

Abstract
The c-Jun N-terminal kinase (JNK) signalling pathway appears to act as a critical intermediate in the regulation of lymphocyte activation and proliferation. The majority of studies on the importance of JNK are focused on its role in T helper responses, with very few reports addressing the mechanisms of JNK in governing CD8 T-cell-mediated immunity. By using a well-defined mousepox model, we demonstrate that JNK is involved in CD8+ T-cell-mediated antiviral responses. Deficiency of either JNK1 or JNK2 impaired viral clearance, subsequently resulting in an increased susceptibility to ectromelia virus in resistant mice. The impairment of CD8 responses in JNK-deficient mice was not directly due to an inhibition of effector T-cell expansion, as both JNK1 and JNK2 had limited effect on the activation-induced cell death of CD8+ T cells, and only JNK2-deficient mice exhibited a significant change in CD8+ T-cell proliferation after acute ectromelia virus infection. However, optimal activation of CD8+ T cells and their effector functions require signals from both JNK1 and JNK2. Our results suggest that the JNK pathway acts as a critical intermediate in antiviral immunity through regulation of the activation and effector function of CD8+ T cells rather than by altering their expansion.
Keywords: activation, c-Jun N-terminal kinase, cytotoxic T cell, poxvirus
Introduction
CD8+ cytotoxic T lymphocytes (CTL) play a pivotal role in immune responses against cancers and intracellular pathogens. During viral infections, CTL exert their effector function through at least two mechanisms; by direct killing of infected cells and through the release of cytokines that induce a systemic antiviral state. CTL responses are initiated by the recognition of virus-derived peptide–MHC class I complexes on professional antigen-presenting cells by the T-cell receptor (TCR) on CD8+ T cells. Additionally, co-stimulatory signals such as ligation of CD28 are necessary for full activation and clonal expansion of T cells.1 TCR and CD28 signalling leads to the activation of a protein kinase cascade and numerous transcription factors, including nuclear factor-κB, nuclear factor of activated T cells, and activator protein-1.2 This is followed by the induction of interleukin-2 (IL-2) synthesis, resulting in autocrine and paracrine T-cell proliferation.3,4 This activation and expansion of virus-immune CTL enables the host to produce large numbers of effector cells which are necessary to successfully control the pathogen.5,6
Cytotoxic T lymphocyte-mediated immunity is particularly important in eliminating or ameliorating viral infections, especially those that demonstrate resistance to other immune responses or cause T-cell dysfunction.7 For this reason, there has been increasing interest in understanding the mechanisms that are involved in the regulation of the effector functions, differentiation and homeostasis of CTL. The mitogen-activated protein (MAP) kinase pathway has been implicated in the proliferation, differentiation and death of mammalian cells.8 The activation of MAP kinases is a common event during infections by various viruses, including cytomegalovirus,9 herpes virus,10,11 rotavirus,12 Epstein–Barr virus13,14 and hepatitis B virus.15 The c-Jun N-terminal kinases (JNK), also known as stress-activated protein kinases, are one of the major components of MAP kinases.16,17 Both of the ubiquitously expressed isoforms, JNK1 and JNK2, have been found to be important mediators of intracellular signalling during immune responses.18 In T lymphocytes, the JNK pathway is synergistically activated by TCR and CD28 ligation. Numerous studies have been conducted to identify the role of JNK in T-cell development and activation over the past decades. The importance of JNK has been best documented for CD4+ T cells after in vitro stimulation (reviewed in ref. 18), while JNK signalling mechanisms in CTL responses have only been investigated in a limited number of studies.19–21
Ectromelia virus (ECTV) is an orthopoxvirus and a natural mouse pathogen that causes an infection termed mousepox; it is the classical animal model for the study of biologically relevant CD8 T-cell responses (ref. 22–26, and reviewed in ref. 27). C57BL/6 (B6) mice are resistant to acute ECTV infection and generate potent cell-mediated responses and a robust T helper type 1 (Th1) response.24,26 Activation of JNK has been shown in recent infection studies using the orthopox virus vaccinia.28,29 Earlier findings indicated that in addition to the T helper response, CTL responses may also be modulated by JNK signalling (reviewed in ref. 18). Considering the very limited information concerning the role of JNK in biologically relevant CTL responses during viral infections,20 we studied in detail whether the JNK pathway within CD8+ T cells is activated in vivo. Additionally, the individual roles of JNK1 and JNK2 in CD8+ T-cell activation and antiviral immunity during ECTV infection were assessed.
Materials and methods
Mice
B6 mice were bred and supplied by the Nankai University Animal Centre (Tianjin, China). JNK1-deficient (JNK1−/−) and JNK2−/− mice have been described previously.30,31 These mice are on the B6 background. JNK1−/−.OT-1 or JNK2−/−.OT-1 mice were generated by intercrossing B6.JNK1−/− or B6.JNK2−/− and B6.OT-1 mice, which express the transgenic TCR (Vα2Vβ5) specific for the SIINFEKL peptide of ovalbumin (OVA) in the context of MHC class I (H-2Kb), as described previously.21 Genotypes of JNK deficiency were verified by PCR, and the expression of Vα2Vβ5 was verified by FACS. Age-matched female mice were used at 6–8 weeks. All mice were maintained under specific pathogen-free conditions and the animal experiments were conducted with approval from the Animal Care and Use Committee of Nankai University.
Viruses
Plaque-purified ECTV Moscow strain [American Type Culture Collection (ATCC) VR1374] and recombinant ECTV-OVA (expressing the SIINFEKL peptide) were propagated in BS-C-1 cells (ATCC CCL26) as previously described.26 Viruses were purified by gradient centrifugation before use.
Infection and viral titration
Mice were inoculated subcutaneously (s.c.) with a single injection of virus into the left hind footpad under anaesthesia as described previously,26 unless noted otherwise. In survival experiments, mice were infected s.c. with either 1 × 103 or 1 × 105 plaque-forming units (PFU) of ECTV. To obtain enough lymphocytes from the popliteal draining lymph nodes (PLN), in some experiments, mice were inoculated s.c. with 1 × 105 PFU of ECTV into both hind footpads (5 × 104 PFU/30 μl into each footpad), and animals were kept for < 8 days to limit suffering. The subcutaneous route was used in most of the experiments to resemble natural infection.32,33 However, an intravenous (i.v.) injection of 1 × 105 PFU ECTV-OVA through the tail vein was chosen in the in vivo proliferation assay to allow stronger and more efficient stimulation of the donor cells. Animals were monitored twice daily, and at different time-points post infection (p.i.), tissue was processed as previously described.26 For virus titration, BS-C-1 cells were cultured under standard tissue culture conditions in minimum essential medium (Gibco Invitrogen, Carlsbad, CA) with 2 mm l-glutamine and 10% heat-inactivated fetal calf serum (Trace Biosciences, Castle Hill, NSW, Australia), and the plaque assay was performed as previously described.26
Flow cytometry
All antibodies used for FACS were purchased from BD Pharmingen (San Jose, CA). Annexin V was purchased from eBioscience (San Diego, CA) and B8R20-27 tetramer was synthesized at the Biomolecular Resources Facility of the Australian National University as described elsewhere.26 Surface and intracellular staining was performed using a standard protocol. For Western blotting, the cell lysates with 30 μg of protein were subjected to 10% SDS–PAGE and transferred onto 0·2-μm PVDF transfer membrane (Millipore, Billerica, MA). After blocking with 5% non-fat milk for 2 hr, blots were incubated overnight at 4° with anti-JNK (1 : 1000) or anti-phospho-JNK (1 : 1000) antibodies followed by horseradish peroxidase-conjugated secondary antibodies (all purchased from Cell Signaling Technology, Danvers, MA). Signals were developed by using the enhanced chemiluminescence method according to the manufacturer's protocol (Pierce, Rockford, IL) and visualized by autoradiography.
