [PubMed] [Google Scholar]
- Rost B., Yachdav G., Liu J. (2004). The PredictProtein server. Nucleic Acids Res 32 (Web Server), W321–W326 10.1093/nar/gkh377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathish N., Yuan Y. (2010). Functional characterization of Kaposi’s sarcoma-associated herpesvirus small capsid protein by bacterial artificial chromosome-based mutagenesis. Virology 407, 306–318 10.1016/j.virol.2010.08.017 [DOI] [PubMed] [Google Scholar]
- Singh P., Nakatani E., Goodlett D. R., Catalano C. E. (2013). A pseudo-atomic model for the capsid shell of bacteriophage lambda using chemical cross-linking/mass spectrometry and molecular modeling. J Mol Biol 425, 3378–3388 10.1016/j.jmb.2013.06.026 [DOI] [PubMed] [Google Scholar]
- Spencer J. V., Newcomb W. W., Thomsen D. R., Homa F. L., Brown J. C. (1998). Assembly of the herpes simplex virus capsid: preformed triplexes bind to the nascent capsid. J Virol 72, 3944–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven A. C., Spear P. G. (1996). Herpesvirus capsid assembly and envelopment. In Structural Biology of Viruses, pp. 312–351 Edited by Burnet R., Chiu W., Garcea R. New York: Oxford University Press [Google Scholar]
- Tatman J. D., Preston V. G., Nicholson P., Elliott R. M., Rixon F. J. (1994). Assembly of herpes simplex virus type 1 capsids using a panel of recombinant baculoviruses. J Gen Virol 75, 1101–1113 10.1099/0022-1317-75-5-1101 [DOI] [PubMed] [Google Scholar]
- Thomsen D. R., Roof L. L., Homa F. L. (1994). Assembly of herpes simplex virus (HSV) intermediate capsids in insect cells infected with recombinant baculoviruses expressing HSV capsid proteins. J Virol 68, 2442–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trus B. L., Heymann J. B., Nealon K., Cheng N., Newcomb W. W., Brown J. C., Kedes D. H., Steven A. C. (2001). Capsid structure of Kaposi’s sarcoma-associated herpesvirus, a gammaherpesvirus, compared to those of an alphaherpesvirus, herpes simplex virus type 1, and a betaherpesvirus, cytomegalovirus. J Virol 75, 2879–2890 10.1128/JVI.75.6.2879-2890.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters J. N., Sexton G. L., McCaffery J. M., Desai P. (2003). Mutation of single hydrophobic residue I27, L35, F39, L58, L65, L67, or L71 in the N terminus of VP5 abolishes interaction with the scaffold protein and prevents closure of herpes simplex virus type 1 capsid shells. J Virol 77, 4043–4059 10.1128/JVI.77.7.4043-4059.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildy P., Russell W. C., Horne R. W. (1960). The morphology of herpes virus. Virology 12, 204–222 10.1016/0042-6822(60)90195-1 [DOI] [PubMed] [Google Scholar]
- Wingfield P. T., Stahl S. J., Thomsen D. R., Homa F. L., Booy F. P., Trus B. L., Steven A. C. (1997). Hexon-only binding of VP26 reflects differences between the hexon and penton conformations of VP5, the major capsid protein of herpes simplex virus. J Virol 71, 8955–8961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakushko Y., Hackmann C., Günther T., Rückert J., Henke M., Koste L., Alkharsah K., Bohne J., Grundhoff A. & other authors (2011). Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome contains a duplication of a long unique-region fragment within the terminal repeat region. J Virol 85, 4612–4617 10.1128/JVI.02068-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z. H., He J., Jakana J., Tatman J. D., Rixon F. J., Chiu W. (1995). Assembly of VP26 in herpes simplex virus-1 inferred from structures of wild-type and recombinant capsids. Nat Struct Biol 2, 1026–1030 10.1038/nsb1195-1026 [DOI] [PubMed] [Google Scholar]
- Zhou F. C., Zhang Y. J., Deng J. H., Wang X. P., Pan H. Y., Hettler E., Gao S. J. (2002). Efficient infection by a recombinant Kaposi’s sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J Virol 76, 6185–6196 10.1128/JVI.76.12.6185-6196.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]