

Abstract
Toll-like receptor (TLR)-8 agonists activate adaptive immune responses by inducing robust production of T helper 1-polarizing cytokines, suggesting that TLR8-active compounds may be promising candidate adjuvants. We recently reported pure TLR8 agonistic activity in a C2-butyl furo[2,3-c]quinoline. We have obtained the structure of human TLR8 ectodomain co-crystallized with the furoquinoline compound, which indicates ligand-induced reorganization of the binding pocket of TLR8. The loss of a key H-bond between the oxygen atom of the furanyl ring of the agonist and Thr574 in TLR8 suggested that the furan ring was dispensable. We employed a disconnection strategy and examined 3- and 4-substituted aminoquinolines. Focused structure-based ligand design studies led to the identification of 3-pentyl-quinoline-2-amine as a novel, structurally simple, and highly potent human TLR8-specific agonist.
Keywords: Vaccines, Adjuvant, Innate immunity, TLR8, Aminoquinolines
The immune system protects the host from infectious agents by first recognizing the presence of the infectious organism and then responding rapidly and appropriately to contain and eliminate the threat. The mobilization of adaptive immune responses involving T-, and B-lymphocytic effector functions are exquisitely pathogen-specific, but are relatively slow, requiring days or weeks.[1] The enormous diversity of infectious organisms and their short generation times led Janeway to postulate that “the immune system has evolved specifically to recognize and respond to infectious microorganisms, and that this involves recognition not only of specific antigenic determinants, but also of certain characteristics or patterns common on infectious agents but absent from the host.”[2]
Janeway's seminal ideas of nonclonal recognition of nonself by “Pattern Recognition Receptors” (PRRs) served as the foundation for the discovery of the sensors of the innate immune system.[3] Among the well-studied of such PRRs initiating innate immune afferent signals include the Toll-like receptors (TLRs),[4] of which there are 10 functional TLRs encoded in the human genome. TLRs -3, -7, -8 and -9 function within the endolysosomal compartment.[5] The activation of TLRs by their cognate ligands leads to production of inflammatory cytokines, and up-regulation of major histocompatibility complex molecules and co-stimulatory signals in antigen-presenting cells as well as activating natural killer cells (innate immune response).[6] These events lead to the priming of naïve lymphocytes and subsequent induction and amplification of antigen-specific T-, and B-cell effector functions (adaptive immune responses).[7]
Our recent efforts on evaluating small molecule agonists of TLR8[8] are primarily aimed at examining such compounds as potential vaccine adjuvants. TLR8 is expressed in myeloid dendritic cells, monocytes, and monocyte-derived dendritic cells. Engagement by TLR8 agonists evokes a dominant proinflammatory cytokine profile including tumor necrosis factor-α (TNF-α, interleukin-12 (IL-12), and IL-18,[9] and appear unique in markedly upregulating the production of Th1-polarizing cytokines TNF-α and IL-12 in neonatal antigen presenting cells.[10] These data, taken together, suggest that TLR8 agonists may be useful as adjuvants for enhancing immune responses in newborns.[11]
A prerequisite for the careful evaluation of TLR8 agonists as potential vaccines adjuvants is the characterization of pure TLR8 agonists with negligible TLR7 activity, for almost all known agonists of TLR8 typified by certain imidazoquinolines such as CL097[12] (2, Fig. 1), and thiazoloquinolines such as CL075[13] (1, Fig. 1), and the 2-aminobenzazepine VTX-2337[14] display mixed TLR7/TLR8-agonism. TLR8-biased agonistic properties have been described for a novel 2-aminobenzazepine derivative (VTX-294),[15] whose complete structure has not been disclosed. We recently reported pure TLR8 agonistic activity in a C2-butyl furo[2,3-c]quinoline 3 (Fig. 1) with IL-12 and IL-18 induction profiles, and yet without IFN-α inducing properties, confirming its selectivity for human TLR8.[8a] Crystal structures of the ectodomain of human TLR8 complexed with mixed TLR7/TLR8-agonistic thiazoloquinolines and imidazoquinolines (including 1 and 2)[16] had allowed a rationalization of our experimentally-determined SAR via induced-fit docking techniques.[8a]
Figure 1.
