

Abstract
The dimeric copper-zinc superoxide dismutase Cu2Zn2SOD1 is a particularly interesting system for biological inorganic chemical studies because substitutions of the native Cu and/or Zn ions by a non-native metal ion cause minimal structural changes and result in high enzymatic activity for those derivatives with Cu remained in the Cu site. The pioneering NMR studies by Ivano Bertini and coworkers of the magnetically coupled derivative Cu2Co2SOD1 are of particular importance in this regard. In addition to Co2+, Ni2+ is also a versatile metal ion for substitution into Cu2Zn2SOD1, showing very little disturbance of the structure in Cu2Ni2SOD1 and a very good mimic of the native Cu ion in Ni2Zn2SOD1. The NMR studies presented here were inspired by and indebted to Professor Bertini's paramagnetic NMR pursuits of metalloproteins. In the current study, we report Ni2+ binding to apo-wild type SOD1 and a time-dependent Ni2+ ion migration from the Zn site to the Cu site and preparation and characterization of Ni2Ni2SOD1, which shows similar coordination properties to those of Cu2Cu2SOD1, namely a different anion binding property from the wild type and a possibly broken bridging His. Mutations in the human SOD1 gene can cause familial amyotrophic lateral sclerosis (ALS), and mutant SOD1 proteins with significantly altered metal binding behaviors are implicated in causing the disease. We therefore conclude by discussing the effects of the ALS mutations on the remarkable stabilities and metal-binding properties of wild type SOD1 proteins and the implications concerning the causes of SOD1-linked ALS.
Introduction
Spectroscopic studies of metal ion-substituted metalloproteins played an important role in the early development of biological inorganic chemistry. At that time, when three-dimensional protein structures were less frequently available, biological inorganic chemists demonstrated that various spectroscopies applied to metal ion-substituted metalloproteins could be powerful tools with which to infer structural properties and to probe structure-activity relationships. A wealth of information about many metalloproteins was made available by such studies.
Copper-zinc superoxide dismutase 1 (Cu2Zn2SOD, SOD1, structure 2 shown in Figure 1A) was one of the metalloproteins studied in great detail using such techniques. Of particular importance in this regard were the pioneering NMR studies by Ivano Bertini and coworkers of the derivative Cu2Co2SOD1 in which the Co2+ in the native Zn site is magnetically exchange coupled to Cu2+.1c, 3, 4 The studies presented here were inspired by and indebted to his paramagnetic NMR pursuits of metalloproteins.
Figure 1.

(A) The structure of bovine SOD1 (PDB 2SOD), showing the Cu (left) and Zn (right) sites and the catalytically important Arg141 (ball-and-stick structure in red); (B) Active site of bovine SOD1, showing the two metal ions M1 = Cu2+ and M2 = Zn2+ in the wild type. The ligands in human SOD1 are H46, H48, and H120 in the Cu site, H71, H80, and D83 in the Zn site, along with the bridging H63.
Starting soon after 1969, when McCord and Fridovich5 announced their discovery of the SOD activity of this protein, wild type Cu2Zn2SOD1 became a particularly interesting system for biological inorganic chemical studies because of its remarkably high thermal stability and versatility in accepting, with high degrees of selectivity, diverse metal ions in place of the native metal ions in the Cu and Zn sites.1b,c Studies of the derivatives in which Zn2+ was replaced by another divalent metal ion, M2+; i.e., Cu2M2SOD1 with M = Co, Ni, Cd, Hg, Cu (M2 site in Figure 1B); were found to be little changed structurally by the metal ion replacement and to retain full enzymatic activity. Studies of the derivatives in which Cu was replaced by another metal ion; i.e., M2Zn2SOD1 with M = Co, Ni, Ag, Cd, Zn (M1 site in Figure 1B); also suggested that non-native metal ion substitutions caused little if any rearrangement of the ligand geometries in the metal binding region of the protein.