Cytotoxic T lymphocytes assay
Antiviral CTL responses were measured ex vivo using lymphocytes from the spleens and PLN of individual animals at different time-points p.i. A Non-Radioactive Cytotoxicity Assay Kit (Promega, Madison, WI) was used according to the manufacturer's instructions. ECTV-infected and uninfected MC57G cells (ATCC CRL-2295) were used as targets to detect the MHC class I-restricted killing.
CD8+ cell enrichment, adoptive transfer and proliferation assay
CD8+ T cells were isolated by negative selection using cell sorting from the spleens of uninfected B6.OT-1, JNK1−/−.OT-1 or JNK2−/−.OT-1 mice as previously described.26 Purified naive CD8+ T cells were then labelled with 5 mm CFSE (Molecular Probes, Eugene, OR), and 1 × 106 cells were transferred into the lateral tail vein of each of the uninfected recipient wild-type (WT), JNK1−/− or JNK2−/− mice. One day after the cell transfer, each recipient was infected intravenously with 1 × 105 PFU of ECTV-OVA. At 24, 48 and 72 hr p.i., the proliferation of donor CD8+ cells within the spleen of recipient mice was quantified based on CFSE dilution as described previously.26
Ex vivo stimulations and cytokine measurement
CD4+ and CD8+ T cells were isolated, respectively, by negative selection using cell sorting from spleens of ECTV-infected mice on day 8 p.i. Syngeneic dendritic cells (SDC) were enriched with a pan-DC Isolation kit (Miltenyi Biotec, Auburn, CA) from spleens of uninfected B6 mice as described previously.34 Purity of the isolated cells was determined by FACS analysis. To assay IL-2 and interferon-γ (IFN-γ), SDC were pulsed with 500 nm of B8R peptide (synthesized at Sangon Co. Ltd., Shanghai, China) for 3 hr. CD8+ T cells (1 × 106 cells) were either co-cultured with B8R-pulsed SDC at a ratio of 10 : 1 (T : DC) for 6 hr or stimulated with anti-CD3 (5 μg/ml; Pharmingen) plus CD28 (1 μg/ml; Pharmingen) for 24 hr. To assay Th cytokines, SDC were infected with ECTV at a multiplicity of infection of 10 per cell for 3 hr. CD4+ T cells were pre-treated with 20 μm SP600125 (Calbiochem, Billerica, MA) or left untreated for 45 min, then 1 × 106 cells were cultured with ECTV-infected SDC at a ratio of 10 : 1 (T : DC) for 24 hr. Supernatants of identical cultures were collected and analysed in triplicate for cytokines using specific ELISA kits (eBioscience) per the manufacturer's instructions.
Statistical analysis
Data were analysed using the GraphPad Prism 5 software (San Diego, CA) as indicated.
Results
Mice deficient in JNK1 or JNK2 are susceptible to ECTV infections
To address the potential effect of JNK in ECTV infection, we initially investigated whether deficiency of JNK1 or JNK2 impacts on the recovery of resistant B6 mice. Groups of JNK1−/−, JNK2−/− and WT mice were infected s.c. with 1 × 103 PFU of ECTV for a preliminary survival assay. Consistent with a previous study,26 all WT mice recovered from infection, whereas a proportion of JNK1−/− and JNK2−/− mice succumbed to the virus (Fig. 1a). To further investigate the susceptibility of JNK-deficient mice to ECTV infection, survival analysis was conducted with a higher dose (1 × 105 PFU) of virus. While only nine of 40 WT mice succumbed to mousepox at this dose, 30 of JNK1−/− and 37 of JNK2−/− mice (out of 60 mice for each strain) died. Significant differences in mortality were observed when comparing WT with either the JNK1−/− (P = 0·0061) or JNK2−/− group (P = 0·0001). Although the mortality of JNK2−/− mice was approximately 10% higher than that of JNK1−/− mice, the difference was not statistically significant (Fig. 1a). These data suggest that deficiency in the JNK signalling pathway causes a defect in the capacity of the host to efficiently control ECTV infection, especially at a higher dose of virus. Therefore, 105 PFU was chosen in our subsequent studies.
Figure 1.
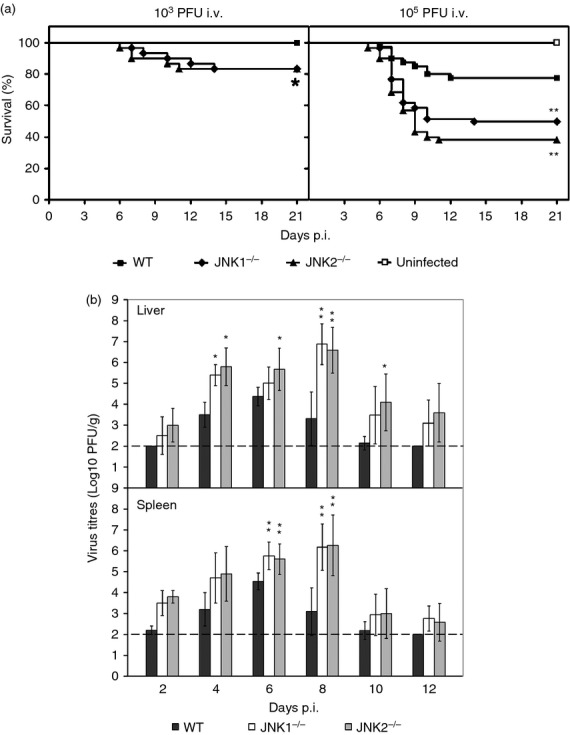
Susceptibility of c-Jun N-terminal kinase (JNK) -deficient mice to ectromelia virus (ECTV) infection. (a) Groups of JNK1−/−, JNK2−/− and wild-type (WT) mice were infected subcutaneously (s.c.) with 103 plaque-forming units (PFU; n = 30 per group) or 105 PFU (JNK1−/− and JNK2−/−, n = 60, respectively; WT, n = 40) of ECTV, and monitored daily for clinical signs of disease and survival. Data are pooled from four separate experiments with similar results. *P < 0·05; **P < 0·01 versus WT, log-rank (Mantel-Cox) test. (b) JNK1−/−, JNK2−/− and B6 WT mice were infected s.c. with 1 × 105 PFU of ECTV. Groups of five mice for each strain were killed on the indicated days post infection (p.i.). Virus titres in the livers and spleens were determined. Results are expressed as mean log10 PFU/g of tissue ± SEM, and the dashed lines indicate the detection limit (2 log10 PFU). *P < 0·05; **P < 0·01 versus WT, Mann–Whitney U-test.