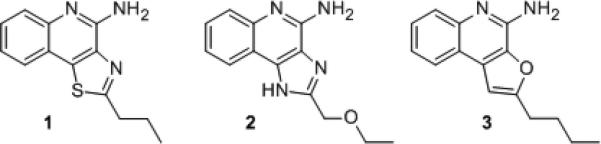
Representative heterocyclic small molecules with TLR8 agonistic activity.
The thiazoloquinoline 1 as well as the furoquinoline 3 were predicted to occupy the same binding pocket formed by both the TLR8 protomers with the binding geometry of the ligands and interacting residues being virtually identical; ionic H-bonds were observed between the C4-amine of both 1 and 3 with the sidechain carboxylate of Asp543 of TLR8, with additional stabilization derived from an H-bond between the β-OH group of Thr574 and either the N2 atom of the thiazole ring of 1 or the oxygen atom of the furanyl ring of 3. Key ππ interactions of the quinoline moieties of 1 and 3 (Phe405/Tyr353), as well as hydrophobic interactions of the C2-alkyl group (Phe346/Ile403/Tyr348) were also observed to occur (Fig. 2).
Figure 2.

Induced-fit docking[8a] of 3 in human TLR8 (PDB ID: 3W3K) showing an salt bridge between the C4-amine and D543*, and an H-bond between the furanyl oxygen atom and T574. Interacting residues in protomers A and B (*) are highlighted in green and cyan, respectively.
Recognizing limitations inherent in docking methods,[17] and cognizant of the crystallographically determined observation of large structural excursions (15 Å) of the top lateral face of TLR8 upon ligand binding,[16] we sought to directly verify and validate our docking results. We were delighted to obtain high-resolution (1.8 Ǻ) structure of human TLR8 co-crystallized with 3 (Fig. S1, Table S1). An examination of the complex confirmed similar binding geometries of 2 and 3 (Fig. S2). The occupancy of TLR8 with 3 induced, as expected, a significant reorganization to form the binding pocket, reflected in significant Cα deviations (Fig. 3A) corresponding primarily to loops of leucine-rich repeats (LRRs) (Fig. 3B). However, the occupancy of 3 in TLR8 is associated with greater excursions of LRR8 and, particularly, of residues 572-574 in LRR18 such that predicted H-bond between Thr574 and the oxygen atom of the furanyl ring of 3 is lost in the crystal structure of the complex (Fig. 3C). This led us to hypothesize that the furan ring in 3 was dispensable, and we envisaged simpler 3- and 4-substituted aminoquinolines via classic disconnection strategies[18] as shown schematically in Fig. 4. We first targeted the 3-alkoxy-2-aminoquinoline series derived by disconnection at C1 in 3. The 3-butoxy analogue 6 was synthesized from commercially-available 3-hydroxy quinoline via O-alkylation and installation of the amine at C2 using reported methods (Scheme 1).[8a, 13b, 19] A homologous series of compounds were also synthesized (Scheme S1). As in other chemotypes that we had previously explored,[8a, 13b, 19b, 20] and consistent with the dimensions of the binding pocket in TLR8,[16] we observed in this homologous series a clear dependence of substituent chain length at C3 with the optimal analogue being 6, which showed maximal agonistic potency in a cell-based TLR8-specific NF-κB transactivation assay (EC50 of 2.2 μM; Figs. 5, S3, Table 1). We reasoned that the electronegativity of the heteroatom at C3 may differentially modulate electron density of the quinoline ring and thus affect both the strength of the salt-bridge of the C2-amine with Asp543, as well as ππ interactions with Phe405. We therefore synthesized N3- butylquinoline and 3-(butylthio)quinoline analogues (9 and 12, respectively), as well as the 3-alkylquinolin-2-amines 14a-f (Scheme 1). Accessing the N3-butylquinoline 9 by conventional strategies via N-oxidation of the commercially available 3-aminoquinoline was problematic, and we found it expedient to utilize as starting material 2-chloro-3-azidoquinoline 7 (derived from commercially available 2-chloro-3-quinolineboronic acid, see Supporting Information). S-alkylation of 3-bromoquinoline 1-oxide obviated the problem of over-oxidation to the sulfone derivative (which was found to be completely inactive) in the synthesis of the 3-(butylthio)quinoline 12.
