

Abstract
Nonlinear optical molecular imaging and quantitative analytic methods were developed to non-invasively assess the viability of tissue-engineered constructs manufactured from primary human cells. Label-free optical measures of local tissue structure and biochemistry characterized morphologic and functional differences between controls and stressed constructs. Rigorous statistical analysis accounted for variability between human patients. Fluorescence intensity-based spatial assessment and metabolic sensing differentiated controls from thermally-stressed and from metabolically-stressed constructs. Fluorescence lifetime-based sensing differentiated controls from thermally-stressed constructs. Unlike traditional histological (found to be generally reliable, but destructive) and biochemical (non-invasive, but found to be unreliable) tissue analyses, label-free optical assessments had the advantages of being both non-invasive and reliable. Thus, such optical measures could serve as reliable manufacturing release criteria for cell-based tissue-engineered constructs prior to human implantation, thereby addressing a critical regulatory need in regenerative medicine.
Keywords: Tissue engineering, Label-free optical molecular imaging, Tissue viability, Oral mucosa, Multiphoton excitation microscopy, Second-harmonic generation (SHG) imaging, Fluorescence lifetime imaging microscopy (FLIM)
1. Introduction
Recent advances in tissue engineering and regenerative medicine (TERM) have become an integral part of reconstructive surgery and tissue/organ regeneration. One such advance is the development of tissue-engineered constructs, which are classified by the United States Food and Drug Administration (FDA) as combination products comprised of an engineered extracellular matrix and a biological component. Biological device manufacturing is strictly regulated by the FDA prior to product release for patient treatment to assure effectiveness and safety. It is essential that the device is objectively (i.e., quantitatively) and non-invasively (i.e., without sectioning or staining) evaluated in real-time to assess cellular viability [1–3], which is the significant technical challenge we address here by developing quantitative methods for tissue-based, label-free nonlinear optical molecular microscopy.
Rather than employing exogenous fluorescent dyes that are bright but invasive to the manufactured tissue, the method described here targets biologically- and metabolically-relevant endogenous tissue fluorophores, primarily nicotinamide adenine dinucleotide (phosphate) (NAD(P)H) and flavin adenine dinucleotide (FAD). NAD(P)H denotes the simultaneous analysis of NADH and NADPH as one fluorophore, grouped because of their similar fluorescence spectrum and their important roles in cellular metabolic function. NADH and FAD are mitochondrial coenzymes that report on cellular metabolism and oxygen consumption (Supplementary Fig. 1), and NADPH is an anabolic coenzyme largely produced by fast growing cells for biosynthesis [4].
NAD(P)H and FAD fluoresce in their reduced and oxidized states, respectively [5,6]. Therefore, their concentrations typically trend oppositely to one another and their relative concentrations can be assessed with fluorescence intensity measurements characterized by a redox ratio (RR) metric, defined as [FAD]/([NAD(P)H]+[FAD]) [7]. Intensity ratios like RR enhance the metabolic variations measured while reducing intensity-based artifacts, including signal variations due to optical loss, which may be difficult to quantify or control in tissues. A complementary approach is fluorescence lifetime imaging microscopy (FLIM). In addition to two-dimensional spatial information, FLIM images contain a third dimension – time [6–8]. FLIM is advantageous for tissue-based sensing because fluorescence lifetime reveals molecular microenvironment (e.g., temperature, pH, and binding), while being insensitive to intensity-based measurement artifacts.
Sensitive, spatially-resolved measures of NAD(P)H and FAD in unstained tissue constructs were achieved by developing quantitative nonlinear optical molecular microscopy methods to optically (non-invasively) section tissue samples into layers as thin as 1 μm [9–12]. Such image resolution enabled the detection of optical signals from a single cellular layer of a three-dimensional engineered tissue. In addition, nonlinear optical techniques enable specific measurements of collagen molecules through second harmonic generation (SHG). Because extracellular collagen and intracellular NAD(P)H and FAD have similar fluorescence characteristics, measuring collagen via SHG enables the spatial separation of intracellular (NAD(P)H and FAD) and extracellular (collagen) fluorescence via image post-processing. Therefore, by combining optical sectioning with fluorescence and SHG measurements, tissue fluorescence arising from cells can be analyzed to quantitatively characterize cellular metabolism and cellular organization for tissue viability assessment from a single cellular layer [7,13].
As a model system for our studies, we employed the engineered cell-based device EVPOME (Ex Vivo Produced Oral Mucosa Equivalent) [1]. EVPOMEs are manufactured by culturing primary human oral keratinocytes atop a dermal equivalent scaffold for tissue formation. EVPOMEs, developed for intraoral grafting procedures for reconstructive surgery of oral and dental soft tissues, were demonstrated to reduce patients’ wound healing time by half [4]. In addition, EVPOMEs were implanted successfully in humans during an FDA approved Phase I clinical trial [14].
2. Materials and Methods
2.1 Procurement of human oral mucosal tissues
Discarded keratinized oral mucosa was collected from patients undergoing minor oral surgical procedures at the University of Michigan (UM) hospital. The UM Medical School Institutional Review Board approved use of the mucosa and patients provided informed consent for research use. The study adhered to the Declaration of Helsinki Guidelines.
2.2 Standard protocols for culturing human oral keratinocytes and manufacturing EVPOMEs
Primary human oral mucosal tissues were harvested from procured discarded keratinized oral mucosa and cultured according to previously described protocols [1,2]. Briefly, primary human oral keratinocytes were enzymatically dissociated from the tissue samples. Oral keratinocyte cultures were established in a chemically-defined, serum-free culture medium (EpiLife and EDGS, Invitrogen/Life Sciences, Carlsbad, CA). The medium contained 0.06 mM calcium, 25 μg/ml gentamicin, and 0.375 μg/ml fungizone (both from Sigma, St.Louis).
For cell culture studies, the oral keratinocytes were seeded onto a 3.5 cm glass bottom dish (MatTek Corp., Ashland, MA) coated with collagen. Calcium concentration in the growth medium was controlled at 0.06 mM for cell proliferation and 1.2 mM to induce cell differentiation.
For EVPOME studies, EVPOME constructs were manufactured by first seeding 200,000 cells/cm2 on 1 cm2 acellular cadaver skin (AlloDerm®, LifeCell, KCI, Branchburg, NJ)) that was pre-soaked in 0.05μg/μL human type IV collagen at 4°C overnight (Sigma-Aldrich, St. Louis, MO). Resulting keratinocytes and AlloDerm® were submerged in medium containing 1.2 mM calcium for 4 days and then raised to an air-liquid phase for an additional 7 days to induce cell stratification and differentiation. Control constructs were cultured in 100 mm dishes (for thermally-stressed) or in 6-well plates (for metabolically-stressed) with inserts in incubators at 37°C with 5% CO2 for all culture days. The day 11 glucose concentration of the culture medium was read by a glucose meter (ACCU-CHEK®, Aviva, Roche, Indianapolis, IN).
2.3 EVPOME stressing protocols
Thermally-stressed constructs were cultured at 43°C for 24 hours beginning on day 9 postseeding and were returned to normal culture conditions starting day 10. To create metabolically-stressed constructs, constructs received no fresh culture medium for 6 days beginning on day 4 post-seeding and were returned to normal culture conditions starting day 10. We note that for the first batch of the metabolic-stress experiment, the construct was starved from day 4 to day 11. As a result, there is no glucose consumption measurement for this patient. In this study, we have grouped optical results from both metabolic-stress protocols because there were no observed differences from construct histology results. For two batches measured in the FLIM thermal stressing study, we note that two of the five batches had no histology or glucose samples measured. Their reported glucose and histology metrics were measured from constructs cultured in parallel with the same primary human cells.
