

Summary
The orchestration of brain function requires complex gene regulatory networks that are modulated, in part, by microRNAs (miRNAs). These noncoding RNAs associate with argonaute (Ago) proteins in order to direct posttranscriptional gene suppression via base pairing with target transcripts. In order to better understand how miRNAs contribute to human-specialized brain processes and neurological phenotypes, identifying their targets is of paramount importance. Here, we address the latter by profiling Ago2:RNA interactions using HITS-CLIP to generate a transcriptome-wide map of miRNA binding sites in human brain. We uncovered ∼7,000 stringent Ago2 binding sites that are highly enriched for conserved sequences corresponding to abundant brain miRNAs. This interactome points to functional miRNA:target pairs across >3,000 genes and represents a valuable resource for accelerating our understanding of miRNA functions in brain. We demonstrate the utility of this map for exploring clinically relevant miRNA binding sites that may facilitate the translation of genetic studies of complex neuropsychiatric diseases into therapeutics.
Introduction
During the past decade, microRNAs (miRNAs; ∼19–22 nucleotides) have emerged as key posttranscriptional regulators of gene expression, having been implicated in nearly all biological processes (Krol et al., 2010). In brain, miRNAs play vital roles in cell-fate specification, neurite projection, and synaptic plasticity, and growing evidence suggests that even slight aberrations in miRNA activities can be detrimental to neuronal function (Im and Kenny, 2012). The human genome encodes nearly 2,000 unique miRNAs, some being highly conserved across species, and others being specific to primate or human lineages. miRNAs are excised from larger stem-loop-containing transcripts and are subsequently incorporated into argonaute (Ago) proteins. This generates functional entities capable of silencing target transcripts via translational repression and mRNA destabilization (Bazzini et al., 2012; Djuranovic et al., 2012). miRNAs typically bind to target mRNA 3′ UTRs containing short stretches of complementarity to the “seed” region of the miRNA, corresponding to positions 2–8 (Lewis et al., 2005). Although this minimal degree of base pairing provides attractive means for miRNAs to coordinate cellular responses by regulating numerous transcripts within common pathways, this poses a significant challenge for elucidating biologically relevant miRNA:target interactions by bioinformatic prediction, an important and common first step toward target identification. Bioinformatic approaches tend to overpredict miRNA binding sites, supporting the need for complementary wet lab data to assist with identifying bona fide interactions. Transcriptional and proteomic profiling studies have aided the search for functional miRNA target sites; however, these approaches are unable to discern whether observed changes in gene expression are the result of direct miRNA:target interactions. To address this, recent work has focused on using high-throughput methods (e.g., high-throughput sequencing of RNA isolated by cross-linking immunoprecipitation [HITS-CLIP], CLIP sequencing [CLIP-seq], photoactivatable ribonucleoside-enhanced CLIP [PAR-CLIP], and crosslinking, ligation, and sequencing of hybrids [CLASH]) (Chi et al., 2009; Hafner et al., 2010; Helwak et al., 2013; Leung et al., 2011; Zisoulis et al., 2010) to biochemically determine Ago:miRNA binding sites on a transcriptome-wide scale. These approaches involve crosslinking RNA binding proteins with RNAs, immunoprecipitating Ago proteins, and sequencing the associated RNAs. Ago proteins (namely, Ago1 and Ago2) are the core components of miRNA-silencing complexes (Krol et al., 2010), and this powerful technique allows for sequencing of both the miRNAs and the mRNA target regions to which they bind. In mice, these approaches have been applied in brain and liver tissues (Chi et al., 2009; Schug et al., 2013); however, human data are only available for cultured cells (Chi et al., 2009; Haecker et al., 2012; Hafner et al., 2010). In order to advance our understanding of miRNA functions in humans, miRNA:target interactions must be further queried in primary tissues. Here, seeking to gain insight into the array of transcripts engaged with miRNAs in human brain, we profiled transcriptome-wide Ago2:RNA interactions in human brain tissues using HITS-CLIP (i.e., CLIP-seq) methodology.
Results
Ago2 HITS-CLIP Yields miRNAs and Their Transcriptomic Target Sites in Human Brain Samples
We performed Ago2 HITS-CLIP on a panel of 11 postmortem human brain samples harvested from adult motor cortex and cingulate gyrus—regions associated with movement and psychiatric disorders (all brain tissues were obtained from males ranging from ages 44–68; Figure 1A; Table S1 available online). Initial analyses confirmed the specific immunoprecipitation of Ago2 bound to radiolabeled cellular RNAs (Figures 1B and 1C). Notably, gel electrophoresis coupled with autoradiography revealed the previously described doublet (Chi et al., 2009), representing Ago2 complexed with miRNAs (lower band) and Ago2 complexed with miRNAs and target RNA tags (upper band). Cloning and high-throughput sequencing of these Ago2-associated RNAs produced nearly 50 million reads for each sample, yielding an average of 200,000 reads mapping to known human mature miRNAs (miRBase) and 15 million uniquely mapped reads within the human genome (Figure 1D). Upon the removal of PCR duplicates on the basis of redundant genomic coordinates, each sample generated roughly 1 million uniquely mapped reads, providing considerable depth for HITS-CLIP analysis.
Figure 1. Workflow for Performing Ago2 HITS-CLIP in Human Brain.
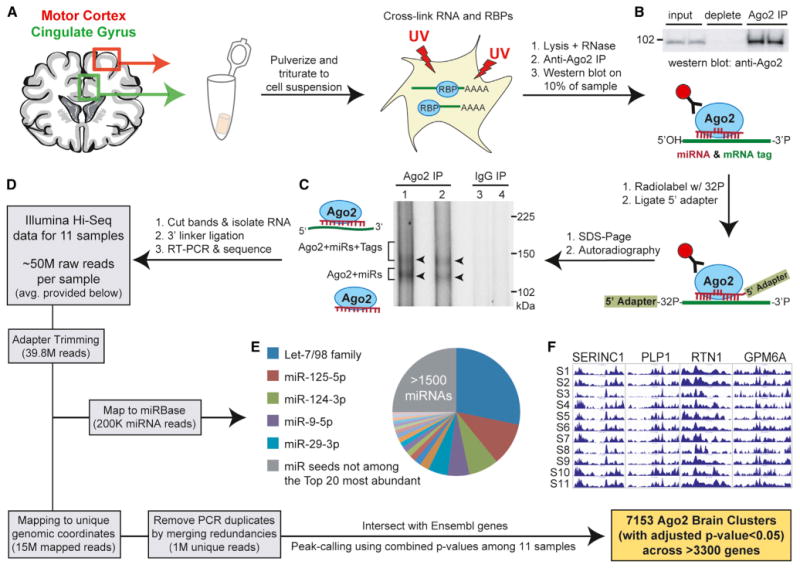
(A) Illustration depicting the regions from which gray matter samples were harvested from human postmortem brains and initial tissue processing steps.
(B) Western blot showing immunoprecipitation efficiency of human Ago2 (94 kDa) from postmortem brain samples.
(C) Autoradiography of 32P-labeled RNA:protein complexes resolved by SDS-PAGE after Ago2 CLIP shows the expected doublet corresponding to the indicated complexes. IgG immunoprecipitation serves as the negative control.
(D) Diagram depicting the workflow for processing high-throughput sequencing data in order to obtain miRNA read information and Ago2 brain clusters. The average number of reads per sample at each filtering step is provided.
(E) Pie chart showing the relative abundance of miRNA reads (grouped into families on the basis of shared seed sequence) for all samples.
(F) UCSC Genome Browser images of positional read coverage for each of the 11 samples (S1–S11) illustrate reproducibility across samples. Images span the 3′ UTRs of the indicated genes, covering > 6,400 nt in total per sample.
See also Figures S1 and S2 and Tables S1–S3.
Reads mapping to mature miRNAs were reproducible across samples (median R2 = 0.79 for all pairwise comparisons; Figure S1A), particularly among the most highly expressed miRNAs (Figure S1B). In total, we detected nearly 1,900 unique miRNAs (Table S2), including an abundance of known brain-enriched miRNAs that were present at relative levels comparable to previous studies profiling human brain miRNAs by small RNA sequencing and microarray (Hu et al., 2011; Shao et al., 2010). When miRNA read counts were grouped into their respective seed families, the top five most abundant Ago2-associated seed sequences (Let-7, miR-125, miR-124, miR-9, and miR-29) comprised nearly 55% of total Ago2 “seed load” present in the samples (Figures 1E and S1B), and the top 20 seeds accounted for 75%. These data are consistent with prior reports citing that highly abundant miRNAs often constitute the vast majority of cellular miRNA pools and are generally the chief miRNAs mediating significant target suppression within cells (Chi et al., 2009; Hafner et al., 2010; Mullokandov et al., 2012).