Figure 3.

A. Cα deviation in TLR8 bound to 3 versus unliganded TLR8. B. Regions undergoing ligand-induced Cα movements of more than 2.5 Ǻ are shown in red for the TLR8 protomers. C. TLR8 (protomers A and B represented in green and cyan, respectively) complexed with 3 showing the loss of H-bond of the furanyl oxygen atom due to reorganization of residues in the binding pocket (PDB code: 3WN4).
Figure 4.
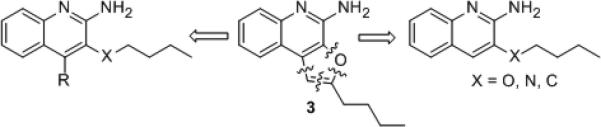
Disconnection strategy of 3 leading to substituted aminoquinolines.
Scheme 1.
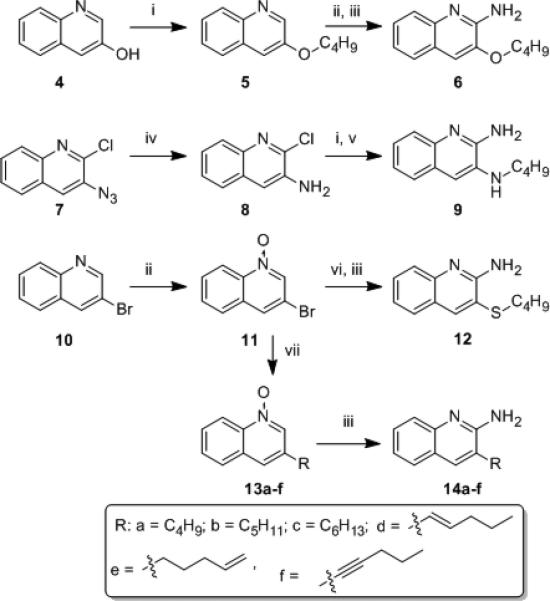
Syntheses of 3-substituted quinolin-2-amine analogues. Reagents: (i) butyl iodide, K2CO3, DMSO; (ii) m-CPBA, CHCl3; (iii) (a) benzoyl isocyanate, CH2Cl2; (b) NaOMe, MeOH; (iv) H2, Pt/C, EtOH; (v) NH3, MeOH; (vi) butylSH, NaH, DMSO; (vii) Pd(PPh3)4, RB(OH)2, K2CO3, 1,4-dioxane, for 14f: Pd(PPh3)4, CuI, 1-pentyne, Et3N:CH3CN (1:3).
Figure 5.

Dose-response profiles of human TLR8 agonistic activities of 3-substituted 2-aminoquinolines. Error bars represent standard deviations obtained on quadruplicates. Compounds 1 and 3 were used as comparators.
Table 1.
EC50 values of human TLR8-specific agonistic activities.
| Compound Number | IUPAC Name | EC50 (μM)a,b |
|---|---|---|
| 6 | 3-butoxyquinolin-2-amine | 2.18 |
| 9 | N3-butylquinoline-2,3-diamine | 4.28 |
| 12 | 3-(butylthio)quinolin-2-amine | 4.16 |
| 14a | 3-butylquinolin-2-amine | 0.41 |
| 14b | 3-pentylquinolin-2-amine | 0.2 |
| 14c | 3-hexylquinolin-2-amine | Inactive |
| 14d | (E)-3-(pent-1-en-1-yl)quinolin-2-amine | 2.67 |
| 14e | 3-(pent-4-en-1-yl)quinolin-2-amine | 0.49 |
| 14f | 3-(pent-1-yn-1-yl)quinolin-2-amine | 12.96 |
| 1 (Ref. 1) | 2-propylthiazolo[4,5-c]quinolin-4-amine | 0.2 |
| 3 (Ref. 2) | 2-butylfuro[2,3-c]quinolin-4-amine | 1.6 |
EC50 values were obtained using hTLR8-specific reporter gene assays.