2.4 Nonlinear optical microscopic imaging
Images were acquired with a Leica TCS SP5 microscope equipped with a Ti:Sapphire laser (Mai Tai, Spectra-Physics). The excitation laser source and the emission light were coupled through an inverted microscope with 40× (1.25 NA) / 63× (1.4 NA) oil immersion objective lenses to image oral keratinocytes in culture and a 25× water immersion objective lens (0.95 NA, 2.5 mm working distance) to image EVPOME constructs. EVPOME constructs were imaged from the top surface down (i.e. through the topmost keratin layer) to mimic post-implantation conditions in vivo. Prior to measurement on an inverted microscope, constructs were flipped over onto measurement dishes. This process was not expected to alter cellular viability or layer thickness.
All measurements were collected in a controlled environment (37°C with 5% CO2) to mimic EVPOME culture. NAD(P)H was excited with excitation at 705 nm; FAD and collagen (to detect SHG) were excited with excitation at 900 nm. Before each measurement, the excitation power at the specimen surface plane at both excitation wavelengths with a 10× objective was calibrated at 20 mW for standard cell cultures and 30 mW for EVPOME constructs to limit photobleaching and non-reversible changes in the sample. The laser power was selected after consulting literature that concluded no short-term cellular damage occurred after 760 or 800 nm exposure with powers of 50 mW (~200 fs pulse width) [15] and 60 mW (~150 fs pulse width) [16], respectively. Currently, there are no FDA approved nonlinear optical microscopy systems for human use. However, a CE-certified device is commercially available in Europe and a preliminary clinical study in the US of this device [17] indicates that such imaging technologies will have medical risk profiles similar to FDA approved clinical imaging modalities, including X-ray and computed tomography instruments [18].
For NAD(P)H intensity detection, the backscattered fluorescence was collected through a band pass filter from 435 to 485 nm; for FAD intensity detection, the backscattered fluorescence was collected through a band pass filter from 500 to 550 nm (Leica Dapi/FITC filter cube). Emission light was collected with short-coupling non-descanned photomultiplier tubes to increase collection efficiency. An internal tunable photomultiplier collected SHG emission from 440 to 460 nm. Detector gain and offset were consistent for each measurement to avoid detector saturation. Images (1024 × 1024 pixels, 0.391× 0.391 μm2 pixel size, 8-bit image depth) were acquired in ~40 seconds with a 200 Hz line scanning speed. The two fluorescence and SHG signals were acquired sequentially, with all three measurements occurring at each site in less than three minutes. To reduce background noise, a line average of eight was employed for all images. For NAD(P)H FLIM detection, emission light was collected through a tunable monochromator from 410 to 490 nm coupled to a photomultiplier tube and photon counting addon (HydraHarp TCSPC, Leica). FLIM measurements of cells in monolayer were collected (1024 × 1024 or 512 × 512 pixels, ~265 × 265 μm2) until 100 counts were reached in the peak channel, lasting an average of ~2 minutes per FLIM image. FLIM measurements of EVPOMEs were collected (512 × 512 or 256 × 256 microns, ~400×400 μm2) for 1.5 minutes.
The image acquisition settings above were applied for both cross-sectional and en-face imaging, except that the cross-sectional images were collected in x-z (vertical) direction and the en-face images were collected in x-y (horizontal) direction. Representative cross-sectional fluorescence images were overlaid with their corresponding SHG images, processed via Photoshop Screen function (Fig. 1 and Fig. 2). No post-processing was performed on representative en-face images in Figure 1, Figures 3–6, Supplementary Figures 1–3 and 5 unless indicated on the figures.
Figure 1.
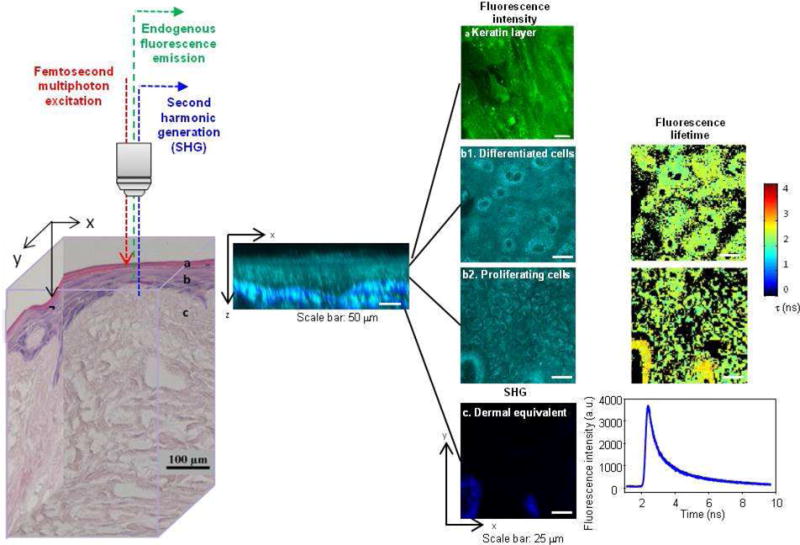
Label-free nonlinear optical molecular imaging non-invasively interrogated a living EVPOME construct in three dimensions, measuring cross-sectional (middle) and en-face (right) images. EVPOME’s layered structure is evident in the histology (left) and in the cross-sectional image (middle) of cellular autofluorescence (NAD(P)H, cyan) with overlaid scaffold SHG (collagen, blue). EVPOMEs were composed of (1) a stratified cellular layer with > 3 cellular layers, including proliferative basal cells and differentiating cells, (2) attachment of basal cells to the dermal equivalent, and (3) a well-defined keratin layer. These three criteria were employed later for histological evaluation of construct viability. Fluorescence intensity, fluorescence lifetime, and SHG microscopic images were acquired to quantitatively assess EVPOME viability (right). Three-dimensional spatially-localized optical measurements interrogated cellular metabolic function and spatial organization (steady-state imaging) as well as cellular microenvironment (FLIM).
Figure 2.
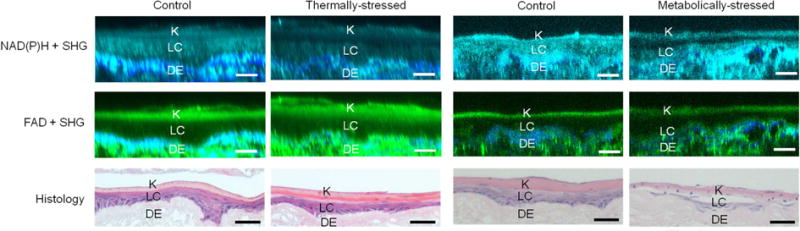
Non-invasive optical characterization of tissue structure. Cross-sectional fluorescence images of EVPOME constructs from NAD(P)H (cyan) and FAD (green) channels with overlaid corresponding SHG images (blue) were compared to sample histology. Control and thermally-stressed (left columns) and metabolically-stressed (right columns)) EVPOMEs exhibit the layered tissue structure, consisting of a top keratin layer (K) ~10–20 μm thick, a middle living cellular layer (LC) ~20–30 μm thick, and a bottom dermal equivalent layer (DE) ~400 μm thick (a portion is shown). While fluorescence images quantitatively and non-invasively characterized EVPOME’s layered structure, histology destroyed the EVPOME constructs. Scale bar: 50 μm.