Unique reads not mapping to known miRNAs were subjected to peak-calling in order to identify Ago2 binding sites (i.e., clusters; see the Experimental Procedures) within Ensemblannotated genes. In brief, we calculated the significance of enrichment for overlapping reads at each genomic position relative to the nearby context and summarized the positional p values across all 11 samples. Positions having a combined Bonferroni adjusted p value < 5% were used for downstream cluster formation, which involved merging nearby coordinates (within 60 nt) and extending all remaining positions to 50 nt in order to account for the Ago2 binding footprint (Chi et al., 2009). This peak-calling strategy resulted in the identification of 7,153 high-confidence human Ago2 brain clusters (Bonferroni adjusted p value < 5%) associating with >3,300 brain-expressed genes (Table S3). We gauged the overall resolution of the Ago2 binding sites by evaluating cluster length distributions. Notably, >80% of the clusters are ≤ 60 nt in length, and only 6% are >100 nt (Table S3). Pairwise correlations of the positional read counts (i.e., wiggle files) for each sample supported moderate to high reproducibility of mapped reads (Figure S2A; median R2 = 0.80). Furthermore, data consistency among samples was visually evident across many genes, as observed in the University of California Santa Cruz (UCSC) Genome Browser (Figures 1F and S2B). As with previous CLIP-based studies, we observed an inherent bias toward identifying Ago2 binding sites in higher-expressed transcripts (data not shown); on average, our studies ascertained roughly two sites per target gene.
Characterization of Ago2 Binding Sites in Human Brain
Next, we characterized the human Ago2 brain clusters. First, we analyzed cluster sequence content by performing unbiased motif enrichment analyses to evaluate the relationship between human Ago2 brain clusters and miRNAs present in the samples. Among the most significantly enriched motifs were sequences corresponding to the top five most abundant miRNA seeds, including 7A1 (reverse complement of miRNA positions 2–7 followed by an adenine base), 7m8 (reverse complement of miRNA positions 2–8), and some noncanonical seed types (e.g., 7m9 [reverse complement of miRNA positions 3–9] and seeds containing G:U wobble pairing or bulged mismatches) (Chi et al., 2012) (Figure 2A and Table S4). Heptamers corresponding to other fairly abundant miRNA seeds were also significantly enriched. Notably, highly enriched miRNA seed motifs showed a positional bias to the immediate right of cluster centers; this corresponds to the expected position of seed sites engaged in reverse complement target recognition and is consistent with data from previous Ago PAR-CLIP studies (Hafner et al., 2010) (Figure 2B). We also performed positional analyses with respect to the miRNAs themselves and found that the enrichment of motifs corresponding to miRNA sequences was primarily constrained to seed regions (Figure S3). In some instances, evidence for significant 3′ compensatory binding was observed (e.g., miR-125-5p and miR-124-3p), whereas other miRNAs showed evidence for shifted binding (miR-103-3p and miR-221-3p). Notably, the latter is not likely the result of miRNA processing heterogeneity, given that the vast majority of small RNA reads mapping to these miRNA stem loops correspond to the annotated mature miRNA (>85%) (data not shown). Furthermore, shifted binding by miR-221 was previously observed in a recent study employing the CLIP-based technique known as CLASH, an advanced approach for identifying noncanonical miRNA binding sites (Helwak et al., 2013).
Figure 2. Characterization of Human Ago2 Brain Cluster Sequences.
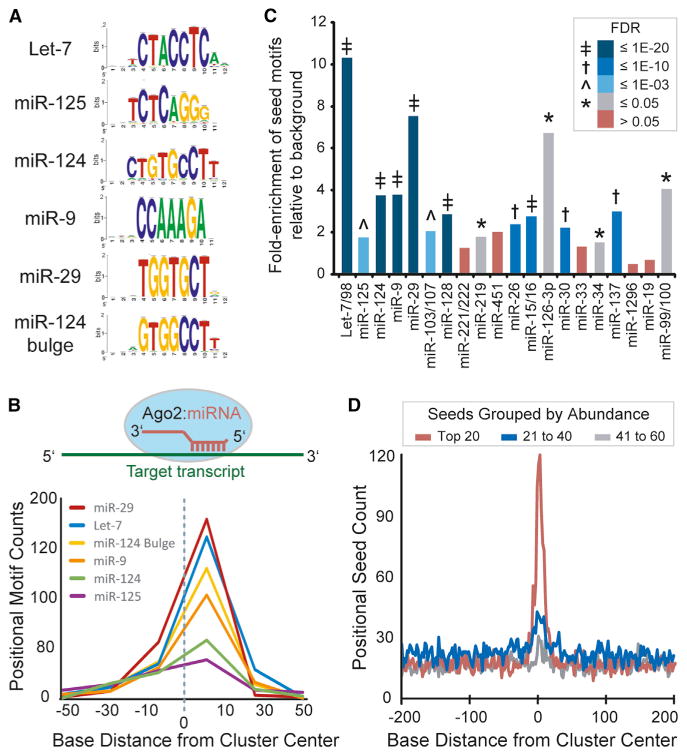
(A) Analysis of cluster sequences with RSAT (peak motifs tool) identified significant enrichment of motifs corresponding to abundant brain miRNAs (logos shown).
(B) Graph summarizing the positional enrichment of motifs identified by RSAT run on Ago2 brain cluster sequences. The enriched motifs corresponding to abundant brain miRNAs are located to the right of center, which is consistent with miRNA target recognition via reverse complementary Watson-Crick base pairing, as illustrated above.
(C) The relative combined enrichment of 7A1 and 7m8 sites corresponding to the top 20 most abundant miRNA seeds is shown. The significance of enrichment (Ago2 clusters versus background; FDR) is indicated by color key and symbol.
(D) The positional seed counts within and near Ago2 brain clusters (centered on zero, median cluster size is 51 nt) are shown for the indicated miRNA seeds grouped by abundance rank.
See also Figures S3 and S4 and Tables S4 and S5.
We also performed a biased approach to determine the combined enrichment of 7A1 and 7m8 sites for each known miRNA seed sequence (Figure 2C and Table S5). Sites corresponding to 15 of the top 20 most abundant miRNA seeds were significantly enriched (false discovery rate [FDR] < 5%), and a general diminishing enrichment among the remaining seeds by rank (e.g., groups 21–40 and 41–60 by ranked seed abundance; Figure 2D) was evident. Notably, significantly enriched motifs among these lesser abundant seeds include those corresponding to miRNAs previously described as brain-enriched (e.g., miR-101, miR-153, and miR-485) or having synaptic (e.g., miR-138) or glial (e.g., miR-23) localization (Cohen et al., 2011; Farh et al., 2005; Lagos-Quintana et al., 2002; Sempere et al., 2004; Siegel et al., 2009; Smirnova et al., 2005), signifying functionality in brain despite moderate-to-lower expression levels.
Second, the intragenic locations of clusters were determined on the basis of Ensembl gene annotations, and 97% resided within mRNA transcripts. The largest proportions of clusters are present in 3′ UTRs and coding regions (41% and 40%, respectively) (Figure 3A). Analogous to the mouse brain Ago HITS-CLIP data (Chi et al., 2009), we observed a positional bias for Ago2 binding sites to be enriched at the 5′ and 3′ ends of 3′ UTRs (Figure 3B), regions associated with increased miRNA functionality (Grimson et al., 2007). Furthermore, >25% of the Ago2 brain clusters in 3′ UTRs overlap with TargetScan-predicted conserved miRNA binding sites (>2-fold enrichment over background expectations, Pearson's chi-square test p < 2.2 × 10−16; Table S3) (Grimson et al., 2007). Overall, Ago2 brain clusters showed increased conservation (PhastCon scores) relative to flanking genomic regions (Figure 3C), lending additional support to site functionality.
Figure 3. Positional Distribution and Conservation of Human Ago2 Brain Clusters.
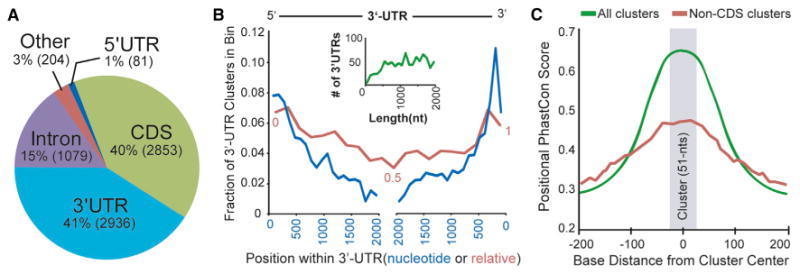
(A) The intragenic distribution of the 7153 human Ago2 brain clusters within mRNAs based on Ensembl gene annotations. The actual number of clusters in each region is provided in parentheses.