In a separate experiment, JNK1−/− and JNK2−/− mice as well as their WT littermates, were infected s.c. with 1 × 105 PFU of ECTV. On the days indicated (Fig. 1b), groups of five mice from each strain were killed and organs were harvested for determination of virus titre. Virus was detectable in the livers and spleens of all three strains as early as day 2 p.i., with the lowest titres in the WT mice. The viral titres in this group plateaued on day 6 p.i. and became undetectable in most WT mice by day 10. Viral clearance was delayed in both JNK1−/− and JNK2−/− mice with consistently increasing titres until day 8 p.i., which were 0·5–3·0 log10 PFU higher than that seen in the WT mice (P < 0·05, Fig. 1b and see Supporting information, Fig. S1). There was no significant difference in virus titres between the JNK1−/− and JNK2−/− groups. The majority of JNK-deficient mice died before day 14, and in those that recovered, the virus was eventually cleared from the target organs between days 16 and 18 p.i.
Cytotoxic T lymphocytes are of primary importance in the clearance of ECTV (reviewed in ref. 35, and our previous studies26). The increased susceptibility of JNK-deficient mice to ECTV infection is possibly a result of defects in CTL responses. However, previous studies have suggested an effect of JNK- or c-Jun-dependent signalling on vaccinia virus infection and antigen expression.28,29 Consequently, we wanted to distinguish whether the increased susceptibility of JNK-deficient mice to ECTV infection is intrinsic to a defect in T cells or related to a failure in viral replication. To address this, we performed a survival analysis on transgenic OT-1 mice infected with ECTV expressing OVA peptide (SIINFEKL).26 Groups of B6.OT-1, JNK1−/−.OT-1 and JNK2−/−.OT-1 mice were infected s.c. with either 103 PFU or 105 PFU of ECTV-OVA (Table 1). Using 103 PFU, all JNK-deficient OT-1 mice recovered from infection of ECTV-OVA in contrast to JNK-deficient mice, which exhibited 20% mortality after ECTV infection (Fig. 1a). At 105 PFU, both JNK1−/−.OT-1 and JNK2−/−.OT-1 mice showed a significantly reduced mortality to ECTV-OVA infection compared with WT JNK-deficient mice given ECTV (Fig. 1a). The mortality of B6 mice infected with ECTV-OVA at 105 PFU was comparable to those infected with ECTV at the same dose (Fig. 1a). However, all OT-1 mice recovered from infection with 105 PFU ECTV-OVA. These results indicate that an impaired CTL response, rather than an increased virus replication, is the principle cause of the increased susceptibility of JNK-deficient mice to ECTV.
Table 1.
Effect of ectromelia virus-OVA on mortality rate and survival time of OT-1 mice with c-Jun N-terminal kinase deficiency
| Dose (PFU/mouse)1 | Strain of mice | Total no. of mice | No. of dead mice | Survival time (days) | Mortality (%) | MTD (days)2 |
|---|---|---|---|---|---|---|
| 103 | B6.WT | 10 | 0 | > 21 | 0 | – |
| B6.OT-1 | 10 | 0 | > 21 | 0 | – | |
| JNK1−/−.OT-1 | 10 | 0 | > 21 | 0 | – | |
| JNK2−/−.OT-1 | 10 | 0 | > 21 | 0 | – | |
| 105 | B6.WT | 15 | 1 | 7 | 27 | 9·5 ± 2·5 |
| 2 | 9 | |||||
| 1 | 13 | |||||
| B6.OT-1 | 15 | 0 | > 21 | 0 | – | |
| JNK1−/−.OT-1 | 15 | 1 | 7 | 20 | 8·0 ± 1·0 | |
| 1 | 8 | |||||
| 1 | 9 | |||||
| JNK2−/−.OT-1 | 15 | 2 | 7 | 27 | 8·0 ± 1·4 | |
| 1 | 8 | |||||
| 1 | 10 |
Infection route, subcutaneous.
MTD, mean time to death. Values represent mean ± SD. Differences in mortality were analysed by chi-square test, and differences in MTD were assessed by Student's t-test.
JNK1 and JNK2 exert distinct roles in the expansion of CD8+ T cells during a primary ECTV infection
An increased JNK activity or phosphorylation of c-Jun has been found primarily in peripheral CD4+ T cells during TCR-mediated activation in vitro.36,37 To address the potential role of JNK in CTL responses to a viral infection, we first analysed the phosphorylation status of JNK in CD8+ T cells upon infection with ECTV in vivo. A striking increase in total and phosphorylated JNK was detected by Western blot analysis using purified CD8+ T cells from virus-infected B6 mice compared with uninfected controls (Fig. 2a). This suggests that JNK plays a role in the induction of the CTL response to ECTV infections.
Figure 2.
