

Background: Levels of Fn14 are increased in skeletal muscle on denervation.
Results: Denervation represses Dnmt3a levels and increases MAPK signaling in skeletal muscle to increase gene expression of Fn14.
Conclusion: Promoter hypomethylation and MAPKs play critical roles in induction of Fn14 levels in denervated muscle.
Significance: Increasing Dnmt3a or blocking MAPK signaling can be an important approach to repress Fn14 levels and denervation-induced muscle atrophy.
Keywords: DNA Methyltransferase, Epigenetics, Mitogen-activated Protein Kinase (MAPK), Muscle Atrophy, Skeletal Muscle, Specificity Protein 1 (Sp1), Transcription Factor, Cytokine, Denervation, TWEAK-Fn14 System
Abstract
The TWEAK-fibroblast growth factor-inducible 14 (Fn14) system is a critical regulator of denervation-induced skeletal muscle atrophy. Although the expression of Fn14 is a rate-limiting step in muscle atrophy on denervation, mechanisms regulating gene expression of Fn14 remain unknown. Methylation of CpG sites within promoter region is an important epigenetic mechanism for gene silencing. Our study demonstrates that Fn14 promoter contains a CpG island close to transcription start site. Fn14 promoter also contains multiple consensus DNA sequence for transcription factors activator protein 1 (AP1) and specificity protein 1 (SP1). Denervation diminishes overall genomic DNA methylation and causes hypomethylation at specific CpG sites in Fn14 promoter leading to the increased gene expression of Fn14 in skeletal muscle. Abundance of DNA methyltransferase 3a (Dnmt3a) and its interaction with Fn14 promoter are repressed in denervated skeletal muscle of mice. Overexpression of Dnmt3a inhibits the gene expression of Fn14 and attenuates skeletal muscle atrophy upon denervation. Denervation also causes the activation of ERK1/2, JNK1/2, and ERK5 MAPKs and AP1 and SP1, which stimulate the expression of Fn14 in skeletal muscle. Collectively, our study provides novel evidence that Dnmt3a and MAPK signaling regulate the levels of Fn14 in skeletal muscle on denervation.
Introduction
Skeletal muscle is a highly plastic tissue of human body that undergoes changes in size and contractility to meet basic functions such as locomotion, metabolism, and respiration. Although physiological remodeling confers adaptation, excessive loss of skeletal muscle mass that occurs in many chronic disease states such as cancer, diabetes, and chronic heart failure leads to morbidity and mortality (1–3). Skeletal muscle also undergoes atrophy when its level of neuromuscular activity is reduced, including in the conditions of immobilization, unloading, and in the settings of “functional denervation” as occurs in the elderly with sarcopenia and in patients with amyotrophic lateral sclerosis (1, 4). In almost all catabolic states, muscle atrophy occurs because of accelerated protein degradation. It is now clear that ubiquitin-proteasome system and autophagy contribute to muscle proteolysis in various conditions including in response to denervation (2, 5–7). However, the triggering events and intracellular pathways leading to the activation of these proteolytic systems in atrophying skeletal muscle remain less understood.
TNF-like weak inducer of apoptosis (TWEAK; gene name TNFSF12)2 cytokine and its receptor Fn14 (gene name TNFRSF12A) have been recently identified as one of the most important mediators of skeletal muscle atrophy in disuse conditions (3, 8). Adult skeletal muscle expresses minimal levels of Fn14 in unchallenged conditions (8, 9). Although the expression of TWEAK does not change significantly, loss of nerve supply causes a rapid and robust increase in the expression of Fn14 in skeletal muscle (8). Elevated activity of TWEAK-Fn14 system in denervated muscle causes activation of transcription factor NF-κB, which subsequently increases the gene expression of E3 ubiquitin ligase, MuRF1, a critical component of ubiquitin-proteasome system in atrophying skeletal muscle (8). The importance of TWEAK-Fn14 dyad in muscle atrophy on denervation has been confirmed by studies employing genetic mouse models. Transgenic mice overexpressing TWEAK in skeletal muscle undergo more rapid atrophy compared with wild-type mice in response to denervation. By contrast, skeletal muscle mass and function are preserved in TWEAK knock-out mice on denervation (8). These findings have highlighted that the inducible expression of Fn14 is a rate-limiting step in denervation-induced muscle atrophy. However, the mechanisms leading to the increased gene expression of Fn14 in skeletal muscle in response to denervation remain completely unknown.
Epigenetic events play important roles in skeletal muscle remodeling in various physiological and pathological conditions (10–12). DNA methylation occurs as 5-methylcytosine mostly in the context of cytosine-guanine dinucleotide, so-called CpG site. It is a well studied epigenetic modification that governs transcriptional regulation and silencing (13). Three catalytically active enzymes, DNA methyltransferase1 (DNMT1), DNMT3a, and DNMT3b, have been identified for the establishment and maintenance of DNA methylation patterns in mammalian cells. Pathologically altered DNA methylation has been described in various cancers (13, 14), and its role is starting to be revealed in many other diseases such as multiple sclerosis (15), Alzheimer's disease (16), and systemic lupus erythematosus (17). However, the role of different DNMTs in regulation of skeletal muscle atrophy program remains completely unknown.
In the present study, we have investigated the mechanisms responsible for the increased expression of Fn14 in skeletal muscle in response to denervation. Our results demonstrate that the gene expression of Fn14 in skeletal muscle on denervation is regulated through DNA methylation. Fn14 promoter contains a CpG island close to its transcription start site (TSS). Denervation of skeletal muscle leads to reduced expression of Dnmt3a and hypomethylation of multiple CpG sites in Fn14 promoter. Overexpression of Dnmt3a inhibits the expression of Fn14 and reduces atrophy in denervated skeletal muscle. Our results also demonstrate that MAPKs such as ERK1/2, JNK1/2, and ERK5 and their downstream phosphorylation targets, AP1 and SP1 transcription factors, play crucial roles in the induction of Fn14 expression in denervated skeletal muscle.
EXPERIMENTAL PROCEDURES
Animals
C57BL6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Sciatic denervation was performed following a protocol as described (8, 18). Briefly, mice were anesthetized with an intraperitoneal injection of Avertin (2,2,2-tribromoethanol); then we shaved the right hind quarters, made a 0.5-cm incision ∼0.5 cm proximal to the knee on the lateral side of the right leg, separated the muscles at the fascia, lifted out the sciatic nerve with a surgical hook or forceps, removed a 2–3-mm piece of sciatic nerve, and finally closed the incision with surgical sutures. At various time points postdenervation, muscle tissue was collected from euthanized mice for biochemical and histological studies. For one experiment, mice were given daily intraperitoneal injection of vehicle alone (100 μl of 30% Me2SO in saline) or with U0126 (10 mg/kg of body weight) or SP600125 (10 mg/kg of body weight) for a total of 3 days. All experimental protocols with mice were approved in advance by the Institutional Animal Care and Use Committee at the University of Louisville.
Bioinformatics
The promoter sequences (approximately −2 kb from start codon) for human (Homo sapiens; NT_010393.16), mouse (Mus musculus; NW_004078085.1), rat (Rattus norvegicus; NW_047333.1), and bovine (Bos taurus; NW_003104571.1) were obtained from the genome database of the National Center for Biotechnology Information. Sequence homology analysis was initially preformed using two align sequences in the basic local alignment search tool (BLAST) and dot matrix at the National Center for Biotechnology Information. Next, the conserved regions between mouse and other mammalian Fn14 promoters were analyzed using ClustalX (version 2.0) and GeneDoc (version 2.7) software. Putative transcription binding sites in Fn14 promoter were identified using TFSEARCH (Searching Transcription Factor Binding Sites, version 1.3), Promo (ALGGEN), and TFbind. Outcomes for transcription factor-binding elements were used for subsequent analysis only if their threshold scores were more than 85%.
Plasmids
Mouse Fn14 promoter fragments were isolated by performing PCR using genomic DNA as template. The sequence for forward primer was as follows: 3-kb Fn14, 5′-GGA TTG GTC GGG CTC TCG AT-3′; 2-kb Fn14, 5′-GAG ACT GCT AAG ATT TCC AGG TCG-3′; 1-kb Fn14, 5′-TTA ATC CCA GCA CTC GGA GGC-3′; 0.7-kb Fn14, 5′-GTT GCC CTT CTC TGG ACT CCA GA-3′; 0.5-kb Fn14, 5′-TCC TGC CTA CAA TCC TAG CAC TTG-3′; and 0.3-kb, 5′-GGT CTC CAC CCC AGA ATT GCA-3′. Reverse primer sequence for all Fn14 constructs was 5′-CAC CCG CAA GCC AGA CTC AG-3′. For the 1-kb Δ175 construct, we used reverse primer with sequence 5′-CAT TCA TTC AAC ACA GTC CCG C-3′. PCR products were isolated and ligated in pCR2.1 TOPO TA cloning vector (Invitrogen) and subsequently placed in front of the firefly luciferase reporter in pGL3 plasmid (Promega). We also cloned 1-kb and 1-kb Δ175 promoter fragments in pGL4.23 [luc2/minP] vector (Promega). Wild-type Dnmt3a construct was obtained from Addgene (Cambridge, MA). An enzymatically inactive mutant of Dnmt3a (Dnmt3aPC→VD) as described (19) was kindly provided by Dr. Taiping Chen of the University of Texas M. D. Anderson Cancer Center. pAP1-SEAP (secreted alkaline phosphatase) reporter plasmid was purchased from Clontech. SP1-luc as described (20) was kindly provided by Dr. Carlos J. Ciudad (University of Barcelona).