Figure 3.
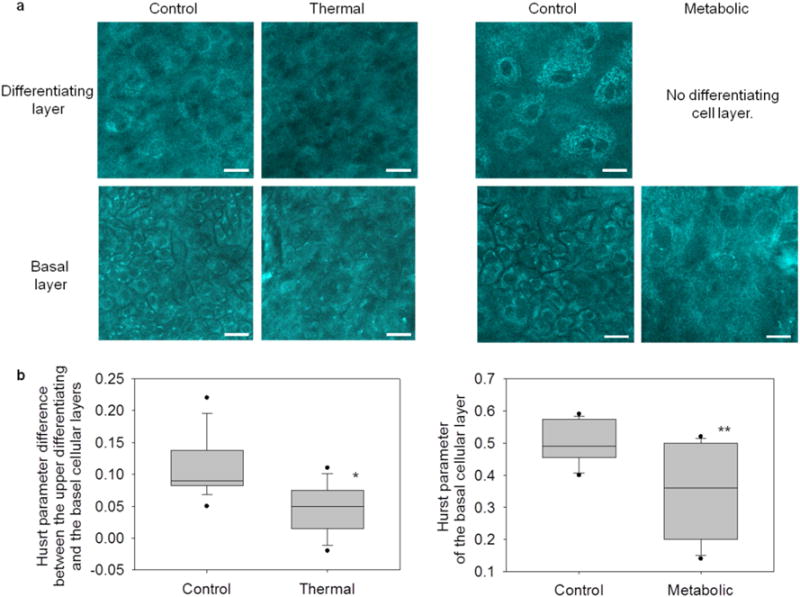
Label-free optical spatial analysis characterized cellular organization in EVPOME constructs and reliably distinguished control from stressed constructs. (a) En-face, optically sectioned NAD(P)H images revealed cellular organization of the EVPOME constructs. In control constructs, differentiating cells (top) were characterized by large, loosely packed cells. Alternatively, basal layer cells (bottom) were characterized by small, closely packed cells. In thermally-stressed constructs, cell morphologies appeared disorganized in both layers. In metabolically-stressed constructs, EVPOMEs grew thin cellular layers (no distinct differentiating and basal layers). Therefore, only the basal layer image was acquired, which appeared disorganized. Scale bar: 20 μm. (b) Cellular organization was quantified by spatial analysis of optical images to extract a Hurst parameter (H). For both stressing experiments, the Hurst parameter significantly distinguished between stressed and control EVPOMEs (*n = 4 batches with 11 H value differences extracted from control and n = 3 batches with 8 from thermally-stressed, P-value = 0.004; ** n = 4 batches with 12 H values extracted from control and n = 3 batches with 10 from metabolically-stressed, P-value <0.001).
Figure 6.
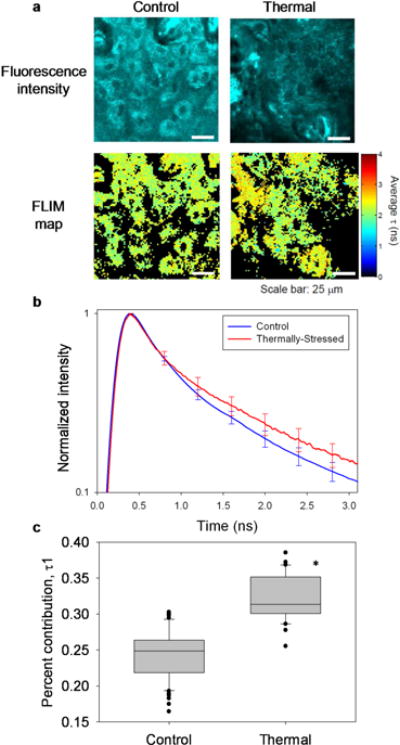
Label-free fluorescence lifetime analysis successfully distinguished control from thermally-stressed EVPOMEs. (a) Representative en-face, optically sectioned NAD(P)H fluorescence intensity images and corresponding FLIM maps for an analyzed region-of-interest in control (left) and thermally-stressed (right) EVPOMEs. Larger mean fluorescence lifetime t was observed in FLIM maps of thermally-stressed constructs relative to controls. (b) Mean of normalized fluorescence decays from regions-of-interest support the observation from FLIM maps that thermally-stressed constructs exhibited greater mean NAD(P)H fluorescence lifetimes than controls. Error bars represent s.e.m. (c) NAD(P)H fluorescence decays measured from constructs were best fit to a two-exponential decay model. Model fits revealed that the percent contribution of τ1 (attributed to bound NAD(P)H) to the measured fluorescence decay was significantly higher in thermally-stressed EVPOMEs than in controls (n = 5 batches with 58 control and 33 thermally-stressed regions analyzed, P-value < 0.001).
2.5 Chemical treatments
MitoTracker®, an exogenous fluorescence dye that only fluoresces within mitochondria, stained the cell culture samples for colocalized fluorescence from both MitoTracker® and NAD(P)H. The metabolic inhibitor, 4 mM KCN, and the uncoupler, 0.5 μM FCCP, (both from Sigma-Aldrich, St. Louis, MO) were employed to vary cellular metabolism. The variation was monitored for two minutes. KCN/FCCP was administered immediately after acquiring the first image.
2.6 Spatial intensity analysis
For spatial analysis, each raw NAD(P)H image was split into an 8×8 grid, totaling 64 sub-images. As a demonstration, ¼ of a NAD(P)H image with 16 of the 64 total sub-images was processed (Supplementary Fig. 2a). Image pre-processing was performed with a 3 × 3 median filter to reduce noise while preserving edges between the cytoplasm, nuclei, and extracellular components. Background fluorescence was removed with the binary mask containing SHG and keratin regions created as described in the previous section. Therefore, analysis was only performed on binary sub-images with < 10% of pixels signals being 1, indicating a sub-image with < 10% of total pixel fluorescence attributed to collagen, elastin, or keratin. A PSD (power spectral density) map was then obtained and filtered with a 5 × 5 mean filter to reduce noise from each binary sub-image. To quantify the PSD characteristics, radial angle-averaged PSD values (ϕ) were calculated and plotted versus spatial frequency (κ) on a logarithmic scale. In the spatial frequency range of 0.14 − 0.5 μm−1, the PSD curve was modeled with an inverse power law, ϕ(κ) ∝ κ−α, and was linearly fit to extract the corresponding power exponent (α). A Hurst parameter (H) was then derived with the equation H = ½(α −1) (Supplementary Fig. 2b). Matlab was employed for all post-processing methods described above.
2.7 Procedure to calculate a redox ratio map
For cells in culture analysis, two raw fluorescence images, one each of NAD(P)H and FAD, were inputs into developed analysis software. Images were pre-processed with a 3 × 3 median filter to increase image contrast. Then, a threshold value was set as the average fluorescence intensity signal from the whole image to create a binary mask which was employed to reduce background fluorescence signals. This binary mask filtered each raw fluorescence image. A redox image was then calculated at each pixel as [FAD]/([NAD(P)H] + [FAD]).