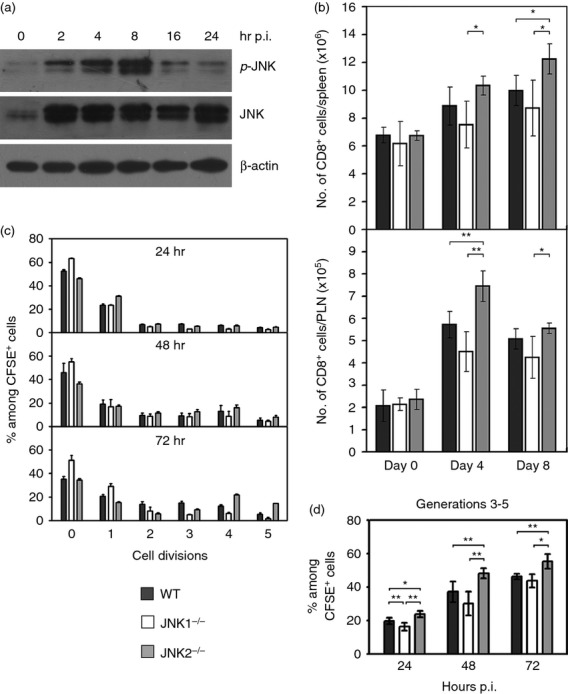
Expansion of CD8+ T cells in c-Jun N-terminal kinase (JNK) -deficient mice after acute ectromelia virus (ECTV) infection. (a) JNK was activated in CD8+ T cells. CD8+ cells were isolated from popliteal lymph nodes (PLN) of virus-infected mice by positive-selection using MACS beads at the indicated time points post-infection (p.i.), and Western blot was performed for phospho-JNK and pan-JNK. Results shown are representative of more than five independent experiments. (b) Compartments of T cells in JNK-deficient mice after acute ECTV infection. JNK1−/−, JNK2−/− and wild-type (WT) mice were infected subcutaneously with ECTV. On day 0, 4 or 8 p.i., spleens and PLN were harvested from groups of five mice for each strain, and T cells were analysed by FACS. Total number of CD8+ T cells in the spleens and PLN are shown. Data represent average value of each group and are representative of three independent experiments. (c, d) Proliferation of CD8+ T cells in JNK-deficient mice after acute ECTV infection. CD8+ cells were isolated from the spleens of uninfected B6.OT-1, JNK1−/−OT-1 or JNK2−/−OT-1 mice. Cells were labelled with CFSE, and 1 × 106 cells were transferred into naive WT, JNK1−/− or JNK2−/− recipients, respectively, followed by intravenous infection with ECTV-OVA. At 24, 48 or 72 hr p.i., spleens were harvested from groups of three recipient mice for each strain. Splenocytes from each recipient were analysed by FACS, and CFSE intensity was used to determine cell proliferation. (c) Mean percentage of each generation (among CD8+ CFSE+ cells). 0, parent generation; 1–5, daughter generations. (d) Summary histograms of CD8+ T-cell proliferation. Only generation 3 and later were considered as specific proliferation cells. Data are representative of two independent experiments with similar results. Results in (b–d) are expressed as mean ± SEM. *P < 0·05; **P < 0·01, Mann–Whitney U-test.
After an ECTV infection, CTL become activated and undergo rapid expansion. To investigate if JNK1 and/or JNK2 play a role in the expansion and activation of virus-immune CTL in vivo during an acute ECTV infection, groups of WT, JNK1−/− and JNK2−/− mice were infected s.c. with ECTV. Lymphocytes from the spleens and PLN of the mice were analysed by FACS on days 0, 4 and 8 p.i. Deficiency of JNK had minimal effects on both total spleen cells (see Supporting information, Table S1) and the ratio of CD8+ and CD4+ subpopulations in the spleens of uninfected mice (Fig. 2b and see Supporting information, Fig. S2). An increase in the number of splenocytes was found in all infected mice. Furthermore, the total number of splenocytes in JNK2−/− mice were slightly higher (P > 0·05) when compared with that in WT and JNK1−/− mice on day 8 p.i. (Table S1). The JNK1−/− mice had fewer CD8+ T cells than WT in the spleens and PLN on day 4 and day 8 p.i., but the differences were not significant. JNK2−/− mice showed a significant increase of CD8+ T cells in the PLN on day 4 p.i. (P < 0·01), and in the spleens on day 8 p.i. (P < 0·05) when compared with WT (Fig. 2b). The number of CD4+ T cells in the spleens of JNK1−/− or JNK2−/− mice was not different compared with WT controls at all time-points p.i. (Fig. S2b). The CD4 : CD8 ratio in the spleens of JNK2−/− mice was significantly decreased (P < 0·05) when compared with both WT and JNK1−/− mice on days 4 and 8 p.i. (Fig. S2c).
JNK1-deficient CD8+ T cells have been reported to exhibit impaired proliferation capacity in vitro after anti-CD3 and anti-CD28 stimulation.19 In the present study, we performed an in vivo proliferation assay to investigate whether JNKs were required for optimal proliferation of CD8+ T cells during viral infections (see Supporting information, Fig. S3). The proliferation of donor CD8+ cells in the spleens of recipient mice at 24, 48 and 72 hr p.i. are summarized in Fig. 2(c). JNK1−/− mice showed remarkably delayed proliferation compared with WT mice. The level of CD8+ T-cell proliferation in JNK1−/− mice was significantly lower (P < 0·01) than WT at 24 hr, but reached control levels after 48 hr p.i. JNK2−/− CD8+ T cells exhibited a dramatic increase in proliferation upon ECTV infection in vivo. As summarized in Fig. 2(d), significantly (P < 0·05) higher numbers of CFSE+ cells were among the divided cell populations (generations 3–5) in JNK2−/− recipients (48% at 48 hr, 55% at 72 hr), whereas fewer dividing cells were found in WT (37% at 48 hr, 46% at 72 hr) and JNK1−/− (30% at 48 hr, 44% at 72 hr) recipients.
Activation-induced cell death (AICD) of T cells is a critical mechanism for the maintenance of peripheral homeostasis in the immune system. It has been reported that the JNK inhibitor SP600125 prevents AICD of CD8+ cells in vitro.38 To distinguish the individual roles of JNK1 and JNK2 in apoptosis of activated CD8+ T cells during an acute infection with ECTV, splenocytes were harvested on day 8 p.i., stained with annexin V and analysed by FACS. We found that both JNK1−/− and JNK2−/− mice had marginally lower numbers of apoptotic cells among the CD8+ CD44hi T-cell cohort compared with their WT littermates (Fig. 3a). To further determine AICD in TCR-mediated activation, CD8+ T cells from acute ECTV-infected mice were re-stimulated with anti-CD3 plus anti-CD28 antibodies ex vivo for 24 hr, and stained with annexin V. Boosted ECTV-activated CD8+ T cells by anti-CD3 plus anti-CD28 showed apoptotic profiles in all three strains (Fig. 3a,c). Interestingly, AICD of ECTV-activated CD8+ T cells in WT mice was significantly reduced by the JNK inhibitor SP600125 (P < 0·01, Fig. 3b,c). A defect of the entire JNK function resulted in significantly lower levels of AICD when compared with either intact JNK or a single deficiency of each JNK isoform.
Figure 3.