Inactive compounds did not show appreciable activity at 500 μM.
A comparison of the activities of these analogues in TLR8 primary screens yielded a clear structure-activity relationship. The TLR8-agonistic potency of the 3-pentyl quinoline 14b was 0.2 μM, i.e., ten-fold greater than that of the 3-butoxy analogue 6b, eight times greater than that of the parent compound 3 (EC50: 1.6 μM), and rivaling that of the reference compound 1 (Fig. 5, Table 1), while the 3-(butylthio)quinoline and N3- butylquinoline analogues were weaker (EC50: 4.2 μM and 4.3 μM for 12 and 9, respectively; Fig. 5, Table 1).
An examination of the dihedral angles indicated a quasi-gauche conformation of the proximal methylenes of the C2-butyl substituent in the crystal structure of 3 bound to TLR8 and, as expected, the introduction of unfavorable geometrical constraints in the (E)-3-(pent-1-en-1-yl)quinoline and 3-(pent-1-yn-1-yl)quinoline analogues 14d and 14f, respectively, diminishes activity while potency is largely spared in compound 14e with a terminal alkene (Fig. 5, Table 1).
Additionally, in examining the interfacial surface topology of the binding site formed by the protomers of TLR8[16] using Voronoi polyhedral modeling,[21] we noticed an accessory hydrophobic groove bounded by Phe346 and Tyr348 which is contiguous with the hydrophobic pocket accommodating the C2-butyl substituent in 3. We were therefore interested in examining whether additional substituents at C4 would further augment the potency of 14b. As shown in Scheme 2, analogues 21a-c were synthesized starting from 3-iodoquinolin-4-ol (15).[8a]
We had initially envisaged a step-wise Suzuki coupling of appropriate alkylboronic acids with the 3-iodo-4-chloroquinoline intermediate 16 which, however, was not optimal because of the formation of a mixture of isomers. We therefore first installed the 3-pentyne substituent by Sonogashira coupling to obtain 17, which proved to be an excellent substrate for subsequent Suzuki coupling, leading to the required analogues 21a-c (Scheme 2). These compounds were feeble in their TLR8-agonistic activity (data not shown), suggesting poor tolerance of steric bulk at C4. In order to confirm that substitutions at C4 are not tolerated, we synthesized congeners of both 4-alkoxy (24a-b) and 4-alkyl quinolin-2-amines (27a-b; Scheme 3) and, as expected, all of these analogues were found to be inactive (Table S2).
Scheme 2.
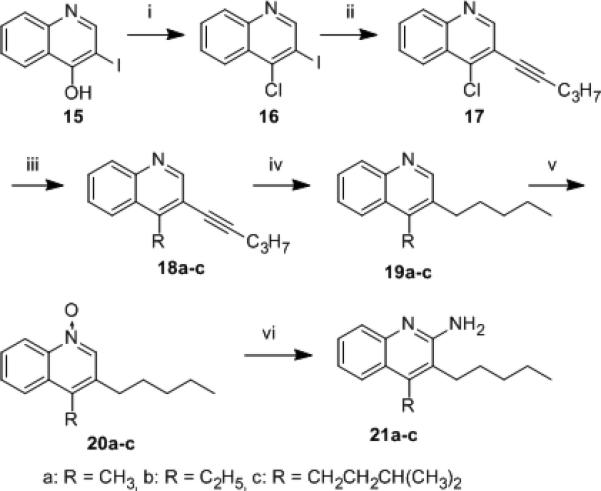
4-alkyl-3-pentylquinolin-2-amines. Reagents: (i) POCl3; (ii) Pd(PPh3)4, CuI, 1-pentyne, Et3N:CH3CN (1:3); (iii) Pd(PPh3)4, RB(OH)2, K2CO3, 1,4-dioxane (iv) H2, Pt/C, EtOH; (v) m-CPBA, CHCl3; (vi) (a) benzoyl isocyanate, CH2Cl2; (b) NaOMe, MeOH.