Supplementary Figure 3 shows the method to calculate en-face redox ratio maps in an EVPOME. The first step removed regions with high background identified as pixels with collagen SHG signal. For basal layer images with collagen fluorescence, three 1024 × 1024 images (400 × 400 μm2), denoised NAD(P)H, denoised FAD, and denoised SHG, were used for analysis. The SHG image, which identified regions with collagen, was further denoised with Markov random field denoising. Markov random field denoising clumped the pixels with strong collagen SHG signals together. Subsequently, pixels having SHG intensity greater than the average SHG signals from the whole image were removed from their corresponding NAD(P)H and FAD fluorescence images. Processed NAD(P)H and FAD fluorescence images were then used to derive a redox ratio at each image pixel.
Image processing was performed in Matlab to remove image pixels with high fluorescence contribution from keratin (attributed to keratin in the keratin layer) in a two-step process. First, images from the differentiating cellular layer that contained keratin fluorescence were processed to create a redox ratio map. Then, Markov random field denoising was applied to the calculated redox ratio map to exclude the pixels with abnormally high redox ratio (attributed to keratin).
2.8 FLIM analysis – exponential fitting procedure
From each acquired FLIM image, two methods of analysis were performed: (1) up to two regions void of collagen and keratin were selected and analyzed with bi-exponential fitting or (2) the region was defined as the entire image field-of-view. All exponential fitting was performed with Leica SymPhoTime software. Composite decays were analyzed after summing all pixels within the defined region. Data was best fit to a bi-exponential decay model, F(t) = a1e−t/τ1 + a2e−t/τ2. The iterative fitting procedure deconvolved an instrument response function (IRF) from each decay, measured as the second harmonic generated response from a collagen sample. Percent contribution from τ1 was calculated as a1/(a1+a2). Measurements from differentiating and basal layers were grouped for analysis per the statistical discussion (see Statistical Analysis in Methods). An alternative average fluorescence lifetime, here called rapid average lifetime determination, was employed to calculate average lifetime without fitting. To calculate this average lifetime, we computed the time for the fluorescence to decay to 1/e of the peak intensity, similar to calculating a half-life in biology.
2.9 Histology assessment
Prior to optical measurements on day 11 post-seeding, a piece of the EVPOME was removed (~20% of total construct area), fixed in 10% formalin, washed with PBS (phosphate buffered saline), and then placed in 70% ethanol solution. All specimens were processed by the histology core at the UM Dental School, cut into 5μm sections and stained with hematoxylin and eosin (H&E). The processed H&E histology slides were assessed by a panel of four blinded expert readers, each with several years’ experience in working with EVPOME constructs. Each histology slide had two to three slices sectioned from one EVPOME. For two of the five batches for FLIM analysis, histology and glucose readings were not directly obtained from the measured constructs, but instead from concurrently cultured constructs from the same primary human cells.
Classification criteria were developed by the expert readers prior to evaluation, where a viability score of 1 (least viable), 2, 3, 4, or 5 (most viable) was used. The classification criteria included the basal cell layer health, the construct cellular organization, and the keratin layer structural quality (Supplementary Fig. 4). The classification criteria are defined as follows.
AlloDerm® with few to no cells, no cellular organization, and an unstructured keratin layer
AlloDerm® with a thin basal layer of cells and sporadic nuclei, flaky separation between the cell layer and keratin layer, and a keratin layer with wispy, not compacted structure
AlloDerm® with a thin basal layer of cells and sporadic nuclei, thin superficial layers with gradually flattening cells, and a top layer of pink, compacted keratin.
AlloDerm® with moderate basal layer of cells with nuclei and rounded cells, moderate superficial layers with gradually flattening light blue cells, and a top layer of pink, compacted keratin.
AlloDerm® with thick basal layer of cells with many nuclei and rounded cells, thick superficial layers with many gradually flattening cells, and a top layer of pink, tightly compacted keratin.
Definitions were agreed upon by all readers. Several representative histology images from each group were selected as a baseline to calibrate readers. The readers then individually assigned a score to each EVPOME histology slide, while blinded to the EVPOME stressing condition and to the other readers’ scores.
The representative histology images in Figure 1 and Figure 2 were acquired with a Nikon Eclipse Ti inverted microscope. A 10× objective (0.25 NA, air) was employed to capture 8-bit bright-field images, resulting in histology images of 2560 × 1920 (pixel size of 0.481 μm) or 1280 × 960 pixels (pixel size of 0.971 μm).
2.10 Statistical analysis
Statistical analysis was performed with a test based on fitting a linear mixed-effects model [19]. The analysis accounts for the data hierarchical nature where, for optics data, measures were taken from multiple sites per EVPOME, and multiple EVPOMEs obtained per patient, and for histology data, multiple histology samples were sectioned per patient and each section was read by four expert readers. For example, for the analysis of standard cell culture data, the mixed-effects model used with patient and cell culture as random intercepts to account for the two levels of nesting where sites were sampled within cultures, and cultures were sampled from within patients. Using these models, we also tested for any differences associated with the differentiated versus proliferated cells or the top versus bottom layer, and when we did not find any differences associated with these with respect to not only the associated statistical significance, but also the magnitude of the associated difference estimate, we dropped these variables from the models. For each parameter, we estimated the mean difference and the corresponding 95% confidence intervals (CI) between stressed versus control conditions based on these models. All tests were conducted using a significance level of 0.01.
3. Results
Optical release criteria were developed from nonlinear optical molecular microscopy images in cross-sectional and en-face geometries. Measured cross-sectional images were non-invasive analogues to a histology section, while measured en-face images preferentially isolated a single cellular layer (Fig. 1 and Supplementary Videos 1–2). Rigorous sample testing ensured the preferential detection of NAD(P)H and FAD fluorescence in both primary human oral keratinocytes in culture and in EVPOME tissue constructs (Supplementary Fig. 1). For each study (RR, spatial, and FLIM), EVPOMEs were cultured from 5 distinct primary human patient donors to account for intrapatient variability. Additionally, up to two stressing conditions were employed to compromise EVPOME viability.
3.1 Non-invasive optical characterization of tissue structure
Control EVPOMEs were compared to stressed EVPOMEs subjected to sub-optimal culture conditions to reduce their viability (5 batches were thermally-stressed for intensity and FLIM studies, 5 batches were metabolically-stressed for intensity studies). The data collected from the EVPOME constructs are summarized in Supplementary Table 1. Optical microscopy captured the layered structure of a living EVPOME construct with cross-sectional NAD(P)H (cyan: greenish-blue) and FAD (green) images overlaid with SHG images (blue) (Fig. 2 and Supplementary Video 1), in good agreement with histology sections. As expected, strong fluorescence signals in the NAD(P)H and FAD channels were detected from the top keratin layer and the bottom dermal equivalent layer (i.e. extracellular matrix). The top layer fluorescence was attributed to keratin, and the bottom layer fluorescence was attributed to collagen and elastin, since each of these species is known to produce strong fluorescence cross-talk in the emission wavelengths ranges studied [20]. The presence of collagen and elastin in the dermal equivalent layer was confirmed previously with a biochemical assay [21].
Representative control and stressed construct images are shown from (left two columns) the thermal-stress experiment and (right two columns) the metabolic-stress experiment (Fig. 2). Consistent with the culture protocols, thermally-stressed EVPOMEs did not always have underdeveloped cellular layer thickness (stratification began after constructs were raised to an air-liquid interface on day 4, stressing starting on day 9), while metabolically-stressed EVPOMEs consistently lacked cellular layer formation (stressing starting on day 4).