Starting in the late 1980s, as the tools of molecular biology because available important studies of site-directed mutant SOD1 proteins added significantly to our understanding of this enzyme by demonstrating the importance of the proper placement of positively charged amino acid residues in guiding the superoxide anion to the active site channel and then to the site of its reactions with either Cu+ or Cu2+ in the active site, thereby achieving nearly diffusion controlled rates of reaction between enzyme and substrate.1c, 6
The picture that emerged from the metal substitution studies was that Cu2Zn2SOD1 possessed a high degree of framework stability and lack of flexibility in its metal binding sites. Nevertheless, despite the apparent rigidity of both metal binding sites, which enforced specific, often non-preferred geometries on the non-native metal ions they bound, some unusual kinetic properties were uncovered by these metal substitution reactions, with examples of slow metal binding reactions and migrations of metal ions from site to site.1b,c It was generally assumed that these characteristics evolved as a consequence of the evolutionary changes that optimized the abilities of SOD1 to function as a superoxide dismutase catalyst. One of the purposes of this report is to reevaluate that assumption in light of more recent studies.
The discovery that mutations in the human SOD1 gene could cause a familial form of the neurodegenerative disease amyotrophic lateral sclerosis (ALS)7 has enabled the preparation of large numbers of physiologically relevant mutant SOD1 proteins, which turn out to have a wide range of diverse properties. For many of these mutant SOD1 proteins, there is considerable evidence that the high degree of rigidity of the metal binding regions and framework stabilization of the entire protein structure have been significantly altered by the mutations.1a, 8 Nevertheless, many of these same ALS-mutant SOD1 proteins are found to retain wild type levels of enzymatic SOD activity.1a Comparisons of the properties of wild type and ALS-mutant SOD1 proteins that retain full SOD activity can now allow us to evaluate which of the unusual characteristics of the SOD1 protein are required for optimum SOD activity, and other explanations can be sought for those that are not.
One of the most versatile of the metal ions substituted into Cu2Zn2SOD1 is Ni2+, which can be introduced into either the Cu or Zn site. When it is bound to Zn site in Cu2Ni2SOD, similar to Co2+, it perturbs the structure very little relative to Cu2Zn2SOD1 while at the same time providing magnetic exchange coupling to the Cu2+ in the Cu site.9 When it is bound to the Cu site in Ni2Zn2SOD, it proves to be a remarkably good mimic of the coordination properties of Cu2+ in that site.10 In previous studies, the Ni derivatives were prepared by addition of Ni2+ to either Cu2E2SOD1 or E2Zn2SOD. In the current study, we report for the first time the direct reaction of Ni2+ with apo-SOD1 and our discovery of a novel time-dependent metal ion migration between metal binding sites. We also report preparation and characterization of Ni2Ni2SOD1 (dubbed “Ni4-SOD1” in this report), which shows similar coordination properties to those of Cu2Cu2SOD1, again supporting the remarkable similarities in the coordination properties of Cu2+ and Ni2+ when bound to either site of this protein. Finally we review the effects of the ALS mutations on the remarkable stabilities and metal ion substitution properties of wild type SOD1 proteins and discuss the implications concerning the causes of SOD1-linked ALS.
Results
The binding of Ni2+ to apo SOD1 was monitored using UV-Vis and NMR spectroscopies, and the resulting spectra were compared to those of the Ni2+- substituted SOD1 derivatives characterized previously,9,10 Addition of 2.0 equivalents of Ni2+ to apo SOD1 in phosphate buffer at pH 6.5 immediately gave a light pink derivative (dubbed here “new Ni2-SOD1”), with an electronic spectrum showing three distinct UV-Vis absorptions at 385 (62 cm–1 M–1 per Ni2+), 480 (48) and 755 (19) nm, and shoulders at 540 and 820 nm (Figure 2). Aside from the absorption at 385 nm, the spectrum is identical to those of Cu+2Ni2SOD1 and Ag+2Ni2SOD1 in which Ni2+ resides in the native Zn sites,9 except that the molar absorptivities of these bands are only about 70% those of the latter derivatives. The absorption at 385 nm, which is not found in the spectra of the M(I)2Ni2SOD1 derivatives, is similar to the absorption at 390 nm found in the derivative Ni2Zn2SOD1 with the Ni2+ in the native Cu site.10 Taken together, these observations suggest that the native Zn sites are about 70% occupied by Ni2+ in new Ni2-SOD1, with the remaining about 30% residing in the native copper sites.