Scheme 3.
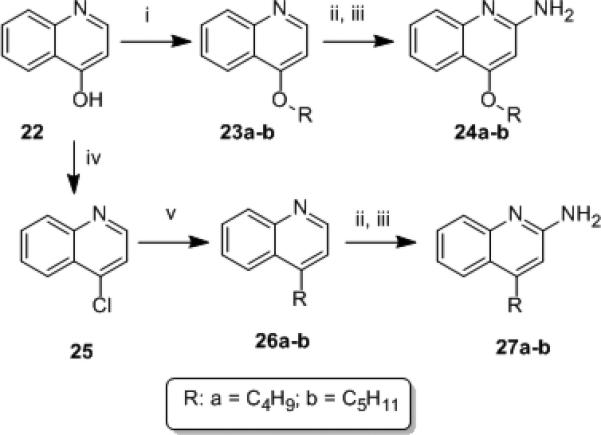
4-substituted quinolin-2-amines. Reagents: (i) butyl iodide, NaH, DMSO; (ii) m-CPBA, CHCl3; (iii) (a) benzoyl isocyanate, CH2Cl2; (b) NaOMe, MeOH; (iv) POCl3; (v) Pd(PPh3)4, RB(OH)2, K2CO3, 1,4-dioxane.
All analogues were counter-screened[8c, 19b, 22] in reporter cell lines specific for human TLR2, TLR3, TLR4, TLR5, TLR7, TLR9, TLR10, Nod1 and Nod2, and compounds 6, 9, 12, and 14a-f were confirmed to be specific for human TLR8. The most potent analogue 14b was characterized further in cytokine/chemokine induction profiles in a panel of secondary screens employing human peripheral blood mononuclear cells[8a] as well as whole human blood.[22c] Consistent with its specificity for TLR8, we observed the induction of a specific set of chemokines and proinflammatory cytokines, including interleukins 12 and 18 (Fig. 6).
Figure 6.

Induction of cytokines (red) and chemokines (blue) in human PBMCs by the lead compound 14b. Means of triplicates are shown.
In summary, we have effectively utilized the structure of TLR8 complexed with ligands in the rational design of a novel TLR8-specific chemotype which retains prominent cytokine-inducing activity profiles in ex vivo human blood assay systems, paving the way for evaluation of this compound as a candidate vaccine adjuvant in appropriate animal models.
Experimental Section
X-ray diffraction data and all synthetic and immunological experimental methods are provided in the Supporting Information.
Supplementary Material
Acknowledgements
This work was supported by NIH/NIAID contract HSN272200900033C (S.D.); a Grant-in-Aid from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (U.O., K.M., and T.S.); the Takeda Science Foundation (U.O. and T.S.); and the Mochida Memorial Foundation for Medical and Pharmaceutical Research (U.O.); and CREST, JST (T.S.).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.20xxxxxxx.