3.2 Spatial assessment of local cellular organization
Spatial assessment consisted of two steps: (1) measurements of cellular layer thickness and (2) quantification of cellular organization. EVPOMEs with average cellular layer thickness less than 15 μm (the average thickness of a cellular monolayer) were not candidates for analysis in step (2) and were excluded from further study. (This included three constructs, one control and two stressed, in each of the thermal-stress and the metabolic-stress experiments. Naturally, the absence of cells detected in step (1) is itself a metric of a construct’s poor viability.) EVPOMEs with a cellular layer > 15 μm were further analyzed in step (2), where cellular organization was quantified via NAD(P)H fluorescence images (Supplementary Fig. 2). NAD(P)H en-face fluorescence images were employed for organizational analysis rather than FAD because of their higher signal intensity and contrast. Spatial intensity analysis employed a PSD map of each image to extract a Hurst parameter (H), related to the cellular organization [13]. The PSD map suggested that the self-affine fractal structure [22] of EVPOMEs was between 2.00 – 7.14 μm, a range consistent with cellular mitochondrial size.
Cellular organization of the differentiating and basal layers of EVPOMEs is shown (Fig. 3a and Supplementary Video 2). In the basal layer of control constructs, keratinocytes were closely packed. As basal cells proliferated and some cells differentiated to the upper keratin layer, keratinocytes became less tightly packed with an irregular cellular organization. Disorganized cells were observed in basal layers of both metabolically-stressed and thermally-stressed EVPOMEs.
Smaller H values corresponded to increased cellular disorganization with non-distinguishable cell-to-cell or cell-to-nucleus borders [22]. In control EVPOMEs, the differentiating layer had a smaller H value than the basal layer. Alternatively in thermally-stressed EVPOMEs, both basal and differentiating layers had small H values. The H value difference between the differentiating and the basal layer was found to be significantly larger in control than in thermally-stressed EVPOMEs (Fig. 3b, after employing the step (1) exclusion criteria above, n = 4 control and n = 3 thermally-stressed batches, P-value = 0.004). In metabolically-stressed EVPOMEs, the H value of the basal layer was significantly lower than that of the basal layer of a control EVPOME (Fig. 3b, after employing the step (1) exclusion criteria above, n = 4 control and n = 3 metabolically-stressed batches, P-value < 0.001).
3.3 Redox ratio metric assessment of local cellular metabolism
A redox ratio, defined as ([FAD] / ([NAD(P)H] + [FAD]), provides a quantitative metric to compare relative metabolic rates of measured samples. A lower RR corresponds to relatively lower FAD fluorescence and/or higher NAD(P)H fluorescence, indicating a higher cellular metabolic rate [7].
En-face fluorescence images were employed to quantify the relative local metabolic rate via analysis of intracellular autofluorescence variations (Supplementary Video 2). The RR metric assessed local cellular metabolism in primary human oral keratinocytes in culture (Fig. 4) and in EVPOME constructs (Fig. 5). Cell culture samples with high proliferation and differentiation were assessed with high resolution fluorescence images and redox ratio maps. Thermal stressing was applied to the cell culture samples from 4 batches. Lower RR was extracted from control compared to thermally-stressed keratinocytes (Supplementary Fig. 5, n = 4 batches, P-value = 0.004).
Figure 4.
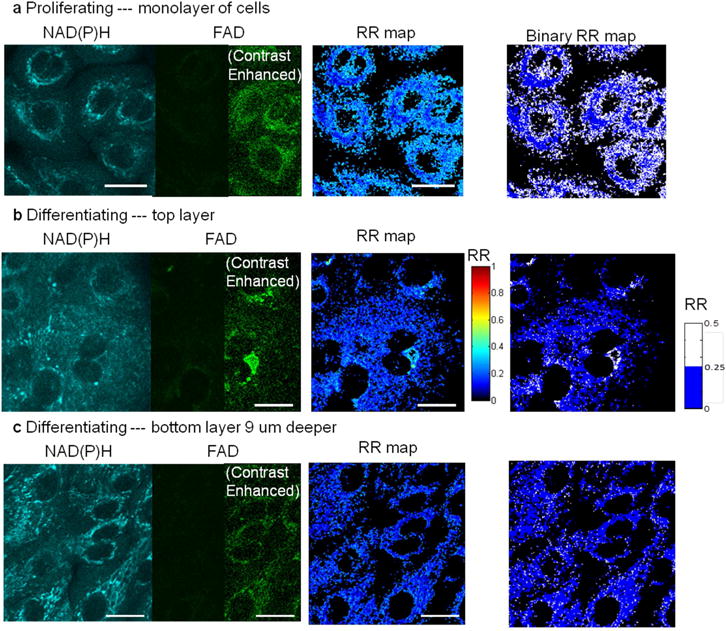
Label-free optical microscopy imaged (section thickness < 1 μm) a single layer of primary human oral keratinocytes in culture. In low-calcium (0.06 mM) medium, keratinocytes proliferated, forming a cellular monolayer. (b) In high-calcium medium (1.2 mM), keratinocytes differentiated into a layered structure, which was sectioned by nonlinear optical microscopy. Proliferating cells were spatially separated as compared to differentiating cells, which tended to crowd together. Shown in (b) and (c) are images of two layers at the same site, and the bottom layer is 9 μm deeper than the top. Both proliferating and differentiating keratinocytes exhibit high NAD(P)H fluorescence but low FAD fluorescence, resulting in a low RR (noted as blue pixels in the RR maps). In particular, proliferating keratinocytes in (a) have low perinuclear RR because highly metabolic mitochondria gathered around the nuclei, shown as the dark blue rings in the binary RR map. Differentiating keratinocytes in (c), on the other hand, homogeneously exhibit dark blue pixels over the binary RR map. In addition, as keratinocytes differentiated upwards, some cells had slightly higher average RR seen as the appearance of light blue pixels in the differentiating layer RR map, indicating their decreasing metabolic activity. Scale bar: 20 μm. Note: For display, contrast enhancement on half of the dim panels was performed by setting image minimum at 10 and maximum at 100 in a scale of 0–255.
Figure 5.
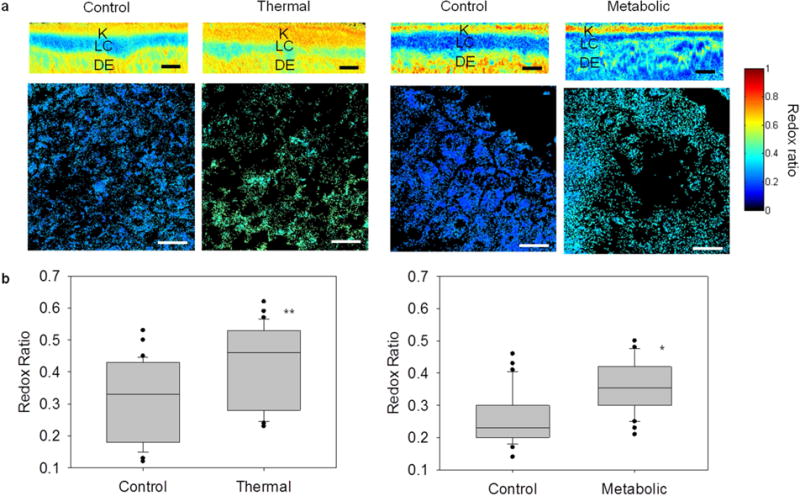
Label-free optical redox ratio mapping characterized cellular metabolism in EVPOME constructs and reliably distinguished control from stressed constructs. (a) The top row shows cross-sectional EVPOME redox ratio maps; the bottom row shows en-face EVPOME redox ratio maps. Control constructs had a lower RR (deeper blue) than stressed constructs, indicative of viable and actively metabolizing cells. (K: keratin; LC: living cellular; DE: dermal equivalent) Scale bar: 25 um. (b) An average RR for each image was extracted to quantify cellular viability of thermally-stressed (left) and metabolically-stressed (right) EVPOMEs. Control EVPOMEs had significantly lower RR than stressed EVPOMEs in both experiments. (n = 5 batches with 30 control and 30 thermally-stressed images analyzed, *P-value = 0.004; ** P-value <0.001).