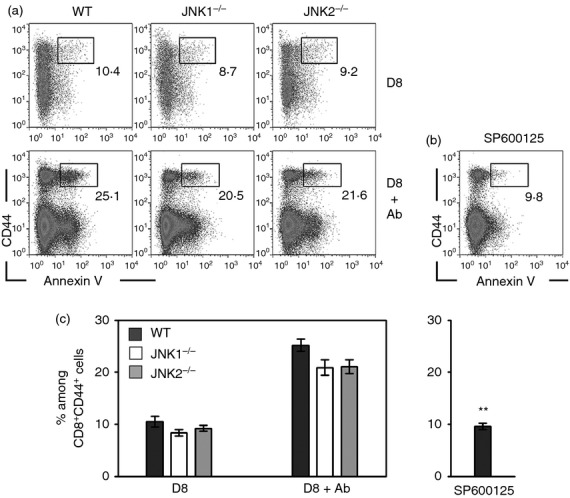
Activation-induced cell death (AICD) of CD8+ T cells in c-Jun N-terminal kinase (JNK) -deficient mice after acute ectromelia virus (ECTV) infection. JNK1−/−, JNK2−/− and wild-type (WT) mice were infected subcutaneously (s.c.) with ECTV. (a) On day 8 post-infection (p.i.), spleens were harvested from groups of five mice for each strain. Lymphocytes were isolated and cultured with either PBS (D8) or CD3 plus CD28 (D8 + Ab) for 24 hr, and then stained with CD8, CD44 and Annexin V. (b) Spleen cells from WT mice in (a) were pre-incubated with SP600125 for 45 min, then cultured with CD3 plus CD28 for 24 hr and stained with CD8, CD44 and Annexin V. The dot plots are gated on CD8+ T cells, and the numbers are the percentages among CD44+ cells. Data represent average values of each group and are representative of three independent experiments. (c) Summary histograms of AICD of CD8+ T cells. Results are expressed as mean ± SEM. **P < 0·01 versus WT in D8 + Ab group, Mann–Whitney U-test.
Both JNK1 and JNK2 are required for effector functions of ECTV-immune CD8+ T cells
Cytotoxic T lymphocyte effector functions are known to be crucial in recovery from acute ECTV infections. Hence, we investigated the role of JNK for inducing the cytotoxic effector function of CD8+ T cells in vivo. Antiviral cytolytic activity was detected as early as day 4 p.i. in the spleen and PLN from WT mice, peaked on day 8 (splenocytes) and day 7 (PLN cells) and persisted up to day 12 p.i. In contrast, cytolytic activity of both JNK1−/− and JNK2−/− splenocytes and PLN was delayed and significantly lower than that of WT (Fig. 4a). The extent of the cytolytic activity corresponds with the changes of virus titres seen in the target organs (Fig. 1b and Fig. S1).
Figure 4.
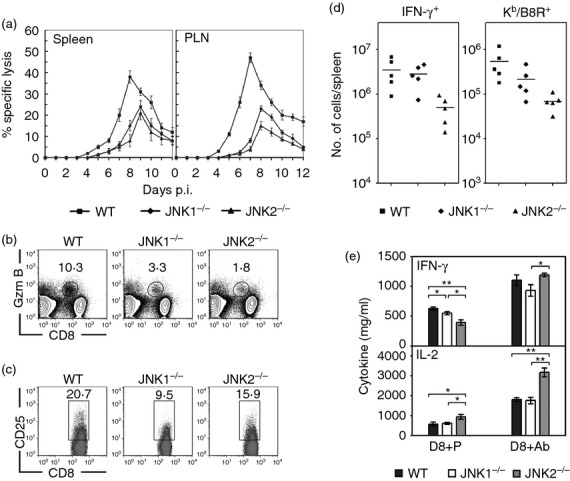
Activation and effector functions of CD8+ T cells in c-Jun N-terminal kinase (JNK) -deficient mice during ectromelia virus (ECTV) infection. (a) JNK1−/−, JNK2−/− and wild-type (WT) mice were infected subcutaneously (s.c.) with ECTV. Virus-specific cytotoxic T lymphocyte (CTL) responses in the spleens and popliteal lymph nodes (PLN) were measured at the indicated time-points. Results are expressed as mean ± SEM of lysis values from five individual mice for each group at an effector : target ratio of 33 : 1. The results are representative of two independent experiments. (b–d) JNK1−/−, JNK2−/− and WT mice (n = 5 per group) were infected s.c. with ECTV. On day 8 p.i., the activation of CD8+ T cells was determined by staining splenocytes with anti-CD8 plus indicated antibodies. The dot plots are gated on total spleen cells (b) or CD8+ T cells (c), and the numbers are the percentages of Granzyme B (Gzm B)- or CD25-positive cells among CD8+ T cells. Data represent average value of each group and are representative of three independent experiments. The symbols in (d) show the total number of specific CD8+ T cells in spleens. Each symbol represents an individual mouse and bars represent the means. (e) Purified CD8+ T cells (1 × 106 cells) from the spleens of mice in (b) were either co-cultured with B8R peptide-pulsed syngeneic dendritic cells for 6 hr (D8 + P) or stimulated with anti-CD3 plus anti-CD28 for 24 hr (D8 + Ab). Cytokines in the supernatants were measured by ELISA. Results are expressed as mean ± SEM (n = 5 per group) and are representative of three independent experiments. *P < 0·05; **P < 0·01, Mann–Whitney U-test.
We then investigated the phenotype of the responding CD8+ T cells from the mice with or without JNK. Splenocytes were analysed by FACS on day 8, the peak of the anti-ECTV CTL response. Similar to the cytolytic activity, striking differences in lytic granule production of CD8+ T cells were found between the spleens of WT and JNK-deficient mice. The percentage of cytolytic CD8+ T cells (Granzyme B+) in the spleens of JNK1−/− mice was significantly lower (P < 0·001) than that of the WT mice, and even lower than in JNK2−/− mice (Fig. 4b). This was consistent with the level of expression of the CD8+ T cells' activation marker CD44 which is highly expressed on effector/memory T cells. A significantly lower percentage of CD8+ T cells from JNK1−/− mice had acquired a CD44hi phenotype on day 8 p.i. compared with those of WT, and the reduction of CD44hi-expressing CD8+ T cells in the JNK2−/− mice was even more pronounced (see Supporting information, Fig. S4). However, a different picture of CD8+ T-cell activation was noted when IL-2 receptor α (IL-2Rα; CD25), another activation marker, was analysed. The percentage of activated CD8+ T cells that expressed CD25 was significantly (P < 0·01) reduced in the JNK1−/− mice, but only moderately reduced (P = 0·065) in JNK2−/− mice, as compared to WT controls (Fig. 4c).
The antigen-specific CD8+ T cells were quantified by MHC I tetramer or intracellular IFN-γ staining (Fig. 4d). The number of IFN-γ+ CD8+ T cells in spleens of JNK2−/− mice were about 10-fold lower than in the other two strains. Quantification by MHC I tetramer binding of the immunodominant ECTV epitope B8R20–2739 also showed a significant decrease (approximately 10-fold, P < 0·05) of virus-specific CTL in JNK2−/− mice when compared with WT, mirroring the IFN-γ expression. Although the proportions of IFN-γ+ CTL were comparable between the JNK1−/− and WT mice, the average number of Kb/B8R-specific CD8+ T cells in the JNK1−/− mice showed a moderate reduction (P > 0·05).