References
- 1.a Plotkin SA. The J. Infect. Dis. 2003;187:1349–1359. doi: 10.1086/374419. [DOI] [PubMed] [Google Scholar]; b Plotkin SA. Clin. Vaccine Immunol. 2009;16:1709–1719. doi: 10.1128/CVI.00290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janeway CA., Jr. Cold Spring Harb. Symp. Quant. Biol. 1989;54:1–13. Part 1. [PubMed] [Google Scholar]
- 3.a Ciechanover AJ, Sznajder JI. Am. J. Respir. Crit. Care Med. 2011;184:i–ii. doi: 10.1164/rccm.201111-1930ED. [DOI] [PubMed] [Google Scholar]; b Medzhitov R. J. Immunol. 2013;191:4473–4474. doi: 10.4049/jimmunol.1302427. [DOI] [PubMed] [Google Scholar]
- 4.a Akira S. Proc. Jpn. Acad. Ser .B Phys. Biol. Sci. 2009;85:143–156. doi: 10.2183/pjab.85.143. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Akira S, Uematsu S, Takeuchi O. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]; Kawai cT., Akira S. Semin. Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 5.a Kawai T, Akira S. Int. Immunol. 2009;21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kawai T, Akira S. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]; c Hoffmann J, Akira S. Curr. Opin. Immunol. 2013;25:1–3. doi: 10.1016/j.coi.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 6.a O'Neill LA, Golenbock D, Bowie AG. Nat. Rev. Immunol. 2013;13:453–460. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]; b Coll RC, O'Neill LA. J. Innate Immunol. 2010;2:406–421. doi: 10.1159/000315469. [DOI] [PubMed] [Google Scholar]
- 7.a Iwasaki A, Medzhitov R. Nat. Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]; b Pasare C, Medzhitov R. Semin. Immunol. 2004;16:23–26. doi: 10.1016/j.smim.2003.10.006. [DOI] [PubMed] [Google Scholar]; c Pasare C, Medzhitov R. Immunity. 2004;21:733–741. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]; d Pasare C, Medzhitov R. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]; e Nemazee D, Gavin A, Hoebe K, Beutler B. Nature. 2006;441:E4. doi: 10.1038/nature04875. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Iwasaki A, Medzhitov R. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Kokatla HP, Sil D, Malladi SS, Balakrishna R, Hermanson AR, Fox LM, Wang X, Dixit A, David SA. J. Med. Chem. 2013;56:6871–6885. doi: 10.1021/jm400694d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kokatla HP, Yoo E, Salunke DB, Sil D, Ng CF, Balakrishna R, Malladi SS, Fox LM, David SA. Org. Biomol. Chem. 2013;11:1179–1198. doi: 10.1039/c2ob26705e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Salunke DB, Yoo E, Shukla NM, Balakrishna R, Malladi SS, Serafin KJ, Day VW, Wang X, David SA. J. Med. Chem. 2012;55:8137–8151. doi: 10.1021/jm301066h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorden KB, Gorski KS, Gibson SJ, Kedl RM, Kieper WC, Qiu X, Tomai MA, Alkan SS, Vasilakos JP, Immunol J. 2005;174:1259–1268. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- 10.Philbin VJ, Dowling DJ, Gallington LC, Cortes G, Tan Z, Suter EE, Chi KW, Shuckett A, Stoler-Barak L, Tomai M, Miller RL, Mansfield K, Levy O, Allergy Clin J. Immunol. 2012 doi: 10.1016/j.jaci.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levy O. Nat. Rev. Immunol. 2007;7:379–390. doi: 10.1038/nri2075. [DOI] [PubMed] [Google Scholar]