(B) The actual nucleotide (blue line) and relative (red line) positional distribution of clusters within binned 3′ UTR locations is shown. The inset graph (green line) depicts the number of 3′ UTRs by length for genes containing 3′ UTR clusters, supporting that the observed decrease in clusters distant from 3′ UTR ends (both 5′ and 3′) is not length dependent.
(C) The positional PhastCon scores within and near Ago2 brain clusters (centered on zero, median cluster size is 51 nt, shaded gray) are shown for all clusters or non-CDS clusters. For the latter, Ago2 brain clusters located in CDS were removed in order to avoid a potential bias for coding regions to show increased conservation, particularly for smaller exons that would be flanked by lesser conserved intronic sequences.
Further characterization of the Ago2 brain clusters identified thousands of putative miRNA:target gene pairs on the basis of cluster sequence content and miRNA abundances. Computational site prediction with TargetScan and PITA was performed on cluster sequences, and the top hits (up to ten per algorithm) corresponding to abundant miRNAs (i.e., top 200 seeds) are provided for each cluster (Table S3) (Grimson et al., 2007; Kertesz et al., 2007). Enriched noncanonical sites for the most highly abundant miRNAs (e.g., Let-7 7m9, miR-124 Bulge, miR-9 G:U, and miR-29 7m9) are also included. To assess the functionality of these miRNA:target pairs, we analyzed microarray data from studies evaluating the effects of miRNA overexpression (ten experiments corresponding to seven miRNAs) or inhibition (pooled depletion of 25 different miRNAs) in cultured human cells (Hafner et al., 2010). As expected, miRNA overexpression resulted in the downregulation of many Ago2 HITS-CLIP-identified targets for the respective miRNA in each independent data set (Figures 4A and S4). Overall, 3′ UTR sites showed greater responsiveness than those in coding regions (CDS; Figure 4B, top). This difference in potency is consistent with prior studies, and more recent data support that miRNA binding to CDS primarily inhibits translation (Hausser et al., 2013). In miRNA inhibition studies, targets identified by Ago2 HITS-CLIP showed derepression (Figure 4B, bottom), and, notably, the magnitude was comparable to that observed for experimentally validated miRNA:target interactions recorded in miRTarBase (Hsu et al., 2011).
Figure 4. Ago2 HITS-CLIP Identifies Functional miRNA:Target Gene Pairs.
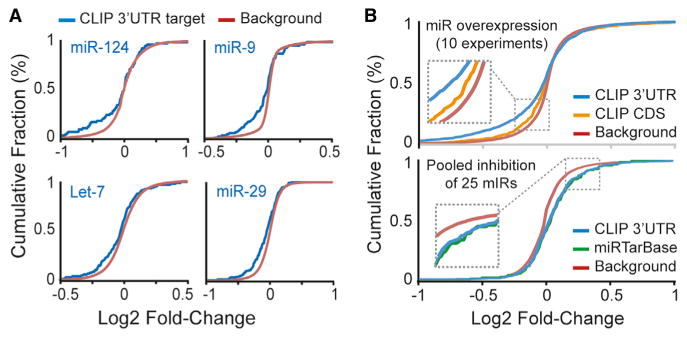
Functionality of miRNA:target gene pairs identified by Ago2 HITS-CLIP was evaluated with microarray data for gene expression changes after miRNA overexpression or miRNA inhibition. Cumulative fraction curves for miRNA:target pairs (e.g., genes with 3′ UTR or CDS Ago2 HITS-CLIP clusters or experimentally validated miRNA:target genes recorded in miRTarBase; each corresponding to the overexpressed or inhibited miRNAs in the microarray experiments) are plotted. Curve displacement to the left or right, relative to background genes (i.e., no site), indicates down- or upregulation, respectively.
(A) Responsiveness of target genes in individual miRNA overexpression data sets (NCBI GEO accession numbers: miR-124, GSM37601; miR-9, GSE33952; Let-7, GSE2918; and miR-29, GSE18713).
(B) Plots for summarized data from 10 miRNA overexpression studies (top) and a single data set for pooled miRNA inhibition (bottom) are shown.
See also Figure S4.
Intersection of Human and Mouse Ago HITS-CLIP Data
To gain insight into the conserved roles of miRNAs in brain, we intersected our data set with orthologous regions corresponding to the mouse brain Ago HITS-CLIP ternary map data (Chi et al., 2009). In the latter, Ago HITS-CLIP was performed in developing mouse brain (i.e., P13 neocortex). We identified 646 shared human and mouse Ago HITS-CLIP regions, representing a significant enrichment relative to background clusters (∼5-fold, Pearson's chi-square test, p < 2.2 × 10−16; Figure 5A and Table S3). Notably, 59% were in coding regions, providing a sizeable resource for investigating the conserved functions of miRNAs outside of 3′ UTRs. Gene ontology analysis with the shared target genes revealed an enrichment for synaptic localization and functions (e.g., neuron projection, synapse assembly, and neurotransmitter and ion transport), supporting a conserved role of miRNAs as key regulators of gene expression at sites distant from the transcriptional activities within the nucleus (Figures 5B and 5C). Concurrently, Ago brain clusters are enriched in genes producing transcripts that localize to the synaptic neuropil (Cajigas et al., 2012) (Figure S5). We also evaluated the regions corresponding to shared human and mouse Ago brain clusters for the presence of TargetScan predicted conserved miRNA binding sites. This analysis produced 101 sites across 56 genes, many of these sites corresponding to brain-enriched miRNAs (Figure 5D).
Figure 5. Intersection of Human and Mouse Brain Ago HITS-CLIP Data.
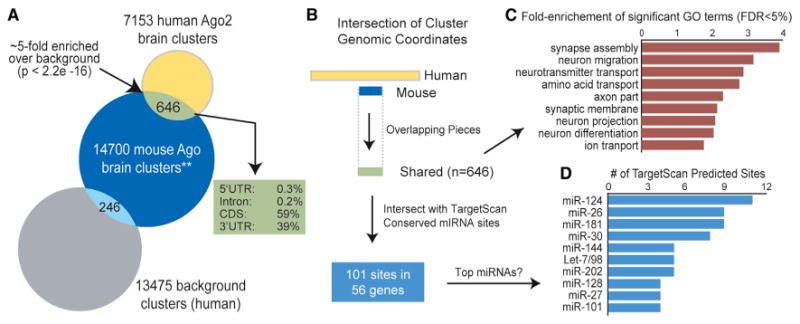
(A) Venn diagram of the number of human Ago2 brain clusters overlapping with the mouse Ago HITS-CLIP brain data (**, ternary map data of miRNA binding sites spanning 7 nt, further described in the Experimental Procedures) (Chi et al., 2009). The number of overlapping human background clusters (i.e., regions up- and downstream of the Ago2 brain clusters; see the Supplemental Information) is provided as a control.
(B) Schematic depicting the processing of overlapping human (≥ 50 nt) and mouse (7 nt) brain Ago HITS-CLIP clusters.
(C) Gene ontology analysis was performed with the genes that harbor “shared” clusters as input relative to all genes containing clusters as background.
(D) The overlapping regions were intersected with TargetScan-predicted conserved miRNA binding sites, and the top represented miRNAs (by number of sites) are shown.
See also Figure S5.
Identification of Clinically Relevant Ago2 Binding Sties in Human Brain
Lastly, we explored the human Ago2 brain clusters for the presence of disease-relevant miRNA:target interactions. We focused our initial efforts on miR-137, which was recently identified as a “hit” in a genome-wide association study among schizophrenia patients and is also being investigated in bipolar disorder (Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium, 2011; Whalley et al., 2012). Our data set harbors 88 putative miR-137 sites (Table S6; based on canonical TargetScan seeds), and 24 of these reside within schizophrenia-related genes (SZGene database, n = 13) and/or genes having correlated expression with prefrontal cortex abnormalities in psychiatric patients (n = 16) (Allen et al., 2008; Kim and Webster, 2010). Notably, TCF4, a previously validated miR-137 target (Kwon et al., 2013), was among these. Here, we tested seven of these genes in order to determine their responsiveness to miR-137. For this, 3′ UTRs of the psychiatric disease-relevant genes harboring miR-137 sites identified by our Ago2 HITS-CLIP studies were cloned downstream of luciferase reporters and tested in cell culture. Remarkably, all seven 3′ UTR reporters were suppressed by miR-137 treatment, relative to negative control miRNA (∼30%–50% silencing; Figure 6A), providing additional support (complementing the above microarray analyses) for the functionality of the miRNA binding sites identified by our HITS-CLIP studies.