To determine whether a deficiency of JNK may impair the production of function-related cytokines by CD8+ T cells during an ECTV infection, we further examined the production of IL-2 and IFN-γ by CD8+ T cells from the mice after ECTV infection. CD8+ T cells were isolated from the spleens of ECTV-infected JNK1−/−, JNK2−/− and WT mice on day 8 p.i. Cells were either co-cultured with B8R peptide-pulsed SDC for 6 hr, or stimulated with anti-CD3 plus anti-CD28 for 24 hr. Yields of IL-2 and IFN-γ in the supernatants were measured by ELISA. In accordance with their inability to undergo ECTV-induced activation, CD8+ T cells from ECTV-infected JNK1−/− mice produced significantly less (P < 0·05) IFN-γ than WT, and there was an even greater reduction in the production of IFN-γ in CD8+ T cells from the JNK2−/− mice (Fig. 4e). Interestingly, IL-2 production by CD8+ T cells was similar in the JNK1−/− and WT mice and significantly higher in the JNK2−/− mice (P < 0·05). Boosting of CD8+ T cells in the presence of CD3 plus CD28 antibodies ex vivo caused a dramatic increase of IFN-γ and IL-2 production in all strains of mice. In particular, the IFN-γ production by JNK2−/− CD8+ T cells was significantly higher than that of JNK1−/− CD8+ T cells (P < 0·05, Fig. 4e).
JNK1 and JNK2 mediate differentiation of Th responses during ECTV infection
Numerous in vitro studies have described an important role for JNK in the differentiation and activation of effector Th1 and Th2 cells upon either mitogenic stimulation or TCR ligation.18,30,31,36 However, the necessity of JNK to the Th responses in vivo is still unknown. Therefore, we investigated the specific role of JNK in the polarization of Th responses in vivo during ECTV infection. JNK2−/− CD4+ T cells exhibited a significant defect in Th1 cytokine production (P < 0·01 for IFN-γ and P < 0·05 for IL-12, respectively), whereas JNK1−/− CD4+ T cells showed normal production when compared with WT controls (Fig. 5a). However, there was a highly significant enhancement in the production of Th2 cytokines (IL-4, IL-5) in the JNK1−/− CD4+ T cells (Fig. 5a). Neither JNK1 nor JNK2 deficiency had any effect on IL-2 production by CD4+ T cells. Ex vivo administration of JNK inhibitor SP600125 significantly reduced IFN-γ and increased IL-4 production by WT CD4+ T cells (P < 0·05), but had no effect on IL-2 production (Fig. 5b).
Figure 5.
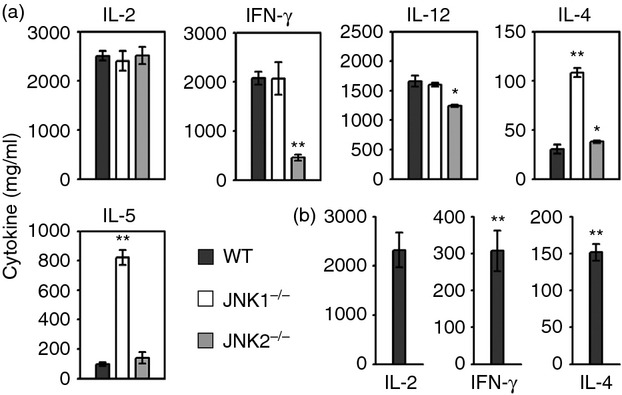
Production of T helper (Th) cytokines in c-Jun N-terminal kinase (JNK) -deficient mice during primary ectromelia virus (ECTV) infection. JNK1−/−, JNK2−/− and wild-type (WT) mice (n = 5 per group) were infected subcutaneously (s.c.) with ECTV. On day 8 post-infection (p.i.), CD4+ T cells were isolated from splenocytes. (a) Splenic CD4+ T cells (1 × 106 cells) were co-cultured with ECTV-infected syngeneic dendritic cells (SDC). Supernatants were collected after 24 hr for cytokine measurement. (b) Splenic CD4+ T cells (1 × 106 cells) from WT mice were pre-incubated with SP600125 for 45 min, then cultured with ECTV-infected SDC for 24 hr. Cytokines in the supernatants were measured by ELISA. Results are expressed as mean ± SEM and are representative of three independent experiments. *P < 0·05; **P < 0·01 versus WT, Mann–Whitney U-test.
Discussion
Initiation of an immune response is partially mediated by the cellular stress due to an infection. Among the stress-activated protein kinases, both JNK1 and JNK2 are involved in the development and maturation of T-cell functions. CTL responses are of primary importance in the recovery of acute viral infections. However, our understanding concerning the role of the JNK pathway in governing antiviral CTL responses is limited. The only other study to address this issue used a mouse model of lymphocytic choriomeningitis virus infections.20 The study found reduced expansion of CD8+ T cells, a decreased virus-immune CTL fraction and increased AICD in JNK1−/− mice during the primary acute but not secondary latent infection. This study also showed an increased expansion of virus-immune CD8+ T cells in JNK2−/− mice during both primary and secondary infections. Although defects of lymphocytic choriomeningitis virus-immune CTL responses were different between JNK1−/− and JNK2−/− mice, both strains showed equivalent CTL-mediated virus-specific killing and viral clearance when compared with WT mice. The authors concluded that JNK signalling may play a role in controlling the number (magnitude) rather than the functionality of CD8+ T cells, and activated CD4+ T cells may be responsible for the enhanced antiviral response in JNK2−/− mice.
Our studies presented here, using the classical ECTV acute viral infection model, clearly implicate the involvement of both JNK1 and JNK2 in the cytotoxic CD8+ T-cell-mediated recovery from a primary infection. We initially attributed the impairment of viral clearance in JNK-deficient mice to an impairment of expansion of CD8+ T cells. However, our subsequent studies indicate that the mechanisms underlying these results were more complex. Unlike the previous study, which observed a hypo-proliferation in JNK1−/− CD8+ T cells after stimulation with anti-CD3 plus anti-CD28 in vitro,19 our in vivo proliferation assays showed that, although the proliferation of CD8+ T cells was delayed, the absolute number of CD8+ T cells in the spleens and PLN of JNK1−/− mice was only slightly less than those in WT mice, 48 hr p.i. JNK2 deficiency resulted in an increased proliferation of CD8+ T cells during ECTV infection, data that are consistent with previous studies pointing to an inhibitory role of JNK2 in CD8+ T-cell proliferation.19,20 Despite the divergent roles that JNK1 and JNK2 play in CD8+ T-cell proliferation, both caused a defect in preventing AICD among CD8+ T cells, either activated in vivo by ECTV or after in vitro CD3 plus CD28 stimulation. Therefore, the impairment of viral clearance in JNK-deficient mice is not determined by the magnitude of peripheral CTL expansion. Our data demonstrate that cytotoxicity was delayed and reduced, and there was a significant reduction of IFN-γ production by CD8+ T cells in both JNK1−/− and JNK2−/− mice. Absence of either JNK1 or JNK2 resulted in a decreased absolute number of virus (B8R) -specific CD8+ T cells, as well as a dramatic reduction of lytic granule-producing CTL in spleens. The changed phenotype of the CTL correlated directly with the impaired viral clearance and increased susceptibility to ECTV infection in JNK-deficient mice.