- 12.a Petricevic B, Wessner B, Sachet M, Vrbanec D, Spittler A, Bergmann M. Shock. 2009;32:484–490. doi: 10.1097/SHK.0b013e3181a5ac8a. [DOI] [PubMed] [Google Scholar]; b Tajuddin T, Ryan EJ, Norris S, Hegarty JE, O'Farrelly C. J. Gastroenterol. Hepatol. 2010;25:1883–1890. doi: 10.1111/j.1440-1746.2010.06281.x. [DOI] [PubMed] [Google Scholar]; c Makni-Maalej K, Boussetta T, Hurtado-Nedelec M, Belambri SA, Gougerot-Pocidalo MA, El-Benna J. J. Immunol. 2012;189:4657–4665. doi: 10.4049/jimmunol.1201007. [DOI] [PubMed] [Google Scholar]
- 13.a Spranger S, Javorovic M, Burdek M, Wilde S, Mosetter B, Tippmer S, Bigalke I, Geiger C, Schendel DJ, Frankenberger B. J. Immunol. 2010;185:738–747. doi: 10.4049/jimmunol.1000060. [DOI] [PubMed] [Google Scholar]; b Kokatla HP, Yoo E, Salunke DB, Sil D, Ng CF, Balakrishna R, Malladi SS, Fox LM, David SA. Org. Biomol. Chem. 2013;11:1179–1198. doi: 10.1039/c2ob26705e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu H, Dietsch GN, Matthews MA, Yang Y, Ghanekar S, Inokuma M, Suni M, Maino VC, Henderson KE, Howbert JJ, Disis ML, Hershberg RM. Clin. Cancer Res. 2012;18:499–509. doi: 10.1158/1078-0432.CCR-11-1625. [DOI] [PubMed] [Google Scholar]
- 15.Dowling DJ, Tan Z, Prokopowicz ZM, Palmer CD, Matthews MA, Dietsch GN, Hershberg RM, Levy O. PLoS ONE. 2013;8:e58164. doi: 10.1371/journal.pone.0058164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanji H, Ohto U, Shibata T, Miyake K, Shimizu T. Science. 2013;339:1426–1429. doi: 10.1126/science.1229159. [DOI] [PubMed] [Google Scholar]
- 17.Davis AM, Teague SJ, Kleywegt GJ. Angew. Chem. Int. Ed. Engl. 2003;42:2718–2736. doi: 10.1002/anie.200200539. [DOI] [PubMed] [Google Scholar]
- 18.Warren SW. Organic Synthesis: The Disconnection Approach. 2nd ed. Wiley; 2008. [Google Scholar]
- 19.a Gerster JF, Lindstrom KJ, Miller RL, Tomai MA, Birmachu W, Bomersine SN, Gibson SJ, Imbertson LM, Jacobson JR, Knafla RT, Maye PV, Nikolaides N, Oneyemi FY, Parkhurst GJ, Pecore SE, Reiter MJ, Scribner LS, Testerman TL, Thompson NJ, Wagner TL, Weeks CE, Andre JD, Lagain D, Bastard Y, Lupu M. J. Med. Chem. 2005;48:3481–3491. doi: 10.1021/jm049211v. [DOI] [PubMed] [Google Scholar]; b Shukla NM, Malladi SS, Mutz CA, Balakrishna R, David SA. J. Med. Chem. 2010;53:4450–4465. doi: 10.1021/jm100358c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoo E, Crall BM, Balakrishna R, Malladi SS, Fox LM, Hermanson AR, David SA. Org. Biomol. Chem. 2013;11:6526–6545. doi: 10.1039/c3ob40816g. [DOI] [PubMed] [Google Scholar]
- 21.a Richards FM. J. Mol. Biol. 1974;82:1–14. doi: 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]; b Gerstein M, Chothia C. Proc. Natl. Acad. Sci. USA. 1996;93:10167–10172. doi: 10.1073/pnas.93.19.10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a Salunke DB, Connelly SW, Shukla NM, Hermanson AR, Fox LM, David SA. J. Med. Chem. 2013 doi: 10.1021/jm400620g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shukla NM, Salunke DB, Balakrishna R, Mutz CA, Malladi SS, David SA. PLoS ONE. 2012;7:e43612. doi: 10.1371/journal.pone.0043612. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hood JD, Warshakoon HJ, Kimbrell MR, Shukla NM, Malladi S, Wang X, David SA. Hum. Vaccin. 2010;6:1–14. doi: 10.4161/hv.6.4.10866. [DOI] [PubMed] [Google Scholar]; d Ukani R, Lewis TC, Day TP, Wu W, Malladi SS, Warshakoon HJ, David SA. Bioorg. Med. Chem. Lett. 2012;22:293–295. doi: 10.1016/j.bmcl.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Agnihotri G, Ukani R, Malladi SS, Warshakoon HJ, Balakrishna R, Wang X, David SA. J. Med. Chem. 2011;54:1490–1510. doi: 10.1021/jm101535e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.