Figure 6. Functional Responsiveness of Disease-Relevant miRNA Binding Sites.
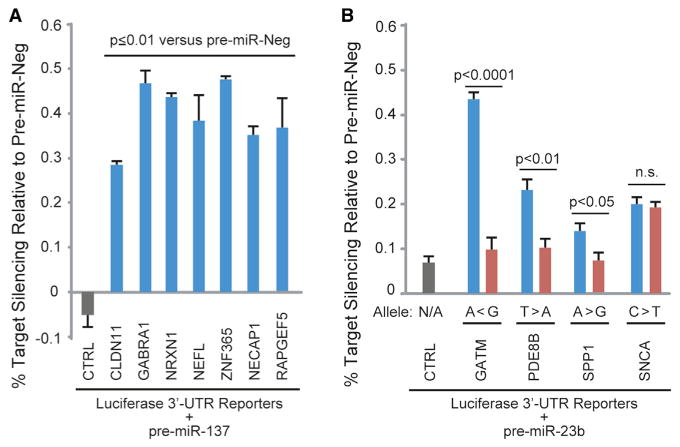
Plasmids (10 ng) coexpressing Firefly luciferase (normalizer) and Renilla luciferase reporters harboring 3′ UTRs corresponding to the indicated genes were cotransfected along with synthetic pre-miRs (50 uM) into HEK 293 cells. Dual luciferase assays were performed at 24 hr posttransfection, and Renilla/Firefly ratios were normalized to the pre-miR-Neg1 treatment for each 3′ UTR construct. CTRL is the “empty” parental plasmid into which 3′ UTRs were cloned.
(A) Silencing activity of miR-137 on schizophrenia-related gene 3′ UTRs harboring miR-137 sites identified by Ago2 CLIP-seq.
(B) Effects of disease-relevant SNPs predicted to alter miR-23 binding sites (refer to Table 1 and Figure S6) on 3′ UTR silencing mediated by pre-miR-23b. Alleles predicted to be silenced are shown in blue, and alleles that disrupt seed pairing are shown in red. Major and minor alleles are denoted by “>” or “<” symbols, respectively. In both panels, each bar depicts the mean ± SEM for n ≥ 3 biological replicates for each treatment. Two-tailed pairwise Student's t tests for the indicated comparisons were performed, and p values are provided. n.s., not significant.
See also Figure S6 and Table S6.
We further queried our data set for clinical relevance by searching for Ago2 binding sites overlapping previously reported disease-associated SNPs. This uncovered prospective clinically-relevant miRNA:target interactions in genes linked to various neuropsychiatric conditions (Table 1). With consideration that such intersections may be due to random chance, we focused this analysis on “hits” for which (1) the SNPs are predicted to alter miRNA:target pairing within seed sites matching abundant brain miRNAs (Figure S6) and (2) data exists to support relationships among the miRNAs, target genes, and disease. For example, in the terminal exon of the TUBA1A gene, weidentified anAgo2cluster overlapping a lissencephaly-causing nonsynonymous mutation, which imparts no obvious detriment to protein function (Tian et al., 2010). Our data indicate that it woulddisrupt a potential miR-124 seed, and the coincident expression of TUBA1A and miR-124 in developing brain suggests that their interaction may be required for proper corticogenesis (Coksaygan et al., 2006; Maiorano and Mallamaci, 2009). We also detected a disease-associated SNP predicted to enhance miR-124 binding within an exonic Ago2 cluster in the IDS gene encoding iduronate 2-sulfatase. The prospect that this may augment the repression of iduronate 2-sulfatase expression is interesting considering that the mutation causes an enzyme deficiency resulting in an attenuated form of Hunter syndrome (Sohn et al., 2012).
Table 1. Summary of Disease-Relevant SNPs in Human Ago2 Brain Clusters.
| Gene | Disease | SNP ID | Alleles (MAFa) | Effect of Minor Allele | Comments | Sourceb |
|---|---|---|---|---|---|---|
| TUBA1A | Lissencephaly | rs137853044 | G/A (rare) | Disrupts miR-124 seed | R402H substitution may be tolerated | HGMD |
| IDS | Hunter syndrome | rs141720810 | C/G (rare) | Enhances mIR-124 site | Disease caused by enzyme deficiency | HGMD |
| MAPT | Tauopathies | rs17652121 | T/C (11%) | Disrupts miR-181 seed | SNP may alter splicing of exon 10 | GWAS-LD |
| rs9468 | T/C (7.8%) | Disrupts miR-128 seed | SNP defines H2 haplotype | Gerrish et al., 2012 | ||
| SNCA | Parkinson's | rs17016074 | C/T (4.4%) | Disrupts miR-23 seed | SNP alters transcript stability | HGMD |
| rs183204610 | C/A (rare) | Disrupts miR-153 seed | miR-153 site shown to be functional | Doxakis, 2010 | ||
| SPP1 | Autoimmune disease | rs1126772 | A/G (17%) | Disrupts miR-23 seed | Spp1 is increased in MS and IBD* | HGMD |
| AAK1 | Parkinson's onset | rs6757825 | G/A (13.3%) | Disrupts miR-22 seed | Earlier onset may be cholesterol related | GWAS-LD |
| PUM2 | ADHD | rs6669 | G/A (3.6%) | Disrupts miR-377 seed | Nearby rs1043419 assoc. with ADHD | miRdSNP |
| PDLIM5 | Schizophrenia | rs12294 | C/T/G (32%, 0.5%) | Disrupts miR-19 seed | Elevated PDLIM5 assoc. with disease | miRdSNP |
| PDE8B | Alzheimer's | rs1580 | T/A (rarec) | Disrupts miR-23 seed | Elevated PDE8B assoc. with disease | miRdSNP |
| PCSK1 | Obesity | rs6235 | G/C (38%) | Disrupts miR-92 seed | Potential miR-9 site within 10 nt | GWAS |
| CELSR2 | Cholesterol | rs629301 | T/G (22%) | Creates miR-10 seed | CELSR2 may play a role in cholesterol efflux | GWAS-LD |
| GATM | Kidney disease | rs17618637 | G/A (5%) | Enhances miR-23 seed | Disease caused by enzyme deficiency | GWAS-LD |
Refer to Figure S6A for miRNA:target base pairing diagrams showing the positions of the respective SNPs. Refer to Figure S6B for reproducibility of Ago2 HITS-CLIP data within regions encompassing these SNPs.
MAF, minor allele frequency; MS, multiple sclerosis; and IBD, inflammatory bowel disease.
HGMD, Human Gene Mutation Database. GWAS-LD refers to SNPs (MAF>5%) in linkage disequilibrium (R2=1) with GWAS SNPs.
PDE8B SNP (rs1580) was originally reported as having a 16% MAF. During the revision of this manuscript, dbSNP135 updated the MAF to <1%.
With respect to more prevalent neuropsychiatric conditions, we identified a robust Ago2 cluster in exon 9 of the MAPT gene encoding Tau. Others have shown that mutations in this region may disrupt splicing inclusion of exon 10, which can contribute to certain tauopathies, including Alzheimer's disease and frontotemporal dementia with Parkinsonism (Hasegawa et al., 1999). MAPT also harbors a 3′ UTR Ago2 cluster with a predicted miR-128 binding site which overlaps a previously described tauopathy-associated SNP (Gerrish et al., 2012). These observations raise the intriguing possibility that Ago2:miRNA interactions could contribute to tauopathies by modulating the levels of pathogenic tau isoforms.
We also identified Ago2 binding sites overlaying SNPs predicted to disrupt miRNA seed sites in the 3′ UTRs of SNCA and SPP—genes with known involvement in neurological conditions. SNCA encodes alpha-synuclein, a component of the pathogenic aggregates observed in Parkinson's disease, and one of these SNPs (rs17016074) increases transcript levels by >30% (Sotiriou et al., 2009), whereas the other resides within a functional miR-153 binding site (Doxakis, 2010). Notably, SNCA duplications or triplications cause familial Parkinson's disease (Chartier-Harlin et al., 2004), and changes in alpha-synuclein levels have been hypothesized to contribute to more common forms of Parkinson's. As for SPP1, this gene encodes for secreted phosphoprotein 1, more commonly known as osteopontin. Osteopontin increases to toxic levels in multiple sclerosis and Crohn's inflammatory bowel disease (Glas et al., 2011; Niino et al., 2003), and the 3′ UTR mutation, previously implicated in both diseases, may contribute to this by disrupting a potential binding site for miR-23b. Interestingly, miR-23b is downregulated in and considered to be a broad suppressor of autoimmune diseases, including lupus, rheumatoid arthritis, and multiple sclerosis (Zhu et al., 2012).