Interleukin-2 has an important role in the survival and expansion of T cells, and its synthesis is a hallmark of T-cell activation. The magnitude and duration of IL-2Rα expression on activated CD8+ T cells controls their ability to ligate IL-2, and so modulates their cytolytic capacity and expansion.40,41 Although IL-2 production by activated CD8+ T cells was comparable between JNK1−/− and WT mice, expression of IL-2Rα was significantly less on JNK1−/− CD8+ T cells than controls. This is consistent with the in vitro study19 and may be associated with the delayed proliferation and impaired effector functions of virus-immune CTLs in JNK1−/− mice after ECTV infection. The loss of JNK2 caused significantly increased production of IL-2 by CD8+ T cells, which may possibly explain the observed hyper-proliferation of these cells. However, in light of the moderately reduced expression of IL-2Rα on JNK2−/− CD8+ T cells, it remains to be determined whether IL-2α does serve a critical function in the control of CTL responses in JNK2−/− mice.
The Th1/Th2 differentiation decides the effector responses against pathogens. During ECTV infection, resistant B6 mice generate an early Th1 response, which is associated with potent cell-mediated immunity leading to effective viral clearance, whereas susceptible mice such as BALB/c generate a Th2 response associated with weak CTL activity and poor virus control.24,26 Our present study demonstrates that the generation of a Th1 response in B6 mice after ECTV infection requires JNK2 but not JNK1. Production of IFN-γ in Th cells was diminished by either JNK2 deficiency or a pan-JNK inhibitor, suggesting that JNK2 exerts most of the control among JNK signalling in Th1 differentiation.31 In synergy with JNK2 signalling, which promotes the type 1 Th responses, JNK1 may act as an inhibitor of the Th2 response during mousepox. Th cells from B6 WT mice produced only small amounts of Th2 cytokines, such as IL-4, IL-5 and IL-10, during ECTV infection (ref. 26 and present study). JNK1 deficiency, either by genetic knockout or application of pharmacological inhibitors, resulted in a remarkable increase of Th2 cytokines. These data would in part explain the dysfunction of CTL in JNK-deficient mice. Given that there were a Th1 diminution in JNK2−/− mice and a Th2 bias in JNK1−/− mice, it was therefore not surprising that deficiency of either JNK1 or JNK2 resulted in significant reductions in CTL responses directed against ECTV.
Altogether, our findings provide further understanding as to the functional diversity of JNK in the regulation of antiviral immunity. Given the importance of JNK signalling in controlling primary CTL responses against viruses, it will be necessary to investigate the role of this pathway in memory T-cell differentiation, maintenance and protective immunity development upon re-infection. Currently, such studies are in progress to address the mechanisms of JNK control for the homeostasis of CD8+ T cells and development of memory CTL.
Acknowledgments
We thank Dr Richard A. Flavell (Yale University School of Medicine) and Dr Zhinan Yin (Nankai University, China) for the JNK-deficient mice; Judy M. Wang (Monash University, Australia) for critical reading of the manuscript; and colleagues at the Biological Experimental Centre (Nankai University) for cell sorting. This study was supported by grants (30972803 and 31070803) from the National Natural Science Foundation of China (to YW).
Glossary
- B6
C57BL/6
- ECTV
ectromelia virus
- JNK
c-Jun N-terminal kinase
- PLN
popliteal lymph node
- SDC
syngeneic dendritic cell
Disclosures
The authors declare no financial or commercial conflict of interest.
Authorship
YW designed the research and wrote the paper. YQW, XM, LZ, XZ and QX performed experiments and analysed data. LL performed the adoptive transfer experiments, and helped with interpreting the results. All authors reviewed and provided input toward the final paper.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Ectromelia virus titres in the spleen and liver. Experiment was performed as described in Fig. 1b.
Figure S2. Compartments of splenic T cells in c-Jun N-terminal kinase (JNK) -deficient mice after acute ectromelia virus infection.
Figure S3. Proliferation of CD8+ T cells in c-Jun N-terminal kinase (JNK) -deficient mice after acute ectromelia virus infection.
Figure S4. Activation of CD8+ T cells in c-Jun N-terminal kinase (JNK) -deficient mice during ectromelia virus infection.
Table S1. Total number of splenocytes in ectromelia virus-infected mice (× 10–8).
References
- 1.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–62. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 2.Cuddapah S, Barski A, Zhao K. Epigenomics of T cell activation, differentiation, and memory. Curr Opin Immunol. 2010;22:341–7. doi: 10.1016/j.coi.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crabtree GR. Contingent genetic regulatory events in T lymphocyte activation. Science. 1989;243:355–61. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- 4.Sabapathy K, Hu Y, Kallunki T, Schreiber M, David JP, Jochum W, Wagner EF, Karin M. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr Biol. 1999;9:116–25. doi: 10.1016/s0960-9822(99)80065-7. [DOI] [PubMed] [Google Scholar]
- 5.Harty JT, Tvinnereim AR, White DW. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol. 2000;18:275–308. doi: 10.1146/annurev.immunol.18.1.275. [DOI] [PubMed] [Google Scholar]
- 6.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–87. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 7.Yewdell JW, Haeryfar SM. Understanding presentation of viral antigens to CD8+ T cells in vivo: the key to rational vaccine design. Annu Rev Immunol. 2005;23:651–82. doi: 10.1146/annurev.immunol.23.021704.115702. [DOI] [PubMed] [Google Scholar]
- 8.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK) – from inflammation to development. Curr Opin Cell Biol. 1998;10:205–19. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 9.Bruening W, Giasson B, Mushynski W, Durham HD. Activation of stress-activated MAP protein kinases up-regulates expression of transgenes driven by the cytomegalovirus immediate/early promoter. Nucleic Acids Res. 1998;26:486–9. doi: 10.1093/nar/26.2.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marsters SA, Ayres TM, Skubatch M, Gray CL, Rothe M, Ashkenazi A. Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-κB and AP-1. J Biol Chem. 1997;272:14029–32. doi: 10.1074/jbc.272.22.14029. [DOI] [PubMed] [Google Scholar]
- 11.McLean TI, Bachenheimer SL. Activation of cJUN N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J Virol. 1999;73:8415–26. doi: 10.1128/jvi.73.10.8415-8426.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holloway G, Coulson BS. Rotavirus activates JNK and p38 signaling pathways in intestinal cells, leading to AP-1-driven transcriptional responses and enhanced virus replication. J Virol. 2006;80:10624–33. doi: 10.1128/JVI.00390-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eliopoulos AG, Young LS. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein–Barr virus-encoded latent membrane protein 1 (LMP1) Oncogene. 1998;16:1731–42. doi: 10.1038/sj.onc.1201694. [DOI] [PubMed] [Google Scholar]