Electroporation of Plasmids in Myoblasts
To overexpress Dnmt3a or Dnmt3aPC→VD protein in C2C12 myoblasts, plasmid DNA was introduced into cells by electroporation (1500 V, 10 ms for duration, three pulses) using a neon transfection system following a protocol suggested by the manufacturer (Invitrogen). After 72 h, the cells were collected for biochemical analysis.
In Vivo Gene Transfer in Skeletal Muscle
Injection of plasmid DNA into TA muscles of mice and electroporation were performed according to a protocol as described (8, 21) with minor modifications. In brief, plasmids were prepared using an endotoxin-free kit (Qiagen) and suspended in sterile saline solution. The mice were anesthetized, and a small portion of TA muscle of both hind limbs was exposed and injected with plasmid DNA (20 μg in 20 μl of saline). After 1 min of plasmid DNA injection, a pair of platinum plate electrodes was placed against the closely shaved skin on either side of the small surgical incision, and electric pulses were delivered transcutaneously. Four 20-ms square wave pulses of 1-Hz frequency at 75 V/cm were generated using a stimulator (model S88; Grass Technologies) and delivered to the muscle. The polarity was then reversed, and a further four pulses were delivered to the muscle. After electroporation, the wound was closed with surgical clips, and mice were returned to their cages and fed a standard diet. After 7 days of plasmid electroporation, the left hind limb was denervated, whereas the right side was only sham operated. Finally, after 3 or 14 days of denervation, the mice were sacrificed, and TA muscle was isolated and used for various biochemical and morphometric analysis.
Morphometric Analysis
Undenervated or 14-day denervated TA muscle of mice electroporated with Dnmt3a plasmid along with enhanced green fluorescence protein (EGFP)-expressing plasmid was isolated and frozen in liquid nitrogen and sectioned in a microtome cryostat. For the assessment of tissue morphology, 10-μm-thick transverse sections were made and examined under Nikon Eclipse TE 2000-U microscope (Nikon). Fiber cross-sectional area (CSA) of EGFP-positive fibers in TA muscle sections was measured using Nikon NIS elements BR 3.00 software (Nikon). For each muscle, the distribution of fiber CSA was calculated by analyzing all EGFP-positive fibers as described (18).
Global DNA Methylation Assay
Genomic DNA was isolated from undenervated and denervated TA muscle of mice or C2C12 myoblasts using DNeasy® blood and tissue kit (Qiagen). Global DNA methylation was assayed using an ELISA-based commercial kit (MDQ1; Imprint methylated DNA quantification kit; Sigma-Aldrich). Two microliters genomic DNA at a concentration of 100 ng/μl was diluted with 28 μl of lysis and binding buffers. After incubation with the detection antibody, absorbance was read at 450 nm. Positive (methylated) and negative (unmethylated) control DNA was supplied with the kit. Global DNA methylation level was calculated as a percentage relative to methylated control DNA. All of the samples were analyzed in duplicate.
DNA Bisulfite Conversion and Sequencing
Genomic DNA from undenervated and 3-day denervated gastrocnemius muscle of mice was purified using the DNeasy blood and tissue kit (Qiagen). Genomic DNA was treated with sodium bisulfite to convert unmethylated cytosine residues to uracil using EZ DNA methylation kit (Zymo Research). PCR amplification of selected region with nested pairs of primers was carried out on the bisulfite converted DNA. PCR conditions were 95 °C for 5 min and 35 cycles of 95 °C for 1 min, 58 °C for 1 min, and 72 °C for 2 min, followed by 15 min at 72 °C. PCR products were cloned into the pCR 2.1 TOPO vector (Invitrogen) and sequenced using M13 forward and reverse primers. Methylation is represented for individual CpG site for ∼18 bacterial clones, and the average methylation of the region was examined. The results of sequencing were analyzed using BIQ software and BISMA.
Cell Culture, Transient Transfection, and Reporter Activity
C2C12 (a mouse myoblastic cell line) cells were purchased from the American Type Culture Collection. The cells were grown in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. Cells plated in 24-well tissue culture plates were transfected with different plasmids using Effectene transfection reagent according to the protocol suggested by the manufacturer (Qiagen). Transfection efficiency was controlled by cotransfection of myoblasts with Renilla luciferase encoding plasmid pRL-TK (Promega). Specimens were processed for luciferase expression using a dual luciferase assay system with reporter lysis buffer per manufacturer's instructions (Promega). Luciferase measurements were made using a luminometer (Berthold Detection Systems). SEAP activity in culture supernatants was measured by a standard assay using para-nitrophenyl phosphate as a substrate.
siRNA-mediated Knockdown
To study the effects of knockdown of c-Jun, SP1, and Dnmt3a, the medium of the cells was changed with fresh growth medium without antibiotics. The cells were transfected with 100 nm of control (scrambled), mouse SP1, mouse c-Jun, or mouse Dnmt3a siRNAs (Santa Cruz Biotechnology) using 9 μl of Lipofectamin RNAiMAX (Invitrogen) and 150 μl of Opti-MEM for 6 h following the manufacturer's instructions (Invitrogen). The medium of the cells was replaced with fresh medium. The cells were harvested after 72 h of transfection.
Electrophoretic Mobility Shift Assay
Skeletal muscle of mice were isolated and analyzed by EMSA as previously described (18). In brief, 25 μg of nuclear extracts prepared from control or 3-day denervated muscle was incubated with 16 fmol of 32P end-labeled consensus oligonucleotides from mouse Fn14 promoter at 37 °C for 30 min, and the DNA-protein complex was resolved on a 7.5% native polyacrylamide gel. The sequences of the oligonucleotides (underlining indicates consensus binding site) used in EMSA are as follows: SP1 (at −80-bp position), AAG GCG GGG GCG GGG GCG GAG C; AP1 (at −167-bp position), TAC TGA ACT GAC TGG GTT TAA C; AP1 (at −391-bp position), GTG GAT ATA ACT CAG TTC TCG C; and AP1 (at −504-bp position), GGC TGA GAT GAG ACA GAA TGT T. The radioactive bands from the dried gel were visualized and quantified by PhosphorImager (GE Healthcare) using ImageQuant TL software.
ChIP Assay
ChIP assay was performed according to a protocol as described (22). Briefly, undenervated and 3-day denervated gastrocnemius (GA) muscle were sliced in small pieces and cross-linked with chromatin using 1% formaldehyde for 10 min. Cross-linking was stopped by adding glycine to a final concentration of 0.125 m for 5 min. After two washes with 0.05 mm PMSF in PBS, the tissue was disrupted by mechanical grinding in PBS containing 0.5 mm PMSF and 2× protease inhibitor mixture. Cells were lysed in 10 mm Hepes, pH 8, 85 mm KCl, 0.5% Igepal, and 2× protease inhibitor mixture. The nuclear fraction was collected by centrifugation and lysed for 20 min in 50 mm Tris-HCl, 10 mm EDTA, 1% SDS at 4 °C. Nuclear extracts were sonicated (25 strokes at 30% for 15 s followed by 60 s of rest) while being kept in an ice-water bath. The lysate was cleared with salmon sperm DNA/protein G-agarose slurry (Cell Signaling Technology). Approximately 200 μl of chromatin fraction was used for each immunoprecipitation assay. After a 5× dilution in ChIP dilution buffer, 20 μl were separated (input), and the remaining diluted chromatin was incubated with the indicated antibody or mouse IgG (negative control). Detection of the immunoprecipitated DNA was performed by PCR with primers specific to Fn14 promoter region. Specifically, to detect Dnmts and SP1 binding at −80 bp in Fn14 promoter, we amplified the 123-bp segment corresponding to region −143 to −20 from ATG (start codon in Exon 1) using primers: CTT ACT CGC AAA GCA GAG TG (forward) and GAA CTG TCT GCT CCC ACC AC (reverse). For the AP1 site at −167 bp, we amplified the region from −243 to −123 using primers: TCG TGG GTA CTC AAG TTT GC (forward) and CCA CTC TGC TTT GCG AGT AA (reverse). For the AP1 site at −391 bp, we amplified the 171-bp segment corresponding to region −417 to −246 using primers: CAA GCA AAC ATA AAG GAT GTG TGG (forward) and TGG AGC GAA GGA AAT CAG TT (reverse). For the AP1 site at the −504-bp position, we amplified the 143-bp segment corresponding to region −610 to −467 using primers: GCC AAC ATC AAG ATC AGC CT (forward) and TCC CAG AGT AGC CTC GAA TT (reverse).