Figure 2.
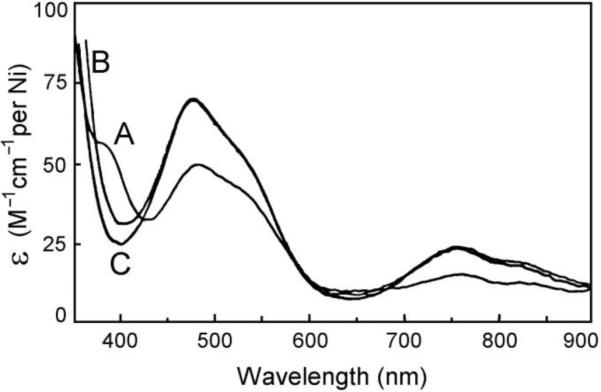
The electronic spectrum of freshly prepared Ni2-SOD1 (A) in 50 mM phosphate at pH 6.5 and room temperature referenced against deionized water. The spectrum is very similar to the spectra of Cu(I)2Ni2SOD1 (B) and Ag2Ni2SOD1 (C).
The binding of paramagnetic Ni2+ to apo SOD1 was studied also using 1H NMR via hyperfine-shifted features.11 The 1H NMR spectrum of new Ni2-SOD1 in D2O (Figure 3B) shows more than ten hyperfine-shifted signals in the range of 90 to –20 ppm. The overall features of the spectrum are quite similar to those of Ag2Ni2-SOD1 (Figure 3A).9 Hyperfine-shifted signals due to the solvent-exchangeable imidazole N–H protons of the coordinated His residues can be readily identified by their disappearance in D2O solutions. The 1H NMR spectra of apo SOD1 with 2.0 and 4.0 equivalents of Ni2+ bound (Figure 3B1 and 3B2, respectively) show at least six solvent exchangeable signals, B, E, H, a, b, and c (marked with dots in Figure 3B1). The spectra are similar except that the intensities of signals a–d are significantly larger relative to those of signals A–I in the four-nickel derivative (dubbed here “Ni4-SOD1“) as compared to those in new Ni2-SOD1.
Figure 3.

Isotropically shifted 1H NMR spectra (200 MHz, 23 °C) of (A) Ag2Ni2SOD, (B) Ni2-SOD1, (C) Ni2-SOD1 + 2Ag+, and (D) aged Ni2-SOD1 + 2Ag+ in D2O with 50 mM phosphate. The residual signals from the original species in (C) and (D) are marked with asterisks. The insets B1 and B2 are the downfield region of the isotropically shifted 1H NMR spectra (500 MHz, 25 °C) of apo-SOD1 in the presence of 2 equivalents (Ni2-SOD1, B1) and 4 equivalents (Ni4-SOD1, B2) of Ni2+ in H2O with 50 mM phosphate at pH 6.5. The signals a, b, B, E, and H are solvent exchangeable signals.
The spin-lattice relaxation times (T1) of the isotropically shifted signals b and d in new Ni2-SOD1, obtained at 90 MHz (9.5 and 9.1 ms), are significantly shorter than those of signals C, D, and G/H/I (21.1, 40.9, and 17.5 ms). NOE measurements were performed on the derivative new Ni2-SOD1 in D2O at 27 °C, and a negative NOE was detected at 11.43 pm when C was irradiated; and at 12.6, 0.88, and –0.77 ppm when G was irradiated. These NOEs are analogous to those observed in Ag2Ni2SOD1 upon irradiation of signals C and G to yield NOEs at 11.70 and 13.48, 0.45, and –2.58 ppm, respectively.