Figure 5a shows cross-sectional RR maps which exhibit the layered EVPOME structure. Construct metabolic function was assessed via the RR of the cellular (middle) layer. Control constructs had lower RRs than stressed constructs, corresponding to higher cellular metabolic rate. After either thermal or metabolic stressing was applied, RRs of stressed EVPOMEs were higher than RRs of control EVPOMEs. The average redox ratio of control and stressed EVPOMEs was compared (Fig. 5b). The RR was significantly higher in the thermally-stressed EVPOMEs than in control EVPOMEs (n = 5, P-value < 0.001). In addition, the RR was significantly higher in the metabolically-stressed EVPOMEs than in the control EVPOMEs (n = 5 batches, P-value = 0.004). We note that NAD(P)H or FAD fluorescence images alone cannot significantly distinguish either thermally-stressed (n = 5 batches, NAD(P)H P-value = 0.43, FAD P-value = 0.02) or metabolically-stressed (n = 5 batches, NAD(P)H P-value = 0.001, FAD P-value = 0.14) from control EVPOMEs, as discussed in Supplementary Table 2.
3.4 FLIM assessment of local cellular viability
Figure 6a shows representative NAD(P)H intensity and corresponding rapid lifetime determination images of control and thermally-stressed EVPOMEs. While intensity images appear comparable in intensity and spatial organization, the average fluorescence lifetimes measured via FLIM are greater for thermally-stressed constructs compared to controls. The average of all time-resolved fluorescence decays analyzed from thermally-stressed constructs is longer than that from control constructs (Fig. 6b), similar to results from primary human oral keratinocytes in culture (Supplementary Fig. 6). Fitting data to multi-exponential decays yielded two primary fluorophore populations, attributed to free (τ2, fast lifetime) and bound (τ1, slow lifetime) NAD(P)H. The percent contribution of τ1 to the measured fluorescence decay was significantly lower in control than in thermally-stressed constructs (Fig. 6c, n = 5 batches, P-value < 0.001). Other fitting parameters are discussed in Supplementary Figure 7.
3.5 Optical assessments of differentiation and basal cell layers
In control constructs, optical measures of cellular spatial organization (the Hurst parameter (H)) detected notable differences between living cells populating the basal layer and those populating the differentiating layer (Fig. 3), whereas optical measures of cellular metabolic function (RR and FLIM metrics) did not detect differences between those layers (see Statistical Analysis in Methods). Metabolic differences between differentiating and basal cell layers might have been anticipated in cultured constructs because of their different functionalities. However, EVPOMEs are produced in vitro in 11 days by seeding hundreds of thousands of proliferating cells (sufficient to create a continuous basal layer) onto the dermal equivalent and the short culture period may be insufficient to produce measurable differences in metabolic function between cell layers. Future optical studies could examine EVPOMEs cultured for longer periods, which exhibit increased cellular maturity (defined as the number of cell layers/increase in stratification) and amplified differences between the differentiating and basal cells layers [1].
3.6 Assessment via histology and glucose consumption assay
Traditional assessment methods, histology and glucose consumption assay, analyzed the control and stressed EVPOMEs studied (Fig. 7). Due to the high patient variability, histology could not distinguish stressed from control EVPOMEs in some cases. Histology assessment showed that thermal stressing and metabolic stressing created two different outcomes, with thermally-stressed EVPOMEs having greater perceived viability than metabolically-stressed EVPOMEs (Fig. 2). However, glucose consumption readings could not distinguish between stressed and control EVPOMEs as discussed in Supplementary Table 2.
Figure 7.
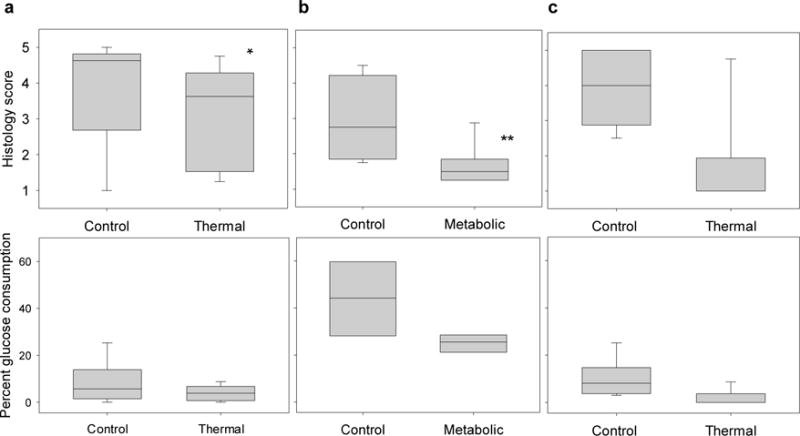
Histology scores assigned by a panel of blinded expert readers and percent glucose consumption measures did not reliably distinguish control and compromised EVPOMEs. Histology scores (with higher scores indicating greater perceived viability; 5 being the most viable) and glucose readings (percent error = 15%) compare control constructs to (a) thermally-stressed and (b) metabolically-stressed EVPOMEs prepared for the fluorescence intensity experiment, and (c) thermally-stressed EVPOMEs prepared for the fluorescence lifetime experiment Histology scores distinguished stressed from control EVPOMEs in some experiments (n = 5 batches, *(a) P-value = 0.002; **(b) P-value < 0.001; (c) P-value = 0.03), but histology is a destructive method for tissue assessment. Percent glucose consumption measures did not distinguish stressed from control constructs in any experiment ((a) n = 5 batches, P-value = 0.1; (b) n = 4 batches, P-value = 0.03; (c) n = 5 batches, P-value = 0.04). Note: The percent glucose consumption measures from the metabolic-stress experiment were larger than those from the thermal-stress experiment because the metabolic-stress culture dishes were smaller, as detailed in Methods.
3.7 Optical measures non-invasively identified stressed EVPOMEs
A robust manufacturing release criterion would identify and eliminate any stressed constructs. Optical redox ratios were compared to histology scoring for the 10 control and 10 stressed constructs assessed by fluorescence intensity (Fig. 8a, 5 thermally-stressed, 5 metabolically-stressed). A RR threshold value of ~0.23 successfully distinguished all 10 stressed EVPOMEs, whereas no such threshold value was possible for histology scoring, indicating that redox ratio is a robust release criterion. Similarly, an optical FLIM measure (the percent contribution of τ1) was compared to histology scoring for the 5 control and 5 thermally-stressed constructs assessed by fluorescence lifetime (Fig. 8b). A percent contribution of τ1 threshold of ~0.28 differentiated all thermally-stressed from all control EVPOMEs, which was not possible using histology scoring.