Western Blot
Quantitative estimation of specific protein was performed following a method as described (8, 18). In brief, protein extracts were prepared from cultured cells or skeletal muscle of mice in lysis buffer (50 mm Tris-Cl, pH 8.0, 200 mm NaCl, 50 mm NaF, 1 mm DTT, 1 mm sodium orthovanadate, 0.3% Igepal, and protease inhibitors). Approximately, 100 μg of protein was resolved on each lane on 10–12% SDS-PAGE, electrotransferred onto nitrocellulose membrane, and probed using anti-Fn14 (Cell Signaling Technology), anti-Dnmt1 (Imgenex), anti-Dnmt3a (Cell Signaling Technology), anti-Dnmt3b (Imgenex), anti-c-Jun (Santa Cruz Biotechnology), anti-SP1 (Santa Cruz Biotechnology), anti-phospho-ERK1/2 (Cell Signaling Technology), anti-ERK1/2 (Cell Signaling Technology), anti-phospho-JNK1/2 (Cell Signaling Technology), anti-JNK1/2 (Cell Signaling Technology), anti-phospho-p38 MAPK (Cell Signaling Technology), anti-p38 MAPK (Cell Signaling Technology), anti-phospho-ERK5 (Cell Signaling Technology), anti-ERK5 (Cell Signaling Technology), anti-phospho-c-Fos (Cell signaling Technology), anti-anti-phospho-c-Jun (Cell Signaling Technology), and anti-tubulin (Cell Signaling Technology) and detected by chemiluminescence. All antibodies were used at a dilution of 1:500 except anti-tubulin, which was used at 1:3000 dilutions. The bands were quantified using National Institute of Health ImageJ software.
RNA Isolation and QRT-PCR
RNA isolation and QRT-PCR were performed using a method as previously described (8, 18). The sequence of primers was as follows: Fn14, 5′-TTG GCG CTG GTT TCT AGT TTC C-3′ (forward) and 5′-TGA ATG AAT GGA CGA CGA GTG G-3′ (reverse); Dnmt1, 5′-GCA AGT CGG ACA GTG ACA-3′ (forward) and 5′-AGT GGG GCC CTT CGT GAA GTG A-3′ (reverse); Dnmt3a, 5′-GCT CTT GCT TAC AAA GAC CAC GGC A-3′ (forward) and 5′-CTC CTG TTC CTC TCC TTC CTT TCG A-3′ (reverse); Dnmt3b, 5′-GCC ATC CGT TCT CGG CTC TCC-3′ (forward) and 5′-ACC AGA GAA CAA AAG TCG AAG ACG C-3′ (reverse); and β-actin, 5′-CAG GCA TTG CTG ACA GGA TG-3′ (forward) and 5′-TGC TGA TCC ACA TCT GCT GG-3′ (reverse).
Statistical Analysis
The results are expressed as means ± S.D. Statistical analyses used Student's t test to compare quantitative data populations with normal distribution and equal variance. A value of p < 0.05 was considered statistically significant unless otherwise specified.
RESULTS
Fn14 Promoter Contains Evolutionary Conserved Region Essential for Its Transactivation
Fn14 (gene name Tnfrsf12a) was originally identified in genetic screen for growth factor-inducible genes in murine NIH 3T3 fibroblasts (23). Levels of Fn14 are dramatically induced in serum-starved NIH 3T3 upon treatment with FGF-1 and PDGF. We first investigated whether the increased expression of Fn14 in denervated muscle is a result of increased levels of FGF-1 and PDGF. Interestingly, our QRT-PCR results showed that the transcript levels of both FGF-1 and PDGF were significantly reduced in skeletal muscle 3 days post-denervation (data not shown), suggesting that these growth factors are not responsible for the up-regulation of Fn14 in denervated muscle.
Previous studies have shown that there is >90% homology between human, mouse, and rat Fn14 at amino acid sequence level (24). To gain insight into the transcriptional regulation of Fn14 gene, we first performed in silico analysis of Fn14 promoters from different species. The TSS is located −29 bp upstream of first ATG in both mouse and rat Fn14 promoters, whereas TSS is at −87 and −23 bp in human and bovine Fn14 promoters, respectively (Fig. 1). The promoter sequence for Fn14 appears to be ∼2 kb because Cldn6 gene is located ∼2 kb upstream of Fn14 gene on the same chromosome. Mouse and rat Fn14 promoter were found to be most homologous, showing ∼61% similarity at the DNA sequence level within the 2-kb promoter region. By contrast, human, mouse, and bovine Fn14 promoter showed ∼36% overall homology. Interestingly, the Fn14 promoter from all the species tested contained a highly conserved region of −170 bp upstream of first ATG in exon 1, suggesting that this evolutionary conserved region may be crucial for transcription of Fn14. Consistent with the previous in silico analysis (24), we found that Fn14 promoter in all the species tested lacks a typical TATA box near TSS (Fig. 1). We also searched for potential transcription factor binding sites by scanning the promoters for published and established consensus sequence. This analysis showed that 2 kb of Fn14 promoter contains conserved consensus DNA sequence for multiple transcription factors such as SP1, AP1, MyoD, GATAs, CEBPα, and NRF2 (Fig. 1).
FIGURE 1.

Schematic of mouse Fn14 promoter and sequence alignment of the proximal regulatory regions from different species. The highly conserved region was displayed at 177 base pairs upstream of the TSS in four mammal Fn14 promoters. Promoter sequence from human, mouse, rat, and bovine Fn14 were aligned using ClustalX and GeneDoc software. Conserved residues are shaded in black. Transcription factor binding elements were determined using TFSearch, Promo (ALGGEN), and TFbind programs. Transcription factor binding sites are indicated in the schematics of the mouse Fn14 promoter (top panel) and boxed in the alignment (bottom panel). GATAs, transcription factors; MyoD, myogenic differentiation 1); CEBPα, CCAAT/enhancer-binding protein. Identical DNA sequences for the indicated regions are shaded in black, gray, and light gray. Red arrowheads indicate the TSS in Fn14 promoter for each species.
Fn14 is expressed at very low to minimal levels in differentiated skeletal muscle of adult mice (8). However, the levels of Fn14 are increased in response to specific catabolic stimuli such as denervation, hind limb unloading, starvation, and muscle injury (8, 21, 25, 26). In contrast, Fn14 is constitutively expressed in many mesenchymal cells including C2C12 myoblasts and mouse primary myoblasts (27, 28), and the levels of Fn14 are repressed upon differentiation of myoblasts into myotubes (27). To find the minimal promoter sequence essential for the expression of Fn14 gene, mouse Fn14 promoter fragments (up to −3 kb from first ATG in exon1) were placed in front of the firefly luciferase reporter plasmid (i.e. pGL3; Promega) and transiently transfected into C2C12 myoblasts. After 48 h of transfection, luciferase activity was measured. As shown in Fig. 2A, pGL3–1kb and pGL3–2 kb constructs were most active in C2C12 myoblasts. Smaller promoter fragments (i.e. pGL3–0.3kb, pGL3–0.5kb, and pGL3–0.7kb) were also active but showed diminished luciferase activity in C2C12 myoblasts compared with pGL3–1kb or pGL3–2kb plasmids (Fig. 2A). To determine whether the conserved region of Fn14 promoter in different species is essential for its transactivation, we deleted 175 bp from pGL3–1kb plasmid and transfected into C2C12 myoblasts. Interestingly, this deletion made the pGL3–1kb promoter construct inactive for the expression of luciferase reporter in C2C12 myoblasts (Fig. 2B).
FIGURE 2.

Characterization of mouse Fn14 promoter in vitro and in vivo. A, a series of 5′-deleted fragments (−3 to −0.3 kb) of the murine Fn14 promoter was generated and cloned at the upstream of the luciferase coding sequence into the promoter-less pGL3 plasmid and transfected into C2C12 myoblasts along with Renilla plasmid (in 1:50 ratio). Schematic representation of the promoter fragments analyzed (left panel). The promoter-driven luciferase activities were measured after 48 h of myoblast transfection. The results were normalized with an empty vector (n = 4). *, p < 0.01; values vary significantly from cells transfected with empty vector (pGL3 alone). B, C2C12 myoblasts were transfected with pGL3–1kb or pGL3–1kbΔ-175bp plasmids along with Renilla plasmid, and luciferase activity was measured 48 h later (n = 4). *, p < 0.01; values vary significantly from C2C12 myoblasts transfected with empty vector. #, p < 0.01; values vary significantly from C2C12 myoblasts transfected with pGL3–1kb plasmid. C, fold change in luciferase activity (normalized using Renilla luciferase) in TA muscle transfected with empty vector (pGL3), pGL3–1kb, or pGL3–1kbΔ-175bp plasmid after 3 days of denervation (n = 3 in each group). *, p < 0.01; values vary significantly from TA muscle transfected with empty vector. #, p < 0.01; values vary significantly from TA muscle transfected with pGL3–1kb plasmid. D, C2C12 myoblasts were transfected with empty pGL3, empty pGL4.23, pGL4.23–1kb, or pGL4.23–1kbΔ-175bp plasmids along with Renilla plasmid and luciferase activity was measured 48 h later (n = 4). *, p < 0.01; values vary significantly from C2C12 myoblasts transfected with empty vectors. Error bars represent S.D.
We next investigated whether the conserved region in Fn14 promoter is essential for its transactivation in denervated muscle. pGL3–1kb or pGL3–1kbΔ-175bp plasmids were electroporated in TA muscle of mice. After 7 days of recovery from electroporation, mice were denervated for 3 days (meaning the sciatic nerve was transected) followed by measurement of luciferase activity. As shown in Fig. 2C, there was ∼8-fold increase in the luciferase activity in TA muscle electroporated with pGL3–1kb plasmid on denervation. By contrast, there was no increase in luciferase activity in denervated TA muscle transfected with pGL3–1kbΔ-175bp (Fig. 2C). Collectively, these results suggest that evolutionary conserved a ∼175-bp sequence upstream of ATG is essential for the transactivation of mouse Fn14 promoter.