The electronic and NMR spectra of new Ni2-SOD1 were unchanged after storage of the solutions at 5 °C for a few weeks. After storage for several months, however, the samples (now dubbed “aged Ni2-SOD1”) exhibited completely different spectra from those of new Ni2-SOD1 (Figures 4 and 5). The electronic spectrum of aged Ni2-SOD1 had absorption bands at 385 and 650 nm (Figure 4B) and resembles that of Ni2Zn2SOD1 with Ni in the native Cu site (390 and 675 nm, Figure 4A).10 The 1H NMR spectrum of the aged Ni2-SOD1 species in H2O buffer shows only four isotropically shifted signals and a broad feature underneath these signals (Figure 5B), of which signals a, b, and c are solvent exchangeable. These NMR features resemble the signals a–d in the new Ni2-SOD1 and Ni4-SOD1 solutions (Figure 3) and the signals in the spectrum of Ni2Zn2SOD1 (Figure 5A).
Figure 4.

The electronic spectra of (A) Ni2Zn2SOD1 in 50-mM phosphate at pH 7.5 and (B) aged Ni2-SOD1 in 50 mM phosphate at pH 6.5 at room temperature.
Figure 5.
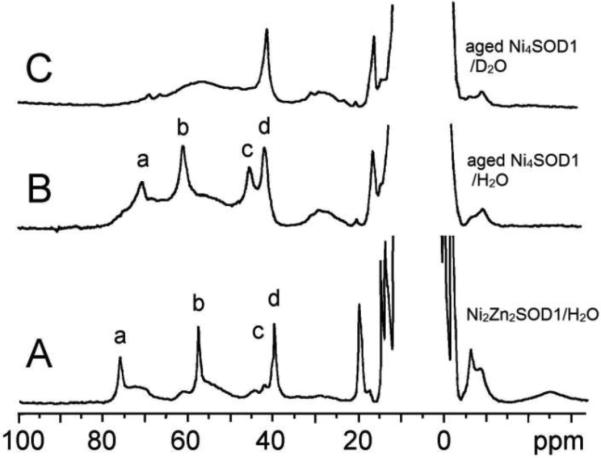
The isotropically shifted 1H NMR spectra of (A) Ni2Zn2SOD1 (200 MHz and 23 °C) in 50-mM phosphate buffer solution, (B) aged Ni2-SOD1 (500 MHz and 25 °C) in 50 mM phosphate at pH 6.5, and (C) aged Ni2-SOD1 in D2O under the same conditions as in (B). The signals a–c are solvent exchangeable.
Addition of 2.0 equivalents of Ni2+ to apo SOD1 in phosphate buffer at pH 7.5 gave the spectra characteristic of aged Ni2-SOD1 directly. When the pH was adjusted down to 6.5, the spectra characteristic of aged Ni2-SOD1 remained unchanged indefinitely.
When 2.0 equivalents Ag+ was added to new Ni2-SOD1, a new derivative formed rapidly with an NMR spectrum (Figure 3C) very similar to that of Ag2Ni2SOD1 (Figure 3A). The same result was obtained but at a much slower rate when 2.0 equivalents Ag+ was introduced into an aged Ni2-SOD1 solution (Figure 3D).
The relaxivities of water proton and phosphate P-31 of 50-mM phosphate buffer at pH 6.5 in the presence of apo SOD1 with increasing amounts of Ni2+ are reported in Figure 6. The relaxation rates are shown to be less affected by the paramagnetic Ni2+ when less than 2.0 equivalents was added, whereas a larger enhancement of the relaxations is revealed when more than 2.0 equivalents was added. The first 2.0 equivalents Ni2+ contributes about ~30% of the relaxivity of that at 4.0 equivalents with two breaking points are observed at 2.0 and 4.0 equivalents, consisting with stoichiometric binding of Ni2+ to the protein determined by electronic spectroscopy (Figure 2).
Figure 6.