As a first step toward evaluating the functional responses of these disease-relevant miRNA:target interactions and the potential effects of the polymorphisms, we performed 3′ UTR reporter luciferase assays for the four miR-23 target genes in Table 1 (i.e., GATM, PDE8B, SPP1, and SNCA). All four showed a repressive response to miR-23b treatment in cell culture, and, notably, a significant impact of the SNP on silencing was observed for three of the targets. These results validate the data set's utility for revealing clinically relevant information and warrant future studies aimed at investigating these interactions in specific brain-cell types. Furthermore, these and other examples in Table 1 corroborate the use of this brain miRNA:target interactome for formulating hypotheses regarding the consequences of disease-associated polymorphisms. To this end, we summarize information for 916 common SNPs (minor allele frequency [MAF] > 1%) present within the human Ago2 brain clusters (Table S7) to serve as a resource for further investigation of polymorphisms that may alter miRNA targeting and influence human disease phenotypes, particularly for the many neurological conditions associated with gene dosage abnormalities.
Discussion
Data support that miRNAs contribute to protective and pathogenic responses to disease; thus, identifying their targets in affected tissues remains a crucial aspect of translational miRNA research. Computational prediction has facilitated these efforts, particularly for the study of canonical seed-based miRNA binding sites. However, there is growing appreciation that biologically relevant miRNA:target interactions can now be broadly surveyed with the use of biochemical approaches, and recent work has demonstrated that CLIP-based methods are inherently better for identifying noncanonical miRNA binding sites (Chi et al., 2012; Helwak et al., 2013). Therefore, we performed Ago2 HITS-CLIP in order to generate a transcriptome-wide map of miRNA binding sites in human brain, a tissue in which miRNAs have been implicated in several diseases (Im and Kenny, 2012). Our work revealed more than 7,000 Ago2 binding sites, many of which harbor sequences corresponding to the most abundant brain miRNAs. These data represent a valuable resource for accelerating our understanding of miRNA functions and how these effectors may interface with human diseases. Here, focusing on the latter, we identify clinically relevant miRNA:target interactions among our HITS-CLIP data and present several exciting examples of how SNPs may contribute to CNS pathologies through the modulation of miRNA activity. Our luciferase validation studies demonstrate that disease-relevant SNPs can influence silencing mediated by miR-23b (Figure 6B). Interestingly, the GATM gene SNP tested showed the most robust differential response, and the minor allele was strongly repressed. GATM encodes for glycine amidinotransferase, a protein involved in creatine synthesis. Although the associated GWAS study implicates this gene in chronic kidney disease, rare GATM mutations have been shown to cause creatine deficiencies in brain, leading to cognitive impairment (Item et al., 2001). Further evaluation of whether the tested SNP affects endogenous GATM levels in order to produce clinical phenotypes will be interesting, particularly for individuals with minor allele homozygosity.
We queried our data set for disease-relevant interactions by identifying targets for the schizophrenia-related miRNA miR-137. In addition to a previously validated interaction of miR-137 with TCF4, our data support seven additional functional targets for miR-137 within genes associated with psychiatric disease (Figure 6A). Intriguingly, there are six common SNPs (>1% MAF) within five of these Ago2 clusters (residing in CLDN11, GABRA1, NEFL, NRXN1, and RAPGEF5; refer to Table S6), highlighting the prospect for future studies to evaluate the effects of these SNPs on miR-137 activity. Altogether, these analyses highlight the utility of this data set for identifying disease-relevant miRNA:target interactions, and future work may help to elucidate additional targeting events involved in neurodegenerative or brain vasculature disease processes.
This resource will also be valuable for querying the basic biological functions of miRNAs. For instance, the field is currently shifting focus to miRNA binding events in regions outside of 3′ UTRs, and our data set includes hundreds to thousands of sites in 5′ UTRs, coding regions, and introns. Future work investigating the functional relevance of these sites will be interesting, particularly studies addressing whether intronic sites are engaged by Ago2 in the nucleus or whether these represent retained intronic sequences targeted in the cytoplasm. Another exciting research avenue would be to test whether CDS-located miRNA binding sites residing within alternatively spliced exons confer isoform-specific silencing; indeed, a preliminary analysis of our data set indicates the presence of nearly 400 clusters within alternatively spliced cassette exons (data not shown). CLIP data sets will also be useful for globally evaluating competitive binding of miRNAs and RNA binding proteins throughout the transcriptome. The possibility of such events involving negative and positive regulators provides attractive means for potent switches in gene regulation mediated at the posttranscriptional level. Alternatively, similar biological switches can be achieved through feedback loops involving miRNAs and transcription factors (Iwama, 2013; Kim et al., 2007), and our Ago2 HITS-CLIP targets include hundreds of transcription factor mRNAs, providing a key resource for ongoing investigations of these types of regulatory circuits.
Future analyses of disease- and biologically relevant miRNA:target interactions in brain could be prioritized by focusing on conserved sites identified in both mouse and human Ago HITS-CLIP studies. Although these data sets are significantly enriched for shared sites, the proportion of nonoverlapping sites is notably larger (Figure 5A). This could be the result of obvious differences between mouse and human brains and/or the varying transcriptomes and miRNA profiles in developing (mouse samples) versus adult (human samples) brains. Alternatively, the mouse data were generated with a pan-Ago antibody, whereas the antibody we employed was specific to Ago2. However, several prior reports have noted that, for the most part, endogenous miRNAs are equivalently sorted into Ago1 and Ago2 (Dueck et al., 2012). Alternatively, disparities between the human and mouse data could reflect the methods and criteria used in peak-calling procedures, given that the mouse Ago ternary map includes many miRNA binding sites with minimal read evidence across few biological replicates. Inevitably, additional studies will be required for determining whether any or all of these factors contribute to the observed discrepancies or whether miRNA targeting in brain can indeed diverge so greatly across species.
Overall, our studies and the accompanying data sets provide a foundation for the further characterization of miRNA:target interactions throughout other brain structures and across normal and diseased human tissues. Expansion into additional brain regions (e.g., midbrain and hindbrain) as well as nonhuman primates will help to clarify how miRNAs contribute spatiotemporally and evolutionarily in higher-order brain and will complement the ongoing efforts to deeply characterize the brain transcriptomes of primate species (Bernard et al., 2012; Somel et al., 2013).
Experimental Procedures
Samples
Postmortem human brain samples were obtained from Juan Troncoso (Brain Resource Center at Johns Hopkins University School of Medicine) with informed consent from families. Gray matter tissues were harvested from the motor cortex (Brodmann's area 4) and cingulate gyrus and stored at −80*C until use. Detailed sample information is provided in Table S1.