Because Fn14 promoter is a TATA-less promoter and deletion of −175 bp rendered Fn14 promoter completely inactive, we next sought to determine whether this conserved region serves as a minimal promoter for the activation of Fn14 gene. We placed −1 kb and −1kbΔ-175bp fragments in the pGL4.23 [luc2/minP] vector (Promega), which contains a minimal promoter having a TATA box and binding sites for general transcription factors. As shown in Fig. 2D, the pGL4.23 empty vector has more basal level of luciferase activity compared with pGL3 vector in C2C12 myoblasts. Interestingly, there was no significant difference in luciferase activity between the cells transfected with pGL4.23–1kb and pGL4.23–1kbΔ-175bp (Fig. 2D). These results suggest that −175 bp may be the minimal promoter essential for the activation of Fn14 locus.
DNA Methylation Controls Inducible Expression of Fn14 Gene
DNA methylation is the classic epigenetic modification in mammalian cells that involves the addition of methyl group to cytosine generally at CpG dinucleotide by DNA methyltransferases (Dnmts), resulting in 5-methyl cytosine (meC). Dense methylation of CpG islands leads to gene silencing, potentially mediated by methyl-CpG binding proteins. Conversely, demethylation often correlates with gene activation (13, 29). One of the unique features of Fn14 promoter is that it contains a GC-rich region close to its transcription initiation sites (Fig. 1). We analyzed a 1-kb Fn14 promoter region for potential CpG islands using online CpG Island Searcher software. The results showed that both mouse and human Fn14 promoters contain a CpG island close to TSS. The CpG island in the mouse Fn14 promoter spans up to −316 bp, whereas in human Fn14 promoter, it is up to −625 bp from first ATG in exon 1 (Fig. 3, A and B). To obtain initial evidence regarding whether DNA methylation plays a role in Fn14 gene expression, C2C12 myoblasts and myotubes were treated with demethylation agent 5-aza-2′-deoxycytidine (5-aza-dC) for 72 h. The medium of the cells was changed every 24 h. Interestingly, 5-aza-dC increased the abundance of Fn14 in both C2C12 myoblasts and myotubes in a dose-dependent manner (Fig. 3, C and D).
FIGURE 3.
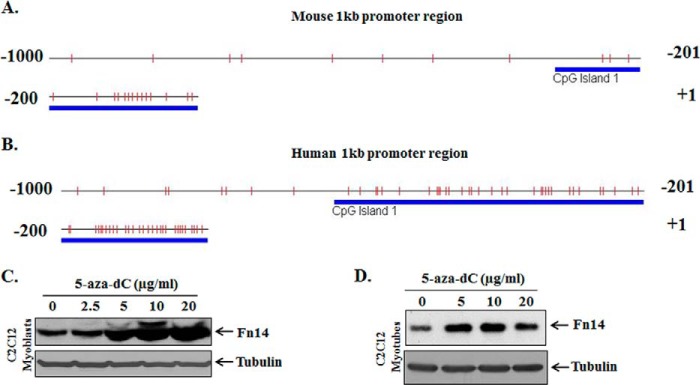
Demethylation agent 5-aza-dC increases the expression of Fn14 in cultured cells. A and B, graphical representation of methylation patterns in the specific CpG islands in mouse (A) and human (B) Fn14 promoter. The vertical bars in each graphic indicate the presence of CpG sites within −1 kb of the Fn14 promoter fragments. The bold lines indicate the presence of putative conserved CpG islands in Fn14 promoter regions evaluated by online software. C and D, C2C12 myoblasts (C) and C2C12 myotubes (D) were treated with indicated concentrations of 5-aza-dC for 72 h, and the cell extracts made were analyzed for Fn14 protein levels. Representative immunoblots presented here demonstrate that treatment with 5-aza-dC increases the amounts of Fn14 in both C2C12 myoblasts and myotubes. The levels of unrelated protein tubulin remained comparable between untreated and 5-aza-dC-treated cells.
Fn14 Promoter Undergoes Hypomethylation in Denervated Skeletal Muscle
Because denervation of skeletal muscle causes a rapid increase in the gene expression of Fn14 (8), we next sought to determine whether the CpG island in Fn14 promoter undergoes demethylation in skeletal muscle upon denervation. To study the methylation status of Fn14 promoter especially in the conserved −170-bp region, we employed methylation-specific PCR and bisulfite sequencing, the commonly used techniques for such studies (30–33). Genomic DNA was extracted from GA muscle of mice using a commercially available kit (Qiagen) and treated with sodium bisulfite to convert unmethylated (but not methylated) cytosine residues to uracil using EZ DNA methylation kit (Zymo Research). The modified DNA was used as a template for PCR. The results showed that mouse Fn14 promoter contains 13 CpG sites within the −170-bp region upstream of ATG (Fig. 4A), and these sites were largely methylated in undenervated muscle (Fig. 4, B and C). Interestingly, many CpG sites were found to be hypomethylated in denervated skeletal muscle compared with undenervated muscle (Fig. 4, B and C). Because cultured C2C12 myoblasts constitutively express Fn14 protein, we also investigated methylation status of 13 CpG sites within −170bp region upstream of ATG. Compared with undenervated muscle of mice, many CpG sites were found to be highly demethylated in C2C12 myoblasts (Fig. 4D). These results suggest that demethylation at specific CpG sites in Fn14 promoter might be responsible for the constitutive expression of Fn14 in cultured cells.
FIGURE 4.

Methylation status of CpG sites within 5′-flanking region of Fn14 gene. A, schematic illustration of CpG sites within conserved region of mouse Fn14 promoter. B, bisulfite sequencing results demonstrating methylated and unmethylated CpG sites in undenervated and 3-day denervated GA muscle of mice. Each row of small boxes represents an individual clone sequenced. Unmethylated (blue boxes) or methylated (red boxes) CpG sites are indicated. C, quantification of methylation on CpG sites in Fn14 promoter in undenervated and 3-day denervated GA muscle of mice (n = 3). The data presented here demonstrate that denervation causes demethylation at specific CpG sites in Fn14 promoter. D, quantification of methylation on CpG sites in Fn14 promoter in undenervated GA muscle of mice and cultured C2C12 myoblasts. The data presented here demonstrate increased demethylation at specific CpG sites in Fn14 promoter in C2C12 myoblasts compared with naïve adult skeletal muscle of mice (n = 3 in each group). E, relative global DNA methylation levels in undenervated and denervated muscle were calculated as percentages relative to methylated control DNA using the Imprint® methylated DNA quantification kit (n = 4 in each group). Error bars represent the S.D. *, p < 0.05; values vary significantly from undenervated skeletal muscle.
Although our analysis showed that Fn14 promoter undergoes hypomethylation, it is not clear whether this phenomenon is specific to Fn14 promoter or whether there is a genome-wide change in the level of DNA methylation in skeletal muscle in response to denervation. To address this issue, we isolated genomic DNA from undenervated and denervated muscle and measured methylation levels using an ELISA-based kit. Intriguingly, overall DNA methylation level was found to be significantly reduced in denervated skeletal muscle compared with contralateral undenervated muscle of mice (Fig. 4E). These results provide initial evidence that DNA methylation might be one of the important epigenetic events that regulates the gene expression of Fn14 in denervated skeletal muscle.
Dnmt3a Regulates the Expression of Fn14 and Fiber Atrophy in Denervated Muscle
In mammalian cells, there are three Dnmts (i.e. Dnmt1, Dnmt3a, and Dnmt3b) that cause DNA methylation. However, it remains unknown how their levels are regulated in skeletal muscle in response to denervation. QRT-PCR analysis showed that the mRNA levels of Dnmt3a are significantly reduced in denervated skeletal muscle compared with contralateral undenervated muscle. In contrast, transcript levels of Dnmt1 and Dnmt3b remained comparable between undenervated and denervated muscle of mice (Fig. 5A). Furthermore, protein levels of Dnmt3a (but not Dnmt1 and Dnmt3b) were found to be considerably reduced in denervated GA muscle compared with contralateral undenervated muscle (Fig. 5B). We next investigated whether Dnmts interact with methylated region of Fn14 promoter in skeletal muscle. ChIP analysis showed that all three Dnmts enrich Fn14 promoter (Fig. 5C). Furthermore, we found that the enrichment of Dnmt3a on Fn14 promoter was reduced (∼65%) in denervated muscle compared with undenervated muscle (Fig. 5C). However, there was no significant change in the enrichment of Dnmt1 or Dnmt3b on Fn14 promoter in denervated muscle compared with undenervated controls (Fig. 5C).
FIGURE 5.