- 14.Wan J, Sun L, Mendoza JW, et al. Elucidation of the c-Jun N-terminal kinase pathway mediated by Epstein–Barr virus-encoded latent membrane protein 1. Mol Cell Biol. 2004;24:192–9. doi: 10.1128/MCB.24.1.192-199.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diao J, Khine AA, Sarangi F, et al. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J Biol Chem. 2001;276:8328–40. doi: 10.1074/jbc.M006026200. [DOI] [PubMed] [Google Scholar]
- 16.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 17.Blonska M, Pappu BP, Matsumoto R, Li H, Su B, Wang D, Lin X. The CARMA1-Bcl10 signaling complex selectively regulates JNK2 kinase in the T cell receptor-signaling pathway. Immunity. 2007;26:55–66. doi: 10.1016/j.immuni.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rincon M, Davis RJ. Regulation of the immune response by stress-activated protein kinases. Immunol Rev. 2009;228:212–24. doi: 10.1111/j.1600-065X.2008.00744.x. [DOI] [PubMed] [Google Scholar]
- 19.Conze D, Krahl T, Kennedy N, et al. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8+ T cell activation. J Exp Med. 2002;195:811–23. doi: 10.1084/jem.20011508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arbour N, Naniche D, Homann D, Davis RJ, Flavell RA, Oldstone MB. c-Jun NH2-terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8+ T cell-mediated antiviral immunity. J Exp Med. 2002;195:801–10. doi: 10.1084/jem.20011481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao Y, Tao J, Li MO, et al. JNK1 is essential for CD8+ T cell-mediated tumor immune surveillance. J Immunol. 2005;175:5783–9. doi: 10.4049/jimmunol.175.9.5783. [DOI] [PubMed] [Google Scholar]
- 22.Kees U, Blanden RV. A single genetic element in H-2K affects mouse T-cell antiviral function in poxvirus infection. J Exp Med. 1976;143:450–5. doi: 10.1084/jem.143.2.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karupiah G, Buller RM, Van Rooijen N, Duarte CJ, Chen J. Different roles for CD4+ and CD8+ T lymphocytes and macrophage subsets in the control of a generalized virus infection. J Virol. 1996;70:8301–9. doi: 10.1128/jvi.70.12.8301-8309.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaudhri G, Panchanathan V, Buller RM, et al. Polarized type 1 cytokine response and cell-mediated immunity determine genetic resistance to mousepox. Proc Natl Acad Sci U S A. 2004;101:9057–62. doi: 10.1073/pnas.0402949101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang M, Sigal LJ. Antibodies and CD8+ T cells are complementary and essential for natural resistance to a highly lethal cytopathic virus. J Immunol. 2005;175:6829–36. doi: 10.4049/jimmunol.175.10.6829. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Chaudhri G, Jackson RJ, Karupiah G. IL-12p40 and IL-18 play pivotal roles in orchestrating the cell-mediated immune response to a poxvirus infection. J Immunol. 2009;183:3324–31. doi: 10.4049/jimmunol.0803985. [DOI] [PubMed] [Google Scholar]
- 27.Mullbacher A. Cell-mediated cytotoxicity in recovery from poxvirus infections. Rev Med Virol. 2003;13:223–32. doi: 10.1002/rmv.381. [DOI] [PubMed] [Google Scholar]
- 28.Santos CR, Blanco S, Sevilla A, Lazo PA. Vaccinia virus B1R kinase interacts with JIP1 and modulates c-Jun-dependent signaling. J Virol. 2006;80:7667–75. doi: 10.1128/JVI.00967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu W, Hofstetter W, Guo W, et al. JNK-deficiency enhanced oncolytic vaccinia virus replication and blocked activation of double-stranded RNA-dependent protein kinase. Cancer Gene Ther. 2008;15:616–24. doi: 10.1038/cgt.2008.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–5. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 31.Yang DD, Conze D, Whitmarsh AJ, Barrett T, Davis RJ, Rincon M, Flavell RA. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 1998;9:575–85. doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- 32.Fenner F. Mouse-pox (infectious ectromelia of mice): a review. J Immunol. 1949;63:341–73. [PubMed] [Google Scholar]
- 33.Tscharke DC, Woo WP, Sakala IG, et al. Poxvirus CD8+ T-cell determinants and cross-reactivity in BALB/c mice. J Virol. 2006;80:6318–23. doi: 10.1128/JVI.00427-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Da'Dara AA, Thomas PG, Harn DA. Dendritic cells activated by an anti-inflammatory agent induce CD4+ T helper type 2 responses without impairing CD8+ memory and effector cytotoxic T-lymphocyte responses. Immunology. 2010;129:406–17. doi: 10.1111/j.1365-2567.2009.03193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanden RV. T cell response to viral and bacterial infection. Transplant Rev. 1974;19:56–88. doi: 10.1111/j.1600-065x.1974.tb00128.x. [DOI] [PubMed] [Google Scholar]
- 36.Weiss L, Whitmarsh AJ, Yang DD, Rincon M, Davis RJ, Flavell RA. Regulation of c-Jun NH2-terminal kinase (Jnk) gene expression during T cell activation. J Exp Med. 2000;191:139–46. doi: 10.1084/jem.191.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabapathy K, Kallunki T, David JP, Graef I, Karin M, Wagner EF. c-Jun NH2-terminal kinase (JNK)1 and JNK2 have similar and stage-dependent roles in regulating T cell apoptosis and proliferation. J Exp Med. 2001;193:317–28. doi: 10.1084/jem.193.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mehrotra S, Chhabra A, Hegde U, Chakraborty NG, Mukherji B. Inhibition of c-Jun N-terminal kinase rescues influenza epitope-specific human cytolytic T lymphocytes from activation-induced cell death. J Leukoc Biol. 2007;81:539–47. doi: 10.1189/jlb.0706479. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Flesch IE, Tscharke DC. Vaccinia virus CD8+ T-cell dominance hierarchies cannot be altered by prior immunization with individual peptides. J Virol. 2009;83:9008–12. doi: 10.1128/JVI.00410-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, Usherwood EJ, Lefrancois L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc Natl Acad Sci U S A. 2010;107:193–8. doi: 10.1073/pnas.0909945107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cox MA, Harrington LE, Zajac AJ. Cytokines and the inception of CD8 T cell responses. Trends Immunol. 2011;32:180–6. doi: 10.1016/j.it.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ectromelia virus titres in the spleen and liver. Experiment was performed as described in Fig. 1b.
Figure S2. Compartments of splenic T cells in c-Jun N-terminal kinase (JNK) -deficient mice after acute ectromelia virus infection.
Figure S3. Proliferation of CD8+ T cells in c-Jun N-terminal kinase (JNK) -deficient mice after acute ectromelia virus infection.
Figure S4. Activation of CD8+ T cells in c-Jun N-terminal kinase (JNK) -deficient mice during ectromelia virus infection.
Table S1. Total number of splenocytes in ectromelia virus-infected mice (× 10–8).