Figure 8.
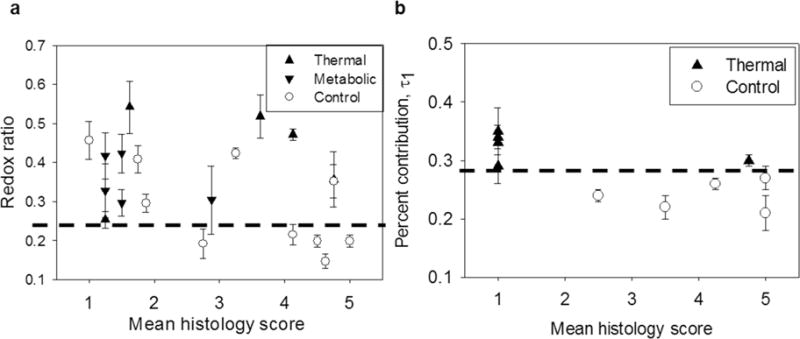
Non-invasive optical metrics obtained from label-free (a) RR and (b) fluorescence lifetime (FLIM) images reliably identified all experimentally stressed tissue constructs, whereas histology scores assigned by a panel of blinded expert readers could not. (a) Mean redox ratio was compared to mean panel histology score for control (n = 10 batches), thermally-stressed (n = 5 batches), and metabolically-stressed (n = 5 batches) EVPOME constructs. An RR threshold (dashed black line) distinguished all 10 experimentally stressed constructs from 5 of the most viable (highest histology score) control constructs. A comparable histology score threshold capable of distinguishing stressed from control constructs was impossible to define. We note that 3 control constructs with poor mean histology score (< 2) were also identified by the optical RR threshold. Therefore, excluding these 3 poorly growing EVPOMEs, the non-invasive optical RR metric successfully identified 5 out of 7 remaining control from all 10 stressed constructs. (b) FLIM analysis was compared to mean panel histology score for control (n = 5 batches) and thermally-stressed (n = 5 batches) EVPOME constructs. A FLIM threshold (dashed black line) distinguished all 5 stressed constructs from the 5 controls. Error bars represent standard deviation of all measurements from one EVPOME. The histology score > 3 in pre-implant assessment of constructs reliably predicted in vivo graft success after 1 week, shown in concurrent studies.
4. Discussion
Perhaps the best indicator of tissue-engineered construct viability is post-implantation graft integration and vascularization. The work reported here demonstrated the ability of label-free optical molecular imaging to reliably and noninvasively identify and map viable constructs prior to implantation. The reported accuracy of this optical approach was compared to standard pre-implantation measures: histological assessment (shown to be reliable) and glucose consumption (shown to be unreliable).
In related implantation studies (data not shown), both histological assessment and cytokine secretion measures of tissue-engineered constructs prior to implantation in 21 mice reliably predicted graft success in vivo, as assessed by post-implantation histology after 1 week. Taken together, these studies and the one reported here indicate that viable pre-implantation grafts initiate continuous tissue development and successful integration and vascularization at the implant site, underscoring the importance of reliable pre-implantation assessment tools.
Importantly, histological examination of thin fixed tissue sections obtained from punch biopsies and read by an expert histology panel is destructive, subjective, and labor intensive [23]. Non-invasive biomolecular assays like glucose consumption and cytokine secretion are global measures that do not provide spatially localized tissue assessment and, in the case of glucose consumption, may be unreliable. In contrast, optical molecular imaging provides a non-invasive assessment tool capable of mapping viability across the engineered construct prior to implantation.
To assure tissue-engineered construct effectiveness and safety, the FDA-approved investigational new drug protocol requires specific and rigorous cell testing and monitoring on the day of product release to assess cell viability [14,24]. Therefore, the development of reliable, label-free technologies that allow real-time, non-invasive assessment on the tissue, cellular, and molecular levels is required to provide a more complete and accurate description of construct viability both in vitro and in situ. Nonlinear optical molecular imaging with quantitative analysis provides a rapid, non-destructive, non-invasive, objective, label-free, and spatially-resolved assessment of tissue-engineered construct viability [25–29]. The constructs employed here, EVPOMEs, contained multiple endogenous sources of molecular optical contrast (including keratin, NAD(P)H, FAD, and collagen) that provided cellular and extracellular information on tissue morphology and function for viability assessments. The laser powers employed for imaging were selected after reviewing multiphoton excitation studies conducted on living tissues, as detailed in Methods. Experimental studies were performed to demonstrate that the majority of detected fluorescence for quantitative viability assessments could be attributed to intracellular metabolic co-factors (Supplementary Fig. 1).
To the best of our knowledge, this is the first report employing label-free nonlinear optical microscopy to characterize in vitro cell-based devices manufactured with primary human cells. Cellular viability information, primarily contained within fluorescence signals from metabolic co-factors NAD(P)H and FAD, was obtained by optically sectioning thin cellular layers via nonlinear optical microscopy. The acquired optical signals enabled tissue viability assessment via spatial, optical redox ratio (RR), and FLIM analysis. Importantly, nonlinear optical imaging with FLIM provided contrast using only a single detection channel (NAD(P)H), unlike RR which required two detection channels (NAD(P)H and FAD).
5. Conclusions
In this work, we demonstrated the potential of non-invasive optical techniques to provide manufacturing release criteria that are more robust than the glucose consumption assay. Importantly, the optical methods described here for quality control of EVPOME could be of value for other TERM applications. Nonlinear optical microscopy has an intrinsic ability to optically section tissues up to depths of ~1 μm (for near-infrared wavelengths). Therefore, the approach is not limited to EVPOME, but is suitable for other engineered cell-based devices, including engineered skin, heart and bladder tissues [30]. Further, a similar approach could be developed to assess in situ the viability and maturation of implanted tissues actively undergoing wound healing. This work demonstrates that technologies based on label-free nonlinear optical molecular imaging could significantly advance TERM applications by enabling non-invasive analysis of engineered tissues in real-time. Using this approach, reliable adjunctive tools could be developed to provide clinicians with quantitative feedback on engineered construct viability, enabling clinicians to select the healthiest engineered constructs for implantation, thereby improving TERM therapy and enhancing patient care.
Supplementary Material
Acknowledgments
We thank R. Wilson and S. Elahi for helpful discussions, S. Elahi, S. Y. Lee, and R. Kim for assisting in histological analysis, S. Y. Lee and K. Zhang for data analysis assistance, C. Edwards and S. Meshinchi (U.M. Microscopy and Image Analysis Lab) for technical assistance. This work is supported in part by U.S. National Institutes of Health (R01-DE-019431, to M.-A.M. and S.E.F.) and U.S. Department of Education (GAANN Fellowship, to W.R.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
C.L.M., S.E.F., and M.-A.M. conceived the study; L.-C.C., W.R.L., and S.K. prepared cell-based devices; L.-C.C. acquired the optical data; L.-C.C., W.R.L., and M.-A.M. analyzed the optical data and developed tissue viability assessment algorithms; L.-C.C., W.R.L., S.K, C.L.M., and S.E.F. performed histological and glucose analysis; L.-C.C., W.R.L., S.K, C.L.M., S.E.F., and M.-A.M. interpreted experimental results; L.-C.C., W.R.L., H.M.K., and M.-A.M. analyzed statistical significance of results. All authors contributed to writing the paper.