Dnmt3a regulates the gene expression of Fn14 in denervated muscle. A, relative mRNA levels of Dnmt1, Dnmt3a, and Dnmt3b in undenervated and 3-day denervated GA muscle of mice measured by QRT-PCR (n = 3 in each group). *, p < 0.01; values vary significantly from contralateral undenervated GA muscle of mice. B, immunoblots showing protein levels of Dnmt1, Dnmt3a, Dnmt3b, and unrelated protein tubulin in undenervated and 3-day denervated GA muscle of mice. C, sheared chromatin prepared from undenervated and denervated GA muscle was subjected to ChIP analysis probing for Dnmt1, Dnmt3a, and Dnmt3b antibody. The data presented here demonstrate that enrichment of Dnmt3a (but not Dnmt1 or Dnmt3b) to Fn14 promoter is reduced upon denervation. D, TA muscle of mice was electroporated with vector alone or with Dnmt3a plasmid followed by denervation for 3 days. Relative mRNA level of Fn14 in vector alone or Dnmt3a transfected undenervated and denervated TA muscle (n = 4). *, p < 0.01; values vary significantly from contralateral undenervated muscle. #, p < 0.01; values vary significantly from denervated muscle transfected with vector alone. E, Western blot analysis demonstrating the protein levels of Fn14 and Dnmt3a in undenervated and denervated TA muscle transfected with vector alone or Dnmt3a plasmid. F and G, C2C12 myoblasts were transfected by electroporation with vector alone, wild-type Dnmt3a, or mutant Dnmt3aPC→VD plasmid for 72 h. Cellular extracts prepared were probed for protein levels of Fn14 and Dnmt3a by Western blot. Representative immunoblots presented here demonstrate that overexpression of wild-type Dnmt3a reduced, whereas overexpression of enzymatically dead Dnmt3aPC→VD mutant increased the protein levels of Fn14 in C2C12 myoblasts. H, C2C12 myoblasts were transfected with control or Dnmt3a siRNA followed by measurement of protein levels of Dnmt3a and Fn14. Representative immunoblots from two independent experiments each done in triplicate show that knockdown of Dnmt3a increases the levels of Fn14 in C2C12 myoblasts. Un, undenervated; Dn, denervated.
To understand whether repression of Dnmt3a is a mechanism for the induction of Fn14 levels in denervated muscle, we studied the effect of overexpression of Dnmt3a on mRNA and protein levels of Fn14. TA muscle of the mice was electroporated with vector alone or Dnmt3a plasmid. The mice were denervated by transection of sciatic nerve, and the mRNA and protein levels of Fn14 in TA muscle were measured 3 days post-denervation by performing QRT-PCR and Western blot, respectively. Interestingly, overexpression of Dnmt3a significantly reduced both mRNA and protein levels of Fn14 in denervated TA muscle (Fig. 5, D and E). To further understand the role of Dnmt3a in Fn14 expression, we overexpressed wild-type Dnmt3a protein in C2C12 myoblasts and studied protein levels of Fn14. As shown in Fig. 5F, forced expression of Dnmt3a drastically reduced the levels of Fn14 protein in C2C12 myoblasts. We also studied the effects of overexpressed Dnmt3aPC→VD mutant, which is enzymatically dead and functions as dominant negative inhibitor of Dnmt3a (19), in C2C12 myoblasts. The results showed that overexpression of Dnmt3aPC→VD increased the levels of Fn14 protein in C2C12 myoblasts (Fig. 5G). Finally, siRNA-mediated knockdown of Dnmt3a significantly increased (control siRNA versus Dnmt3a siRNA: 1.0 ± 0.78 versus 2.84 ± 0.37) the abundance of Fn14 protein in C2C12 myoblasts (Fig. 5H). Collectively, these results suggest that Dnmt3a represses the levels of Fn14 in myogenic cells.
Because Dnmt3a regulates gene expression of Fn14 in skeletal muscle on denervation, we also evaluated whether Dnmt3a has a role in denervation-induced skeletal muscle atrophy. TA muscle of wild-type mice was electroporated with vector alone or Dnmt3a plasmid. Efficiency of gene delivery was monitored by co-electroporation with EGFP-expressing plasmid as described (21). The mice were then denervated for 14 days followed by quantification of fiber CSA. Interestingly, overexpression of Dnmt3a significantly inhibited the denervation-induced loss in fiber CSA (Fig. 6, A and B). Collectively, these results suggest that denervation represses the expression of Dnmt3a, which leads to its reduced enrichment at CpG sites in Fn14 promoter leading to Fn14 promoter hypomethylation and activation.
FIGURE 6.
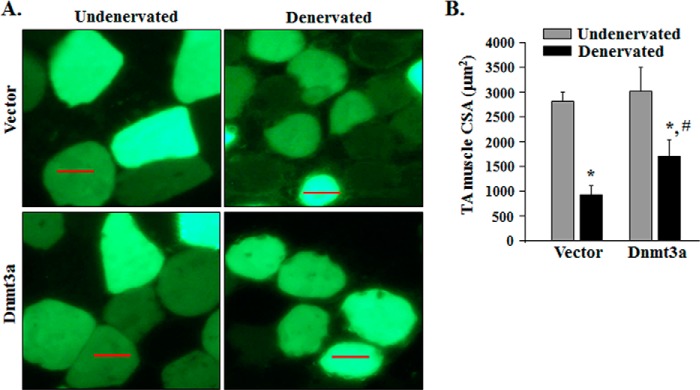
Forced expression of Dnmt3a inhibits denervation-induced muscle atrophy. A, TA muscle of mice was electroporated with vector alone or Dnmt3a plasmid along with an EGFP expressing plasmid. Representative photomicrographs of undenervated, and 14-day denervated TA muscle sections are presented here. Scale bars, 50 μm. B, average fiber CSA in vector alone and Dnmt3a-transfected TA muscle after 14 days of denervation (n = 4 in each group). *, p < 0.01; values vary significantly from undenervated TA muscle transfected with vector alone or Dnmt3a. #, p < 0.01; values vary significantly from denervated TA muscle transfected with vector alone. Error bars represent the S.D.
SP1 and AP1 Transcription Factors Bind to Fn14 Promoter in Denervated Skeletal Muscle
Although Fn14 is a TATA-less promoter, it contains a consensus DNA sequence for several transcription factors including SP1 near its TSS. It has been suggested that the SP1-mediated TATA-less mechanism causes preferential responsiveness to specific activators for many genes (34). In addition to SP1, our in silico analysis showed that the 1-kb region of Fn14 promoter contains consensus sequence for many other transcription factors such as NRF2, MyoD, AP1, GATAs, and C/EBPα (Fig. 1). Because mere presence of consensus sequence does not always guarantee that transcription factor will bind, we first performed EMSA to study the binding of specific transcription factors to their consensus sequence in Fn14 promoter. As shown in Fig. 7A, DNA binding activity of SP1 to its consensus sequence at −80 bp upstream of ATG was significantly increased in denervated muscle. Furthermore, EMSA showed that AP1 binds to three consensus AP1 binding sites (at −167, −391, and −504 bp upstream of ATG) in Fn14 promoter (Fig. 7A). By contrast, there was no increase in DNA binding activity of NRF2, GATAs, and C/EBPα transcription factors to their consensus sequence in Fn14 promoter in denervated muscle (data not shown). SP1 also bound to another consensus sequence (at −177 bp upstream of ATG) in Fn14 promoter. However, there was no difference in the level of binding at this sequence between undenervated and denervated muscle (data not shown).
FIGURE 7.

SP1 and AP1 transcription factors bind to mouse Fn14 promoter in denervated skeletal muscle. A, EMSA gels presented here demonstrate that DNA binding activity of SP1 and AP1 to their consensus sequence at indicated positions upstream of ATG in Fn14 promoter is increased in GA muscle of mice after 3 days of denervation. B and C, undenervated and 3-day denervated GA muscle of mice was processed for ChIP assay to study in vivo binding of SP1 (B) and c-Jun (C) to their consensus sequence in mouse Fn14 promoter. The data presented here demonstrate that enrichment of SP1 and c-Jun to Fn14 promoter is drastically increased in denervated GA muscle of mice. PCR was performed with primer sets amplifying SP1 (−80 bp) or AP1 (−167, −391, and −504 bp) consensus sequence containing fragments within the −1-kb region of mouse Fn14 promoter. Total input (10%) was used as a positive control, whereas the isotype-matched IgG was used as a negative control. Myoblasts were transfected with control, SP1, or c-Jun siRNA for 48 h, and the protein extracts made were probed for Fn14, SP1, and c-Jun. D and E, representative immunoblots from two independent experiments each done in triplicate presented here demonstrate that knockdown of SP1 (D) or c-Jun (E) decreased the protein levels of Fn14 in myoblasts.
To further ascertain whether SP1 and AP1 interact with Fn14 promoter in vivo, we performed ChIP assay. As shown in Fig. 7B, enrichment of SP1 at the −80-bp sequence in Fn14 promoter was significantly increased (∼18-fold) in denervated muscle compared with contralateral undenervated controls. Similarly, enrichment of AP1 was also increased by ∼300-fold at −167, ∼50-fold at −391, and ∼75-fold at −504 consensus binding sites (Fig. 7C). To determine whether SP1 and AP1 transcription factors play a role in Fn14 expression, we investigated the effects of knockdown of SP1 and c-Jun (a subunit of AP1) on the levels of Fn14 protein in cultured myoblasts. Interestingly, siRNA-mediated knockdown of SP1 significantly reduced protein levels of Fn14 (control siRNA versus SP1 siRNA, 1.0 ± 0.34 versus 0.4 ± 0.09) in myoblasts (Fig. 7D). Similarly, knockdown of c-Jun significantly diminished the levels of Fn14 protein (control siRNA versus c-Jun siRNA, 1.0 ± 0.36 versus 0.37 ± 0.23) in myoblasts (Fig. 7E), suggesting that both SP1 and AP1 are involved in the gene expression of Fn14 in myogenic cells.