Ago2 HITS-CLIP
CLIP was performed as previously described (Chi et al., 2009) with modifications. Samples (∼60 mg) of human postmortem brain tissue were triturated in cold PBS, UV crosslinked, lysed, and treated with DNase and a low concentration of RNase. Immunoprecipitation was performed by incubating lysed samples with Protein A Dynabeads (Invitrogen) complexed with anti-Ago2-3148 (Grey et al., 2010). Then, beads were washed, treated with polynucleotide kinase 32P-γ-ATP in order to 5′-end label the associated RNAs, and washed again. 5′ linker ligation was performed with a PAGE-purified DNA-RNA hybrid linker containing 5′ Amino Modifier C6, RNA bases (underlined) and ambiguous bases (Ns): 5′-(5AmMC6)-CCTCTCTAUGGGCAGUCGGU GAUNNNNG-3′. Then, samples were treated with calf intestinal alkaline phosphatase in order to remove 3′ phosphates. Immunoprecipitated complexes were resolved on a NuPAGE Novex 3–8% Tris-Acetate Gel with Tris-Acetate running buffer as per the manufacturer's instructions (Invitrogen). Products were transferred to BA-85 nitrocellulose (Whatman) with the Mini-PROTEAN 3 cell and Mini Trans-Blot module (Bio-Rad), and film autoradiography was performed. The bands corresponding to Ago2 complexed with miRNAs (∼110 kDa), and Ago2:miRNA:target complexes (∼130 kDa) were cut, placed in 1.5 ml Eppendorf tubes, and treated with proteinase K(Qiagen). RNAs were extracted with phenol and chloroform and overnight precipitation in 3M NaOAc (pH 5.2) in a 1:1 EtOH:isopropanol solution, and ligated to a PAGE-purified 3′ RNA linker (note that the 3′ RNA linker was 5′ phophorylated and 3′ puromycin blocked; 5′-P-GUUACGCUCGCCGUAGAGG-Pmn-3′). After phenol and chloroform extraction and precipitation, samples were subjected to RT-PCR with a reverse primer (5′-AGTTCAGACGTGTGCTCTTCCG ATCCTCTACGGCGAGCGTAAC-3′) for the RT reaction followed by PCR with the reverse and forward(5′-CCTCTCTATGGGCAGTCGGTGAT-3′) primers (95°C for 2 min followed by 25–30 cycles of 95°C for 20 s, 59°C for 25 s, 72°C for 20 s, and lastly a 5 min incubation at 72°C). Products corresponding to miRNAs and target RNA tags were PAGE purified with traditional methods and subjected to a second round of PCR in the presence of custom Illumina Hi-Seq fusion primers (see below; 95°C for 2 min followed by 10–15 cycles of 95°C for 20 s, 61°C for 25 s, 72°C for 30s, and lastly a 5 min incubation at 72°C). The primers were ILMN-F1 DNA primer (5′-AATGATACGGC GACCACCGACAGGTCCTCTCTATGGGCAGTCGGTGAT-3′) and an example of the ILMN barcode primer (ILMN-BCn; barcode underlined, 5-CAAGCA GAAGACGGCATACGACGTGATGTGACTGGAGTTCAGACGTGTGCTC TTCCGATC-3′). Eight barcodes were used: (1) CGTGAT, (2) ACATCG, (3) GCCTAA, (4) TGGTCA, (5) CACTGT, (6) ATTGGC, (7) GATCTG, and (8) TCAAGT. Products were PAGE purified, and a quality assessment of the Ago2 HITS-CLIP libraries was performed by the University of Iowa DNA Facility with the High Sensitivity DNA Assay on the Agilent Bioanalyzer DNA-1000. Library concentration was measured with a KAPA Library Quantification Kit for Illumina (Kapa Biosystems) according to manufacturer's instructions. Libraries were concentrated (by speed-vac) or diluted to 10 nM as needed. High-throughput sequencing was carried out by the University of Illinois, Roy J. Carver Biotechnology Center on the Illumina Hi-Seq 2000. Libraries were pooled in order to allow sequencing of four samples per lane (miRNAs and target RNA tags for each sample; eight total barcodes). A 9:1 ratio of tags:miRNA libraries was used, and pooled libraries (10 nM) were submitted to the sequencing facility along with a custom Read1 primer (100 uM; custom Read1 sequencing primer [DNA], 5′-CAGGTCCTCTCTATGGGCAGTCGG TGAT-3′). We obtained 75–100 nt reads with Read1, and the barcode was sequenced by standard means.
Western Blot
Protein sampled during the CLIP procedure was separated on a NuPAGE Novex 3–8% Tris-Acetate Gel (Invitrogen) before being transferred to a 0.45 mm polyvinylidene fluoride membrane (Millipore). The membrane was blocked with 2% milk in 1 × PBS with 0.05% Tween 20. Mouse anti-Ago2 (Wako Pure Chemicals Industries) diluted 1:2,000 in blocking buffer was added and incubated overnight at 4°C followed by horseradish-peroxidase-conjugated goat anti-mouse (Jackson ImmunoResearch) diluted 1:10,000 in blocking buffer. Ago2 protein was detected by ECL Plus substrate (Amersham Biosciences) and film.
Preprocessing and Mapping of Ago2 HITS-CLIP Tags
We removed the first 5 nt, which corresponded to the 3′ end of the 5′ linker, from the 5′ end of reads. Then, we scanned the reads from 5′ end to 3′ end by the adaptor sequence (GTTACGCTCGCCGTAGAGG), allowing up to two mismatches. Because it is possible that the adaptor that is attached to 3′ end of the read is not completely sequenced, we also scanned the 3′ end of reads that require at least 10 nt by the 5′ end of adaptor, allowing up to two mismatches. After adaptor trimming, we excluded reads with length less than 10 nt from downstream analysis. Then, adaptor-trimmed mRNA tags were mapped to the human genome (hg19) with Bowtie (Langmead et al., 2009), allowing up to two mismatches. Gene coordinates were downloaded from Ensembl (version 57).
Estimation of miR Expression Levels
We downloaded the human miRNA sequences from miRBase (release 18) (Kozomara and Griffiths-Jones, 2011) and merged redundant miRNA sequences from the same miRNA family. Then, we used Bowtie to map the miRNA tags to the miRBase, allowing up to one mismatch. The miRNA expression levels are quantified as the number of reads mapped to individual miRNAs normalized by the total number of mapped reads in miRBase. The expression levels from different samples were further normalized by quantile normalization in order to control for batch effect.
Peak Calling by Zero-Truncated Negative Binomial Model
We used the zero-truncated negative binomial model (ZTNB) to measure the significance of read coverage height for each mapped position (Uren et al., 2012). For each gene in a given sample, we assume that the height of each position follows the Poisson distribution, yi ∼Poisson (λi), yi > 0. λi is the expected height for position 1, and it is a random variable following a gamma distribution, λi ∼Gamma (1/α, α), where α is the overdispersion parameter. If α = 0, then it is reduced to Poisson. The ZTNB distribution has density
where μi = λi (as in Poisson).
The α parameter is estimated by maximum likelihood function with numerical method. For each height (Hi), the p value (Pi) is calculated by summing the probabilities for finding Hi, or higher height. The calculation is performed by R packages MASS and pscl.
To call HITS-CLIP peaks with information from multiple replicates, we used Fisher's method to combine p values of replicates. For each position, we combined the p values from all biological replicates into one test statistic (χ2) using the formula
where pi is the p value for the jth replicates. χ2 has a chi-square distribution with 2k degrees of freedom, where k is the number of replicates. The combined p value for each position was adjusted by Bonferroni correction in order to account for multiple hypothesis testing.
Identification of Statistically Significant Ago2 Brain Clusters
We used Bonferroni adjusted p value < 0.05 as the cutoff in order to define statistically significant positions. Then, we merged those significant positions into continuous regions for any positions within 60 nt of each other. Given that the average Ago-mRNA footprint is 45–62 nt (Chi et al., 2009), we extended the continuous regions with length less than 50 nt to 50 or 51 nt symmetrically from the 5′ and 3′ ends. We defined those regions as Ago2 brain clusters. Furthermore, we defined the specific positions of Ago2 brain clusters with respect to annotated gene structures. The gene structure information (e.g., exon, intron, CDS, and UTRs) was downloaded from Ensembl (version 57) in order to determine the intragenic locations of the Ago2 brain clusters. Intragenic clusters with ambiguous locations (i.e., those falling in more than one category; for example, both CDS and 3′ UTR) were assigned positions on the basis of the prioritization scheme (3′ UTR > CDS > 5′ UTR > intron) for the data shown in Figure 3A. As an alternative, we also assigned ambiguous clusters partial values (e.g., 0.5 cluster to CDS and 0.5 cluster to 3′ UTR). This yielded the following positional distributions: 3% 5′ UTR, 38% CDS, 33% 3′ UTR, 23% intron, and 3% other. The values for this latter scoring scheme are provided in the cluster summary (Table S3).
Correlation of Ago2-Associated RNAs across Samples
Wiggle files were generated with the use of an in-house program to determine the read coverage at each genomic position for each sample data set on a per-genomic-strand basis. Wiggle files for plus and minus strands for each sample were combined across all genomic intervals. Overlapping intervals between the two files were merged by taking the maximum height at each genomic position. Then, all pair-wise correlations were performed with the wigCorrelate script available from the UCSC Genome Browser (Rhead et al., 2010).
PhastCon Score
Positional conservation within the human Ago2 brain clusters was evaluated with the Conservation Plot (version 1.0.0) tool available as part of the Cistrome Analysis Pipeline Module (Liu et al., 2011). A window size of 600 nt was used.