Competing financial interests
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Izumi K, Song J, Feinberg SE. Development of a tissue-engineered human oral mucosa: from the bench to the bed side. Cells Tissues Organs. 2004;176:134–152. doi: 10.1159/000075034. [DOI] [PubMed] [Google Scholar]
- 2.Hellman KB. Tissue engineering: translating science to product. Ashammakhi N, Reis R, Chiellini F, editors. Topics in Tissue Engineering. E-book. 2008:1–28. Available from URL: http://www.oulu.fi/spareparts/ebook_topics_in_t_e_vol4/
- 3.Haycock JW. 3D cell culture: a review of current approaches and techniques. Methods Mol Biol. 2011;695:1–15. doi: 10.1007/978-1-60761-984-0_1. [DOI] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pitts J, Sloboda R, Dragnev K, Dmitrovsky E, Mycek M-A. Autofluorescence characteristics of immortalized and carcinogen-transformed human bronchial epithelial cells. J Biomed Opt. 2001;6:31–40. doi: 10.1117/1.1333057. [DOI] [PubMed] [Google Scholar]
- 6.Lakowicz JR. Principles of Fluorescence Spectroscopy. New York: Springer; 2009. [Google Scholar]
- 7.Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc Natl Acad Sci U S A. 2007;104:19494–9. doi: 10.1073/pnas.0708425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L-C, Lloyd WR, Chang C-W, Sud D, Mycek MA. Fluorescence lifetime imaging microscopy for quantitative biological imaging. In: Sluder G, Wolf DE, editors. Methods in Cell Biology Digital Microscopy. 4. San Diego: Elsevier; 2013. pp. 457–88. [DOI] [PubMed] [Google Scholar]
- 9.Mycek M-A, Pogue BW. Handbook of Biomedical Fluorescence. New York: CRC; 2003. [Google Scholar]
- 10.Wang BG, König K, Halbhuber KJ. Two-photon microscopy of deep intravital tissues and its merits in clinical research. J Microsc. 2010;238:1–20. doi: 10.1111/j.1365-2818.2009.03330.x. [DOI] [PubMed] [Google Scholar]
- 11.Huang S, Heikal AA, Webb WW. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys J. 2002;82:2811–25. doi: 10.1016/S0006-3495(02)75621-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scanlon CS, Van Tubergen EA, Chen L-C, Elahi SF, Kuo S, Feinberg S, et al. Characterization of squamous cell carcinoma in an organotypic culture via subsurface nonlinear optical molecular imaging. Exp Biol Med. 2013;238:1233–41. doi: 10.1177/1535370213502628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levitt JM, Hunter M, Mujat C, McLaughlin-Drubin M, Münger K, Georgakoudi I. Diagnostic cellular organization features extracted from autofluorescence images. Opt Lett. 2007;32:3305–7. doi: 10.1364/ol.32.003305. [DOI] [PubMed] [Google Scholar]
- 14.Izumi K, Neiva RF, Feinberg SE. Intraoral grafting of tissue-engineered human oral mucosa. Oral Craniofac Tissue Eng. 2011;1:103–11. doi: 10.11607/jomi.te11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dela Cruz JM, McMullen JD, Williams RM, Zipfel WR. Feasibility of using multiphoton excited tissue autofluorescence for in vivo human histopathology. Biomed Opt Express. 2010;1:1320–30. doi: 10.1364/BOE.1.001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer F1, Volkmer B, Puschmann S, Greinert R, Breitbart W, Kiefer J, et al. Risk estimation of skin damage due to ultrashort pulsed, focused near-infrared laser irradiation at 800nm. J Biomed Opt. 2008;13:413201–8. doi: 10.1117/1.2960016. [DOI] [PubMed] [Google Scholar]
- 17.Balu M, Mazhar A, Hayakawa CK, Mittal R, Krasieva TB, König K, et al. In vivo multiphoton NADH fluorescence reveals depth-dependent keratinocyte metabolism in human skin. Biophys J. 2013;104:258–67. doi: 10.1016/j.bpj.2012.11.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.König K. Clinical multiphoton tomography. J Biophotonics. 2008;1:13–23. doi: 10.1002/jbio.200710022. [DOI] [PubMed] [Google Scholar]
- 19.Fitzmaurice GM, Laird NM, Ware JH. Applied Longitudinal Analysis. 2. Hoboken: John Wiley & Sons; 2011. [Google Scholar]
- 20.Wu YC, Xi P, Qu JAY, Cheung TH, Yu MY. Depth-resolved fluorescence spectroscopy reveals layered structure of tissue. Opt Express. 2004;12:3218–23. doi: 10.1364/opex.12.003218. [DOI] [PubMed] [Google Scholar]
- 21.Hotta T, Yokoo S, Terashi H, Komori T. Clinical and histopathological analysis of healing process of intraoral reconstruction with ex vivo produced oral mucosa equivalent. Kobe J Med Sci. 2007;53:1–14. [PubMed] [Google Scholar]
- 22.Gouyet J-F, Mandelbrot B. Physics and fractal structures. New York: Springer-Verlag; 1996. [Google Scholar]
- 23.Chandra M, Vishwanath K, Fichter GD, Liao E, Hollister SJ, Mycek MA. Quantitative molecular sensing in biological tissues: an approach to non-invasive optical characterization. Opt. doi: 10.1364/oe.14.006157. [DOI] [PubMed] [Google Scholar]
- 24.Izumi K, Feinberg SE, Iida A, Yoshizawa M. Intraoral grafting of an ex vivo produced oral mucosa equivalent: a preliminary report. Int J Oral Maxillofac Surg. 2003;32:188–97. doi: 10.1054/ijom.2002.0365. [DOI] [PubMed] [Google Scholar]; Express. 2006;14:6157–71. doi: 10.1364/oe.14.006157. [DOI] [PubMed] [Google Scholar]
- 25.Dittmar R, Potier E, van Zandvoort M, Ito K. Assessment of cell viability in three-dimensional scaffolds using cellular auto-fluorescence. Tissue Eng Part C Methods. 2012;18:198–204. doi: 10.1089/ten.tec.2011.0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Georgakoudi I, Rice WL, Hronik-Tupaj M, Kaplan DL. Optical spectroscopy and imaging for the noninvasive evaluation of engineered tissues. Tissue Eng Part B Rev. 2008;14:321–40. doi: 10.1089/ten.teb.2008.0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloyd WR, Chen L-C, Mycek MA. Fluorescence spectroscopy. In: Morgan SP, Rose FR, Matcher SJ, editors. Optical Techniques in Regenerative Medicine. Boca Raton: CRC; 2013. pp. 171–203. [Google Scholar]
- 28.Lloyd WR, Chen L-C, Wilson RH, Mycek MA. Biophotonics: clinical fluorescence spectroscopy and imaging. In: Moore J, Maitland DJ, editors. Biomedical Technology and Devices. Boca Raton: CRC; 2013. pp. 267–92. [Google Scholar]
- 29.Appel AA, Anastasio MA, Larson JC, Brey EM. Imaging challenges in biomaterials and tissue engineering. Biomaterials. 2013;34:6615–30. doi: 10.1016/j.biomaterials.2013.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Yamato M, Shimizu T, Sekine H, Ohashi K, Kanzaki M, et al. Reconstruction of functional tissues with cell sheet engineering. Biomaterials. 2007;28:5033–43. doi: 10.1016/j.biomaterials.2007.07.052. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.