MAPKs Are Involved in the Increased Levels of Fn14 in Denervated Skeletal Muscle
We next investigated the mechanisms by which denervation increases the activity of AP1 and SP1 transcription factors in muscle cells. Several published studies have shown that MAPKs such as ERK1/2 and JNK1/2 phosphorylate subunits of AP1 transcription factor, leading to its nuclear translocation and up-regulation of target genes (35). Furthermore, ERK5 has been recently found to increase the activity of SP1 in myogenic cells (36). Protein extracts prepared from undenervated and 3-day denervated GA muscle of mice were immunoblotted with phospho-ERK1/2, phospho-JNK1/2, phospho-p38, and phospho-ERK5 antibody. Interestingly, the phosphorylation of ERK1/2, JNK1/2, and ERK5 was found to be significantly up-regulated in skeletal muscle upon denervation (Fig. 8, A and C). Furthermore, the phosphorylation of c-Jun subunit of AP1 was also found to be drastically increased in denervated muscle compared with undenervated controls (Fig. 8, B and C). In contrast, the phosphorylation levels of p38 MAPK remained comparable between undenervated and denervated GA muscle (Fig. 8A).
FIGURE 8.

Activation of MAPK in skeletal muscle upon denervation. A, representative immunoblots presented here demonstrate phosphorylated and total ERK1/2, JNK1/2, p38 MAPK, and ERK5 protein in undenervated and 3-day denervated GA muscle of mice. Black lines indicate that intervening lanes have been spliced out. B, representative immunoblots presented here demonstrate phosphorylated and total c-Jun and c-Fos protein in undenervated and 3-day denervated GA muscle of mice. C, densitometry quantification of immunoblots of undenervated and denervated GA muscle (n = 3 in each group). *, p < 0.05; values vary significantly from undenervated muscle of mice. Error bars represent the S.D. D, C2C12 myoblasts grown in growth medium were treated with vehicle alone, U0126 (20 μm), PD184352 (2 μm), or SP600125 (50 μm) for 24 h, and cellular extracts made were probed for Fn14. Representative immunoblots presented here show that protein levels of Fn14 are reduced in C2C12 myoblasts upon treatment with U0126, PD184352, and SP600125 compounds. E, C2C12 myoblasts were transfected with pAP1-SEAP or pSP1-luc plasmids for 24 h followed by treatment with U0126 (20 μm) or SP600125 (50 μm) for an additional 24 h and measurement of reporter gene activity in cell extracts. The data presented here show that treatment with U0126 significantly reduced the activity of AP1 and SP1 reporter, whereas SP600125 reduced activity of AP1 reporter. *, p < 0.05; values vary significantly from those treated with vehicle alone. F and G, protein levels of Fn14 in undenervated and denervated TA muscle of mice after treatment with vehicle alone, U0126 (10 mg/kg of body weight), or SP600125 (10 mg/kg of body weight) for a total 3 days (n = 3 in each group). Error bars represent the S.D. *, p < 0.01; values vary significantly from undenervated muscle of mice.
To understand whether MAPKs are involved in the expression of Fn14, we performed both in vitro and in vivo studies. We used two different MEK inhibitors, U0126 and PD184352, which have different specificities. U0126 inhibits both ERK1/2 and ERK5 pathways, whereas PD184352 specifically inhibits ERK1/2 without affecting ERK5 pathway (36). As shown in Fig. 8D, treatment with U0126 reduced (∼90%) the levels of Fn14 protein in C2C12 myoblasts. Moreover, treatment with PD184352 and SP600125 (an inhibitor of JNK1/2) also diminished (∼50%) protein levels of Fn14 in C2C12 myoblasts (Fig. 8D). Before testing them in vivo, we first sought to determine whether U0126 and SP600125 affect the activity of AP1 and SP1 using reporter assays. C2C12 myoblasts were transiently transfected with pAP1-SEAP or pSP1-luc reporter plasmids followed by treatment with U0126 or SP600125. As shown in Fig. 8E, treatment with U0126 significantly reduced the transactivation of both AP1 and SP1 reporters in C2C12 myoblasts. By contrast, SP600125 inhibited only AP1 without having any significant effect on the transactivation of SP1 (Fig. 8E). These results suggest that MAPKs regulate the activity of SP1 and AP1 in myogenic cells. To evaluate the role of MAPK-AP1 signaling in denervation-induced increase in the levels of Fn14, we treated mice with U0126 or SP600125 and studied the levels of Fn14 in skeletal muscle 3 days post-denervation. The results showed that treatment with either U0126 or SP600125 reduced the protein levels of Fn14 in denervated skeletal muscle (Fig. 8, F and G). Interestingly, whereas U0126 was more effective than SP600125 in reducing Fn14 levels in cultured myoblasts, in denervated muscle SP600125 was more effective than U0126 in reducing the levels of Fn14. This discrepancy could be attributed to difference in their effective concentration in vivo or bioavailability of these compounds after administration in mice. Nevertheless, because both U0126 and SP600125 reduced the levels of Fn14 in denervated muscle, it is evident that MAPK signaling is essential for the up-regulation of Fn14 in denervated skeletal muscle.
DISCUSSION
Loss of nerve supply causes progressive skeletal muscle atrophy. Recent studies have identified a few transcription factors, such as Foxo, myogenin, NF-κB, and AP1, that cause skeletal muscle atrophy through augmenting the expression of various atrogenes (37–42). It has been also reported that the expression of TWEAK receptor Fn14 is rapidly increased in various disuse conditions including denervation and that TWEAK-Fn14 dyad drives a muscle atrophy program through the activation of NF-κB and ubiquitin-proteasome system (8, 26). TWEAK receptor Fn14 is quite unique among the members of TNF receptor superfamily (TNFRSF). It is the smallest member of the TNFRSF and lacks a death domain in its cytoplasmic tail. Although most members of TNFRSF are constitutively expressed, the expression of Fn14 is highly inducible in many organs including skeletal muscle, kidney, and liver in response to injury (43, 44). However, the mechanisms that regulate the inducible expression of Fn14 have not been yet identified.
Fn14 promoter lacks the typical TATA box sequence important for mammalian transcription initiation; however, it contains a consensus SP1 sequence close to the TSS (Fig. 1). Although the biological significance of TATA-less promoters remains debatable, it has been argued that SP1-mediated TATA-less mechanism causes preferential responsiveness to specific activators (34). Methylation of cytosine within CpG sites is a major form of DNA modification that plays an important role in the regulation of gene expression (45). DNA methylation silences gene expression by sterically hindering transcription factors from binding to their promoter or by providing binding sites for methyl-binding domain proteins that recruit histone deacetylase (HDAC)-containing repressor complexes to modify chromatin structure (29, 46). Sequential deletion of mouse Fn14 promoter revealed that ∼1 kb sequence upstream of the first ATG site in Fn14 exon 1 is sufficient for its full transcriptional activation in both cultured myoblasts and denervated skeletal muscle of mice (Fig. 2). Moreover, Fn14 promoter contains a conserved region (∼170 bp) close to TSS, which is critical for the transactivation of Fn14 promoter in both cultured cells and denervated skeletal muscle of mice. Intriguingly, this evolutionarily conserved region also contains a CpG island, and many potential CpG sites within the CpG island are methylated in undenervated skeletal muscle. On denervation, the CpG sites within this region undergo hypomethylation with concomitant expression of Fn14 in skeletal muscle, indicating that demethylation of Fn14 promoter is an important mechanism for the inducible expression of Fn14 during denervation-induced muscle atrophy (Figs. 4 and 5). It is also notable that one of the CpG sites (i.e. #6 in Fig. 4A) that are highly demethylated upon denervation is within the consensus SP1 sequence close to TSS (Figs. 1 and 4). Indeed, our EMSA and ChIP analyses confirmed that the binding of SP1 to this DNA consensus sequence is enhanced in denervated skeletal muscle, and SP1 is required for the expression of Fn14 in cultured muscle cells (Fig. 7). It is notable that although this conserved region is essential and appears to be the minimal promoter, it is not sufficient for the full transcriptional activation of Fn14. The Fn14 promoter is approximately −1 kb and contains consensus binding sites for multiple transcription factors.
Although the role of histone modifiers such as HDACs in skeletal muscle atrophy has been identified to some extent (37), it remains unknown whether DNA methylation also plays a role in denervation-induced muscle atrophy. Our study provides initial evidence that overall genome-wide methylation (Fig. 4E) and the levels of Dnmt3a are repressed in denervated skeletal muscle of mice (Fig. 5, A and B). Because forced expression of Dnmt3a reduced the expression of Fn14 in denervated skeletal muscle and in cultured myogenic cells (Fig. 5), it is likely that Dnmt3a mediates the methylation of CpG sites within Fn14 promoter in skeletal muscle. Furthermore, reduced amounts and/or interaction of Dnmt3a may also lead to an inability to re-establish DNA methylation at demethylated Fn14 locus, shifting the equilibrium toward the demethylated state in denervated muscle. Our study also demonstrates that forced expression of Dnmt3a inhibits skeletal muscle atrophy upon denervation (Fig. 6). Although Fn14 promoter is one of potential methylation targets of Dnmt3a, it is also possible that Dnmt3a causes methylation of several other promoters whose gene products are involved in denervation-induced muscle atrophy. Identification of additional targets of Dnmt3a-mediated methylation in denervated skeletal muscle is a key area for future research.