Motif Analysis
The Regulatory Sequence Analysis Tools (RSAT) peak motifs package was used to analyze the Ago2 brain cluster sequences by inputting 100 nt peak sequences centered on the clusters and using default parameters to identify ten motifs per algorithm on single strands (Thomas-Chollier et al., 2012). Data from this analysis was used for Figures 2A and S2B. As an alternative, we also performed motif analysis in order to identify enriched motifs within Ago2 brain clusters relative to background clusters. For the latter, we selected upstream (−100 nt − cluster_length, −100 nt)and downstream (100nt + cluster_length, +100 nt) coordinates and excluded regions overlapping with Ago2 brain cluster regions. We implemented a sliding window scan (window size = 7 nt) of each region (brain clusters or background) with a 1 nt increment in order to identify the incidences of all possible heptamers, including miRNA seeds. Enrichment data for all possible heptamers was used to generate Figure S3 and Table S4. To determine the enrichment of motifs corresponding to miRNAs, we summarized the data matching 7m8 and 7A1 site types for all known miRNA seeds (Grimson et al., 2007). The p value for any given miRNA seed was calculated from the number of windows that matched miRNA seeds in all Ago2 brain cluster and background regions with a Fisher's exact test. The p values were transformed to FDRs with the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995). Summarized seed data were used to produce Figure 2C and Table S5.
Functional Validation of miRNA:Target Gene Pairs
Microarray data for the indicated miRNA overexpression and inhibition studies were obtained from the NCBI Gene Expression Omnibus (GEO). Data were processed with GEO2R or Partek Genomics Suite (Partek) in order to determine log2 fold changes in gene expression between experimental and control conditions. For miRNA overexpression studies, the cumulative fractions of Ago2 HITS-CLIP target genes (with 3′ UTR- or CDS-located clusters harboring the relevant miRNA seed sites) and background genes (i.e., no site) were generated for each of the 10 miRNA overexpression microarray data sets (individual plots shown in Figures 4A and S4). Data for these analyses were grouped by discrete intervals (0.01 log2 fold-change increments). The data for all ten experiments were summarized by averaging the cumulative fraction values for targets and background sets at each interval (Figure 4B, top).For the miRNA inhibition study, Hafner et al. (2010) performed a pooled depletion of 25 highly expressed miRNAs in human embryonic kidney (HEK) 293 cells. Among these, seven corresponded to highly abundant brain miRNAs present in our data, including the following seed families: Let-7, miR-15/16, miR-30, miR-101, miR-103/107, miR-124, and miR-27. Genes containing 3′ UTR-located Ago2 HITS-CLIP clusters harboring sites matching any of these seven miRNA seeds were used for this analysis (Figure 4B, bottom, “CLIP 3′ UTR”), as were experimentally validated target genes for any of these seven miRNAs, on the basis of miRTarBase entries (Hsu et al., 2011).
For luciferase-based validation studies, 3′ UTRs for the indicated target genes were PCR amplified from human genomic DNA (isolated from HeLa and HEK 293 cells) with the DNA primers provided in Table S8. Products were digested with XhoI and NotI and ligated into these same sites within the dual luciferase expression plasmid psiCheck2 (Promega). This positions the 3′ UTRs downstream of Renilla luciferase, creating an experimental reporter, whereas the Firefly luciferase serves as the normalizing control. Site-directed mutagenesis was performed with standard methods in order to engineer the relevant SNP alleles into 3′ UTR reporter constructs. HEK 293 cells grown in 24-well plates were cotransfected in triplicate with 10 ng of luciferase reporter plasmids and 50 uM of commercially available synthetic pre-miRNAs corresponding to miR-137, miR-23b, or a negative control (miR-Neg1). Transfections were performed with Lipofectamine 2000 (Invitrogen), and Firefly and Renilla luciferase activities were assessed at 24 hr posttransfection with the Dual-Glo Luciferase Assay System (Promega) per the manufacturer's instructions. Luminescent readings were measured for each sample in duplicate using a GloMax Microplate Reader (Promega), and results were calculated as the quotient of Renilla/Firefly luciferase activities with subsequent normalization to the pre-miR-Neg1 control for each 3′ UTR tested. Biological replicates were increased to n = 6 by repeating the transfections and downstream analyses.
Intersection to Mouse Ago HITS-CLIP Data
For the intersection of human Ago2 brain clusters with mouse Ago HITS-CLIP data, we used the downloadable ternary map data from http://ago.rockefeller.edu/AgoTernaryTable_mm9.txt/ (Chi et al., 2009) and converted the coordinates to human hg19 with the Galaxy lift-over function (Goecks et al., 2010). Note that the mouse Ago ternary map consists of all sites (i.e., uncurated in terms of robustness) identified by Ago HITS-CLIP in P13 mouse neocortex (refer to Chi et al., 2009, for a description).
Gene Ontology Analysis
Gene ontology analysis was performed with the GOrilla web server (Eden et al., 2009). Official gene symbols for genes containing human and mouse shared Ago brain clusters were input as the “target set,” and genes containing human Ago2 brain clusters were input as “background set” in order to control for bias of HITS-CLIP approaches toward higher expressed transcripts.
Evaluation of SNPs
Genomic coordinates of human Ago2 brain clusters were intersected with coordinates of GWAS SNPs (Hindorff et al., 2009) and accompanying SNPs in complete linkage disequilibrium (R2 = 1) having minor allele frequencies > 5% (dbSNP 135). Additional disease-relevant SNPs or mutations were curated from RegulomeDB (Boyle et al., 2012), miRdSNP (Bruno et al., 2012), Human Gene Mutation Database (Stenson et al., 2009), literature review, and the UCSC Genome Browser (Rhead et al., 2010). Note that rs9468 and rs629301 were identified in Ago2 brain clusters with Bonferroni adjusted p value < 25%, while all others overlap with Bonferroni adjusted p value < 5% clusters. Putative miRNA target sites within human Ago2 brain clusters encompassing disease-relevant SNPs were identified with TargetScan (Grimson et al., 2007), PITA (Kertesz et al., 2007), RegRNA (Huang et al., 2006), or miRdSNP (Bruno et al., 2012), and miRNA:target hybridization was evaluated with the RNAhybrid web server (Krüger and Rehmsmeier, 2006).
Supplementary Material
Acknowledgments
We thank members of the B.L.D. laboratory for insightful discussion, Clayton Oakley for assistance with molecular cloning, Kevin Knudtson and his staff of the University of Iowa DNA Facility and Alvaro Hernandez and his staff at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign, for sequencing-related services, Lan Lin for performing library quantitation, Edgardo Rodriguez for manuscript review, and Juan C. Troncoso and Olga Pletnikova and the Brain Resource Center, Division of Neuropathology, Department of Pathology, Johns Hopkins University School of Medicine for providing brain samples. This work was funded by the Roy J. Carver Trust (University of Iowa) and the National Institutes of Health (NS50210, DA025132, HL07121, and GM088342). Y.X. is supported by an Alfred Sloan Research Fellowship.
Footnotes
Accession Numbers: Relevant data sets, including raw data, have been deposited in the NCBI Gene Expression Omnibus under accession number GSE52084.
Supplemental Information: Supplemental Information contains six figures and eight tables and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2013.10.062.
Contributor Information
Yi Xing, Email: yxing@ucla.edu.
Beverly L. Davidson, Email: beverly-davidson@uiowa.edu.