Our results show that MAPK signaling and AP1 transcription factor are also important regulators of gene expression of Fn14 in denervated skeletal muscle (Figs. 7 and 8). AP1 is composed of homodimers and heterodimers of Jun and Fos family proteins (47). It is also one of the most important transcription factors activated through MAPK signaling cascades (47). A plethora of literature now exists suggesting that the activation of MAPK-AP1 is responsible for various biological responses including cellular proliferation in response to growth factors and serum stimulation (35). Indeed, Fn14 was identified in a genetic screen as one of the highly inducible genes in serum-stimulated cells (23, 48). Our results demonstrate that denervation causes rapid activation of ERK1/2 and JNK1/2 in skeletal muscle (Fig. 8A). Furthermore, the phosphorylation of c-Jun subunit of AP1 is increased in denervated skeletal muscle of mice (Fig. 8B), indicating that muscle denervation causes the activation of AP1 through stimulating MAPK signaling pathways in skeletal muscle. Fn14 promoter contains multiple AP1 binding sequences, and our results validate that AP1 binds to at least three AP1 consensus DNA sequence within the −1-kb promoter region of Fn14 (Fig. 7, A and C). Furthermore, siRNA-mediated knockdown of c-Jun or pharmacological inhibition of MAPKs reduced the levels of Fn14 protein in cultured myoblast or denervated muscle, confirming that MAPKs and AP1 are required for the expression of Fn14 (Figs. 7E and 8, D, F, and G).
The mechanisms by which denervation causes the activation of MAPKs and AP1 in skeletal muscle remain enigmatic. However, a previous study has demonstrated that the expression of HDAC4 is highly up-regulated in denervated muscle, and it mediates atrophy through augmenting the expression of muscle-specific E3 ubiquitin ligases, MAFBx and MuRF1 (37). More recently, it has been reported that HDAC4 binds and promotes the deacetylation and activation of MEKK2, which leads to the activation of downstream MAPK cascades and AP1 transcription factor (42). The role of HDAC4-MAPK-AP1 signaling cascade in denervation-induced muscle atrophy has been validated by the findings that knockdown of HDAC4 or the components of AP1 rescues muscle atrophy to similar extents (42). The results of the present study suggest that Fn14 is one of the important downstream targets of HDAC4-MAPK-AP1 signaling during denervation-induced muscle atrophy.
Intriguingly, we have also found that the activation of ERK5 is increased in skeletal muscle in response to denervation (Fig. 8, A and C). Although the role of ERK5 in muscle atrophy remains unknown, ERK5 phosphorylates SP1 transcription factor, leading to its binding to the consensus DNA sequence in the promoters of Klf2/4 genes in myogenic cells (36). Because binding of SP1 to Fn14 promoter is increased in denervated skeletal muscle, it is possible that activation of ERK5 stimulates SP1 activity, leading to the increased expression of Fn14. Previous studies have demonstrated that SP1 also gets activated and plays an important role in muscle atrophy in response to dexamethasone treatment (49, 50). SP1 binds to promoter region and augments gene expression of ubiquitin, leading to increased activation of ubiquitin-proteasome system. Interestingly, U0126, which inhibits both ERK1/2 and ERK5, has been shown to repress the dexamethasone-induced gene expression of ubiquitin in cultured myotubes potentially through inhibiting the activity of SP1 (49). Taken together, these findings suggest that the activation of SP1 causes muscle atrophy through augmenting the gene expression of various proteins including Fn14 and ubiquitin.
Our results also demonstrate that Fn14 promoter is hypomethylated in C2C12 myoblasts, which may facilitate the binding of AP1 and SP1 to their consensus sequence in Fn14 promoter (Fig. 4D). Constitutive expression of Fn14 in cultured cells may be a result of promoter hypomethylation and increased levels of growth factors and mitogens in serum-containing medium that facilities the activation of MAPKs. Previous studies have also demonstrated that several growth factors such as FGF-1 and PDGF induce the gene expression of Fn14 in serum-starved fibroblasts (23, 51), further suggesting that growth factors are some of the important stimuli for the increased levels of Fn14 in cultured cells. It is also notable that Fn14 is expressed at high levels in developing skeletal muscle of neonatal/young mice. However, Fn14 levels are dramatically reduced in skeletal muscle of adult mice (9). Although it is possible that growth factors play a role in the expression of Fn14 in skeletal muscle during development, they are not the key regulators of Fn14 expression in denervated muscle of adult mice because the expression of some of these factors (e.g. FGF and PDGF) is indeed reduced in denervated muscle of mice (data not shown).
In summary, our study provides novel evidence that promoter demethylation and MAPK signaling cascades play a critical role in the increased gene expression of Fn14 during denervation-induced muscle atrophy. Our study has also identified a role for Dnmt3a in muscle atrophy in response to denervation. Although our results demonstrate that Dnmt3a and promoter demethylation are critical regulators, at present, we cannot rule out the possibility that other epigenetic regulators involved in chromatic remodeling also promote the activation of Fn14 locus in skeletal muscle on denervation. Based on the results of this study, we propose a putative model for the inducible expression of Fn14 in skeletal muscle in response to denervation (Fig. 9). In undenervated skeletal muscle, Dnmt3a (and potentially other Dnmts) hypermethylates CpG sites in Fn14 promoter, which prevents binding of AP1 and SP1 to their consensus DNA sequence. Moreover, MAPKs and AP1 and SP1 are inactive in undenervated skeletal muscle. In response to denervation, the levels of Dnmt3a and its enrichment to Fn14 promoter are reduced leading to promoter hypomethylation. Denervation also stimulates ERK1/2, JNK1/2, and ERK5 pathways, which cause the activation and nuclear translocation of AP1 and SP1 and their interaction with hypomethylated promoter to initiate the gene expression of Fn14 (Fig. 9).
FIGURE 9.

Schematic model describing regulation of gene expression of fn14 in denervated skeletal muscle. In undenervated muscle, Dnmt3a is constitutively expressed, which keeps Fn14 promoter in methylated state. MAPK pathways are also inactive in undenervated muscle. Denervation inhibits the gene expression of Dnmt3a leading to hypomethylation of CpG island in Fn14 promoter region. Denervation also stimulates MAPK signaling and downstream activation of AP1 and SP1 essential for Fn14 gene expression. Me, methylation.
Acknowledgments
We are grateful to Dr. Taiping Chen (University of Texas M. D. Anderson Cancer Center, Smithville, TX) for providing enzymatically inactive Dnmt3a mutant and Dr. Carlos J. Ciudad (University of Barcelona, Barcelona, Spain) for providing pSP1-luc reporter plasmid. We also sincerely thank Dr. Junaith S. Mohamed (West Virginia University, Morgantown, WV) for valuable help in standardization of chromatin immunoprecipitation assays.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 AG029623 and R01 AR059810 (to A. K.) and R01 NS073584 (to M. H.).
- used are: TWEAK
- tumor necrosis factor-like weak inducer of apoptosis
- 5-aza-dC
- 5-aza-2′-deoxycytidine
- AP1
- activator protein 1
- CSA
- cross-sectional area
- Dnmt
- DNA methyltransferase
- EGFP
- enhanced green fluorescent protein
- Fn14
- fibroblast growth factor-inducible 14
- GA
- gastrocnemius
- HDAC
- histone deacetylase
- QRT-PCR
- quantitative real time PCR
- SP1
- specificity protein 1
- TA
- tibial anterior
- TSS
- transcription start site
- SEAP
- secreted alkaline phosphatase
- TNFRSF
- TNF receptor superfamily.