References
- Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, Tanzi RE, Bertram L. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, Giraldez AJ. Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science. 2012;336:233–237. doi: 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser A Stat Soc. 1995;57:289–300. [Google Scholar]
- Bernard A, Lubbers LS, Tanis KQ, Luo R, Podtelezhnikov AA, Finney EM, McWhorter MM, Serikawa K, Lemon T, Morgan R, et al. Transcriptional architecture of the primate neocortex. Neuron. 2012;73:1083–1099. doi: 10.1016/j.neuron.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno AE, Li L, Kalabus JL, Pan Y, Yu A, Hu Z. miRdSNP: a database of disease-associated SNPs and microRNA target sites on 3′UTRs of human genes. BMC Genomics. 2012;13:44. doi: 10.1186/1471-2164-13-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajigas IJ, Tushev G, Will TJ, tom Dieck S, Fuerst N, Schuman EM. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron. 2012;74:453–466. doi: 10.1016/j.neuron.2012.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, Darnell RB. An alternative mode of microRNA target recognition. Nat Struct Mol Biol. 2012;19:321–327. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JE, Lee PR, Chen S, Li W, Fields RD. MicroRNA regulation of homeostatic synaptic plasticity. Proc Natl Acad Sci USA. 2011;108:11650–11655. doi: 10.1073/pnas.1017576108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coksaygan T, Magnus T, Cai J, Mughal M, Lepore A, Xue H, Fischer I, Rao MS. Neurogenesis in Talpha-1 tubulin transgenic mice during development and after injury. Exp Neurol. 2006;197:475–485. doi: 10.1016/j.expneurol.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, Green R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. 2012;336:237–240. doi: 10.1126/science.1215691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285:12726–12734. doi: 10.1074/jbc.M109.086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueck A, Ziegler C, Eichner A, Berezikov E, Meister G. microRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 2012;40:9850–9862. doi: 10.1093/nar/gks705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Gerrish A, Russo G, Richards A, Moskvina V, Ivanov D, Harold D, Sims R, Abraham R, Hollingworth P, Chapman J, et al. The role of variation at AβPP, PSEN1, PSEN2, and MAPT in late onset Alzheimer's disease. J Alzheimers Dis. 2012;28:377–387. doi: 10.3233/JAD-2011-110824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glas J, Seiderer J, Bayrle C, Wetzke M, Fries C, Tillack C, Olszak T, Beigel F, Steib C, Friedrich M, et al. The role of osteopontin (OPN/SPP1) haplotypes in the susceptibility to Crohn's disease. PLoSONE. 2011;6:e29309. doi: 10.1371/journal.pone.0029309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J Galaxy Team. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey F, Tirabassi R, Meyers H, Wu G, McWeeney S, Hook L, Nelson JA. A viral microRNA down-regulates multiple cell cycle genes through mRNA 5′UTRs. PLoS Pathog. 2010;6:e1000967. doi: 10.1371/journal.ppat.1000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haecker I, Gay LA, Yang Y, Hu J, Morse AM, McIntyre LM, Renne R. Ago HITS-CLIP expands understanding of Kaposi's sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog. 2012;8:e1002884. doi: 10.1371/journal.ppat.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M. FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett. 1999;443:93–96. doi: 10.1016/s0014-5793(98)01696-2. [DOI] [PubMed] [Google Scholar]
- Hausser J, Syed AP, Bilen B, Zavolan M. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Res. 2013;23:604–615. doi: 10.1101/gr.139758.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SD, Lin FM, Wu WY, Liang C, Huang WC, Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al. miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011;39(Database issue):D163–D169. doi: 10.1093/nar/gkq1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HY, Guo S, Xi J, Yan Z, Fu N, Zhang X, Menzel C, Liang H, Yang H, Zhao M, et al. MicroRNA expression and regulation in human, chimpanzee, and macaque brains. PLoS Genet. 2011;7:e1002327. doi: 10.1371/journal.pgen.1002327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HY, Chien CH, Jen KH, Huang HD. RegRNA: an integrated web server for identifying regulatory RNA motifs and elements. Nucleic Acids Res. 2006;34(Web Server issue):W429–W434. doi: 10.1093/nar/gkl333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im HI, Kenny PJ. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012;35:325–334. doi: 10.1016/j.tins.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Item CB, Stöckler-Ipsiroglu S, Stromberger C, Mühl A, Alessandrì MG, Bianchi MC, Tosetti M, Fornai F, Cioni G. Arginine:glycine amidinotransferase deficiency: the third inborn error of creatine metabolism in humans. Am J Hum Genet. 2001;69:1127–1133. doi: 10.1086/323765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwama H. Coordinated networks of microRNAs and transcription factors with evolutionary perspectives. Adv Exp Med Biol. 2013;774:169–187. doi: 10.1007/978-94-007-5590-1_10. [DOI] [PubMed] [Google Scholar]
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- Kim S, Webster MJ. Correlation analysis between genome-wide expression profiles and cytoarchitectural abnormalities in the prefrontal cortex of psychiatric disorders. Mol Psychiatry. 2010;15:326–336. doi: 10.1038/mp.2008.99. [DOI] [PubMed] [Google Scholar]
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39(Database issue):D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34(Web Server issue):W451–W454. doi: 10.1093/nar/gkl243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon E, Wang W, Tsai LH. Validation of schizophrenia-associated genes CSMD1, C10orf26, CACNA1C and TCF4 as miR-137 targets. Mol Psychiatry. 2013;18:11–12. doi: 10.1038/mp.2011.170. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Young AG, Bhutkar A, Zheng GX, Bosson AD, Nielsen CB, Sharp PA. Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol. 2011;18:237–244. doi: 10.1038/nsmb.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiorano NA, Mallamaci A. Promotion of embryonic corticocerebral neuronogenesis by miR-124. Neural Dev. 2009;4:40. doi: 10.1186/1749-8104-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, Brown BD. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods. 2012;9:840–846. doi: 10.1038/nmeth.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niino M, Kikuchi S, Fukazawa T, Yabe I, Tashiro K. Genetic polymorphisms of osteopontin in association with multiple sclerosis in Japanese patients. J Neuroimmunol. 2003;136:125–129. doi: 10.1016/s0165-5728(03)00004-3. [DOI] [PubMed] [Google Scholar]
- Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, et al. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38(Database issue):D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug J, McKenna LB, Walton G, Hand N, Mukherjee S, Essuman K, Shi Z, Gao Y, Markley K, Nakagawa M, et al. Dynamic recruitment of microRNAs to their mRNA targets in the regenerating liver. BMC Genomics. 2013;14:264. doi: 10.1186/1471-2164-14-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao NY, Hu HY, Yan Z, Xu Y, Hu H, Menzel C, Li N, Chen W, Khaitovich P. Comprehensive survey of human brain microRNA by deep sequencing. BMC Genomics. 2010;11:409. doi: 10.1186/1471-2164-11-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel G, Obernosterer G, Fiore R, Oehmen M, Bicker S, Christensen M, Khudayberdiev S, Leuschner PF, Busch CJ, Kane C, et al. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat Cell Biol. 2009;11:705–716. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova L, Gräfe A, Seiler A, Schumacher S, Nitsch R, Wulczyn FG. Regulation of miRNA expression during neural cell specification. Eur J Neurosci. 2005;21:1469–1477. doi: 10.1111/j.1460-9568.2005.03978.x. [DOI] [PubMed] [Google Scholar]
- Sohn YB, Ki CS, Kim CH, Ko AR, Yook YJ, Lee SJ, Kim SJ, Park SW, Yeau S, Kwon EK, et al. Identification of 11 novel mutations in 49 Korean patients with mucopolysaccharidosis type II. Clin Genet. 2012;81:185–190. doi: 10.1111/j.1399-0004.2011.01641.x. [DOI] [PubMed] [Google Scholar]
- Somel M, Liu X, Khaitovich P. Human brain evolution: transcripts, metabolites and their regulators. Nat Rev Neurosci. 2013;14:112–127. doi: 10.1038/nrn3372. [DOI] [PubMed] [Google Scholar]
- Sotiriou S, Gibney G, Baxevanis AD, Nussbaum RL. A single nucleotide polymorphism in the 3′UTR of the SNCA gene encoding alpha-synuclein is a new potential susceptibility locus for Parkinson disease. Neurosci Lett. 2009;461:196–201. doi: 10.1016/j.neulet.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Chollier M, Herrmann C, Defrance M, Sand O, Thieffry D, van Helden J. RSAT peak-motifs: motif analysis in full-size ChIP-seq datasets. Nucleic Acids Res. 2012;40:e31. doi: 10.1093/nar/gkr1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Jaglin XH, Keays DA, Francis F, Chelly J, Cowan NJ. Disease-associated mutations in TUBA1A result in a spectrum of defects in the tubulin folding and heterodimer assembly pathway. Hum Mol Genet. 2010;19:3599–3613. doi: 10.1093/hmg/ddq276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren PJ, Bahrami-Samani E, Burns SC, Qiao M, Karginov FV, Hodges E, Hannon GJ, Sanford JR, Penalva LO, Smith AD. Site identification in high-throughput RNA-protein interaction data. Bioinformatics. 2012;28:3013–3020. doi: 10.1093/bioinformatics/bts569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalley HC, Papmeyer M, Romaniuk L, Sprooten E, Johnstone EC, Hall J, Lawrie SM, Evans KL, Blumberg HP, Sussmann JE, McIntosh AM. Impact of a microRNA MIR137 susceptibility variant on brain function in people at high genetic risk of schizophrenia or bipolar disorder. Neuropsychopharmacology. 2012;37:2720–2729. doi: 10.1038/npp.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Pan W, Song X, Liu Y, Shao X, Tang Y, Liang D, He D, Wang H, Liu W, et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat Med. 2012;18:1077–1086. doi: 10.1038/nm.2815. [DOI] [PubMed] [Google Scholar]
- Zisoulis DG, Lovci MT, Wilbert ML, Hutt KR, Liang TY, Pasquinelli AE, Yeo GW. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat Struct Mol Biol. 2010;17:173–179. doi: 10.1038/nsmb.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.