REFERENCES
- 1. Bonaldo P., Sandri M. (2013) Cellular and molecular mechanisms of muscle atrophy. Dis. Model Mech. 6, 25–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glass D. J. (2010) Signaling pathways perturbing muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 13, 225–229 [DOI] [PubMed] [Google Scholar]
- 3. Bhatnagar S., Kumar A. (2012) The TWEAK-Fn14 system: breaking the silence of cytokine-induced skeletal muscle wasting. Curr. Mol. Med. 12, 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jackman R. W., Kandarian S. C. (2004) The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 287, C834–C843 [DOI] [PubMed] [Google Scholar]
- 5. Glass D. J. (2005) Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 37, 1974–1984 [DOI] [PubMed] [Google Scholar]
- 6. Lecker S. H., Goldberg A. L., Mitch W. E. (2006) Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 17, 1807–1819 [DOI] [PubMed] [Google Scholar]
- 7. Sandri M. (2010) Autophagy in skeletal muscle. FEBS Lett. 584, 1411–1416 [DOI] [PubMed] [Google Scholar]
- 8. Mittal A., Bhatnagar S., Kumar A., Lach-Trifilieff E., Wauters S., Li H., Makonchuk D. Y., Glass D. J., Kumar A. (2010) The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J. Cell Biol. 188, 833–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dogra C., Changotra H., Wedhas N., Qin X., Wergedal J. E., Kumar A. (2007) TNF-related weak inducer of apoptosis (TWEAK) is a potent skeletal muscle-wasting cytokine. FASEB J. 21, 1857–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Proserpio V., Fittipaldi R., Ryall J. G., Sartorelli V., Caretti G. (2013) The methyltransferase SMYD3 mediates the recruitment of transcriptional cofactors at the myostatin and c-Met genes and regulates skeletal muscle atrophy. Genes Dev. 27, 1299–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guasconi V., Puri P. L. (2008) Epigenetic drugs in the treatment of skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 11, 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alamdari N., Aversa Z., Castillero E., Hasselgren P. O. (2013) Acetylation and deacetylation: novel factors in muscle wasting. Metabolism 62, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Law J. A., Jacobsen S. E. (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jones P. A., Baylin S. B. (2007) The epigenomics of cancer. Cell 128, 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Casaccia-Bonnefil P., Pandozy G., Mastronardi F. (2008) Evaluating epigenetic landmarks in the brain of multiple sclerosis patients: a contribution to the current debate on disease pathogenesis. Prog. Neurobiol. 86, 368–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mastroeni D., McKee A., Grover A., Rogers J., Coleman P. D. (2009) Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer's disease. PLoS One 4, e6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Javierre B. M., Fernandez A. F., Richter J., Al-Shahrour F., Martin-Subero J. I., Rodriguez-Ubreva J., Berdasco M., Fraga M. F., O'Hanlon T. P., Rider L. G., Jacinto F. V., Lopez-Longo F. J., Dopazo J., Forn M., Peinado M. A., Carreño L., Sawalha A. H., Harley J. B., Siebert R., Esteller M., Miller F. W., Ballestar E. (2010) Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. 20, 170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paul P. K., Gupta S. K., Bhatnagar S., Panguluri S. K., Darnay B. G., Choi Y., Kumar A. (2010) Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell Biol. 191, 1395–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen T., Tsujimoto N., Li E. (2004) The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol. Cell Biol. 24, 9048–9058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nicolás M., Noé V., Jensen K. B., Ciudad C. J. (2001) Cloning and characterization of the 5′-flanking region of the human transcription factor Sp1 gene. J. Biol. Chem. 276, 22126–22132 [DOI] [PubMed] [Google Scholar]
- 21. Paul P. K., Bhatnagar S., Mishra V., Srivastava S., Darnay B. G., Choi Y., Kumar A. (2012) The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol. Cell Biol. 32, 1248–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pardo P. S., Mohamed J. S., Lopez M. A., Boriek A. M. (2011) Induction of Sirt1 by mechanical stretch of skeletal muscle through the early response factor EGR1 triggers an antioxidative response. J. Biol. Chem. 286, 2559–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meighan-Mantha R. L., Hsu D. K., Guo Y., Brown S. A., Feng S. L., Peifley K. A., Alberts G. F., Copeland N. G., Gilbert D. J., Jenkins N. A., Richards C. M., Winkles J. A. (1999) The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J. Biol. Chem. 274, 33166–33176 [DOI] [PubMed] [Google Scholar]
- 24. Zheng T. S., Burkly L. C. (2008) No end in site: TWEAK/Fn14 activation and autoimmunity associated end-organ pathologies. J. Leukocyte Biol. 84, 338–347 [DOI] [PubMed] [Google Scholar]
- 25. Mittal A., Bhatnagar S., Kumar A., Paul P. K., Kuang S., Kumar A. (2010) Genetic ablation of TWEAK augments regeneration and post-injury growth of skeletal muscle in mice. Am. J. Pathol. 177, 1732–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu C. L., Kandarian S. C., Jackman R. W. (2011) Identification of genes that elicit disuse muscle atrophy via the transcription factors p50 and Bcl-3. PLoS One 6, e16171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dogra C., Hall S. L., Wedhas N., Linkhart T. A., Kumar A. (2007) Fibroblast growth factor inducible 14 (Fn14) is required for the expression of myogenic regulatory factors and differentiation of myoblasts into myotubes. Evidence for TWEAK-independent functions of Fn14 during myogenesis. J. Biol. Chem. 282, 15000–15010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Girgenrath M., Weng S., Kostek C. A., Browning B., Wang M., Brown S. A., Winkles J. A., Michaelson J. S., Allaire N., Schneider P., Scott M. L., Hsu Y. M., Yagita H., Flavell R. A., Miller J. B., Burkly L. C., Zheng T. S. (2006) TWEAK, via its receptor Fn14, is a novel regulator of mesenchymal progenitor cells and skeletal muscle regeneration. EMBO J. 25, 5826–5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 [DOI] [PubMed] [Google Scholar]
- 30. Chang X., Monitto C. L., Demokan S., Kim M. S., Chang S. S., Zhong X., Califano J. A., Sidransky D. (2010) Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer Res. 70, 2870–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kerkel K., Schupf N., Hatta K., Pang D., Salas M., Kratz A., Minden M., Murty V., Zigman W. B., Mayeux R. P., Jenkins E. C., Torkamani A., Schork N. J., Silverman W., Croy B. A., Tycko B. (2010) Altered DNA methylation in leukocytes with trisomy 21. PLoS Genet. 6, e1001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu H., Dong H., Robertson K., Liu C. (2011) DNA methylation suppresses expression of the urea cycle enzyme carbamoyl phosphate synthetase 1 (CPS1) in human hepatocellular carcinoma. Am. J. Pathol. 178, 652–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Acharyya S., Sharma S. M., Cheng A. S., Ladner K. J., He W., Kline W., Wang H., Ostrowski M. C., Huang T. H., Guttridge D. C. (2010) TNF inhibits Notch-1 in skeletal muscle cells by Ezh2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy. PLoS One 5, e12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smale S. T. (1997) Transcription initiation from TATA-less promoters within eukaryotic protein-coding genes. Biochim. Biophys. Acta 1351, 73–88 [DOI] [PubMed] [Google Scholar]
- 35. Chang L., Karin M. (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37–40 [DOI] [PubMed] [Google Scholar]
- 36. Sunadome K., Yamamoto T., Ebisuya M., Kondoh K., Sehara-Fujisawa A., Nishida E. (2011) ERK5 regulates muscle cell fusion through Klf transcription factors. Dev. Cell 20, 192–205 [DOI] [PubMed] [Google Scholar]
- 37. Moresi V., Williams A. H., Meadows E., Flynn J. M., Potthoff M. J., McAnally J., Shelton J. M., Backs J., Klein W. H., Richardson J. A., Bassel-Duby R., Olson E. N. (2010) Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 143, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cai D., Frantz J. D., Tawa N. E., Jr., Melendez P. A., Oh B. C., Lidov H. G., Hasselgren P. O., Frontera W. R., Lee J., Glass D. J., Shoelson S. E. (2004) IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell 119, 285–298 [DOI] [PubMed] [Google Scholar]
- 39. Mourkioti F., Kratsios P., Luedde T., Song Y. H., Delafontaine P., Adami R., Parente V., Bottinelli R., Pasparakis M., Rosenthal N. (2006) Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J. Clin. Invest. 116, 2945–2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sandri M., Sandri C., Gilbert A., Skurk C., Calabria E., Picard A., Walsh K., Schiaffino S., Lecker S. H., Goldberg A. L. (2004) Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 117, 399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao J., Brault J. J., Schild A., Cao P., Sandri M., Schiaffino S., Lecker S. H., Goldberg A. L. (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 6, 472–483 [DOI] [PubMed] [Google Scholar]
- 42. Choi M. C., Cohen T. J., Barrientos T., Wang B., Li M., Simmons B. J., Yang J. S., Cox G. A., Zhao Y., Yao T. P. (2012) A direct HDAC4-MAP kinase crosstalk activates muscle atrophy program. Mol. Cell 47, 122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Winkles J. A. (2008) The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat. Rev. Drug. Discov. 7, 411–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burkly L. C., Michaelson J. S., Zheng T. S. (2011) TWEAK/Fn14 pathway: an immunological switch for shaping tissue responses. Immunol. Rev. 244, 99–114 [DOI] [PubMed] [Google Scholar]
- 45. Ordway J. M., Curran T. (2002) Methylation matters: modeling a manageable genome. Cell Growth Differ. 13, 149–162 [PubMed] [Google Scholar]
- 46. Bird A. (2001) Molecular biology: methylation talk between histones and DNA. Science 294, 2113–2115 [DOI] [PubMed] [Google Scholar]
- 47. Karin M. (1995) The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270, 16483–16486 [DOI] [PubMed] [Google Scholar]
- 48. Wiley S. R., Cassiano L., Lofton T., Davis-Smith T., Winkles J. A., Lindner V., Liu H., Daniel T. O., Smith C. A., Fanslow W. C. (2001) A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity 15, 837–846 [DOI] [PubMed] [Google Scholar]
- 49. Marinovic A. C., Zheng B., Mitch W. E., Price S. R. (2002) Ubiquitin (UbC) expression in muscle cells is increased by glucocorticoids through a mechanism involving Sp1 and MEK1. J. Biol. Chem. 277, 16673–16681 [DOI] [PubMed] [Google Scholar]
- 50. Zheng B., Ohkawa S., Li H., Roberts-Wilson T. K., Price S. R. (2010) FOXO3a mediates signaling crosstalk that coordinates ubiquitin and atrogin-1/MAFbx expression during glucocorticoid-induced skeletal muscle atrophy. FASEB J. 24, 2660–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Feng S. L., Guo Y., Factor V. M., Thorgeirsson S. S., Bell D. W., Testa J. R., Peifley K. A., Winkles J. A. (2000) The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. Am. J. Pathol. 156, 1253–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]