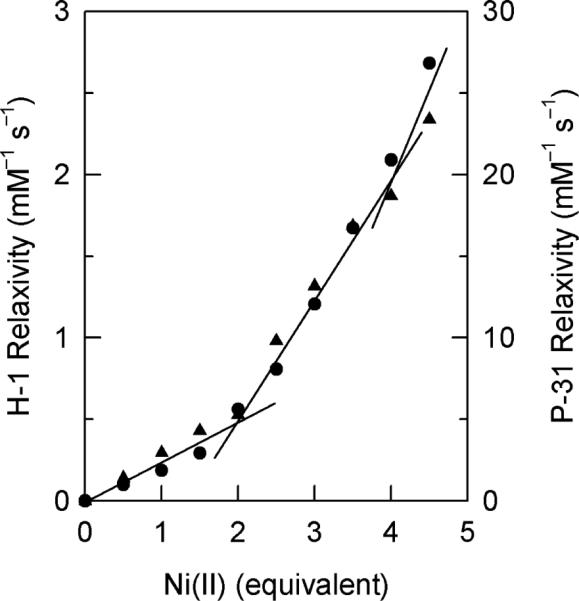
1H NMR relaxivity of water (200 MHz, sh=utrif ) and 31P NMR relaxivity of phosphate (202.5 MHz, •) of 50 mM phosphate buffer at pH 6.5 and 23 °C in the presence of apo SOD1 with different amounts of Ni2+.
The 1H NMR signals of new Ni2-SOD1 decreased gradually upon addition of increasing amounts of azide until they disappeared completely in the presence of 123 mM azide, with concomitant appearance of some new signals (Figure 7A and 7B). When the spectrum of new Ni2-SOD1 was acquired in D2O buffer solution in the presence of a saturating amount of azide, only one sharp signal was left along with some residual signals from Ni2-SOD1 (Figure 7C).
Figure 7.

The isotropically shifted 1H NMR spectra (200 MHz and 23 °C) of (A) Ni2-SOD1 and (B) Ni2-SOD1 in the presence of 123 mM N3– in H2O with 50 mM phosphate at pH 6.5 wherein the solvent exchangeable signals are marked; (C) Ni2- SOD1 in D2O with saturating amounts of azide after reaching equilibrium; and (D) new Ni2-SOD1 and (E) Ni2-SOD1 + 6 CN– in D2O with 50 mM phosphate at pD 6.5 (360 MHz and 23 °C) . The marked signals in (E) decrease in intensity with increasing amount of cyanide, and disappear after addition of excess amounts of cyanide.
Upon addition of up to six equivalents of cyanide, the signals in the spectrum of new Ni2-SOD1 in D2O, except for the signal at 41.7 ppm, decreased with concomitant appearance of three new signals (marked with arrows in Figure 7E). These three new signals disappeared in the presence of even larger amounts of cyanide.
To gain further information about cyanide binding to Ni2+, addition of cyanide to the simple Ni2+-His complex was monitored with NMR. The hyperfine-shifted signals of the complex were not significantly affected by the presence of only 1.0 equivalent of cyanide, but disappeared when the complex became diamagnetic upon addition of 2.0 equivalents of cyanide.
Discussion
Ni2+ binding to the zinc site
The electronic (Figure 2) and NMR (Figure 3A,B) spectral features of new Ni2-SOD1 and of Ni4-SOD1 can be separated into two sets, the first set is comprised of the electronic absorptions at 480, 755, 540(sh), and 820(sh) nm and the 1H NMR signals A–I. These spectral features strongly resemble those of M2Ni2SOD1 (M = Cu+ or Ag+, Figure 3A)9 and are thus consistent with Ni2+ bound to the native Zn site of apo SOD1. We conclude that Ni2+ initially binds rapidly and preferably to the native Zn site of apo SOD1 at pH 6.5 (I, Scheme 1). The relatively slow relaxivities of water and phosphate when less than 2.0 equivalents of Ni2+ had been added to apo SOD1 (Figure 6) also support the conclusion that Ni2+ binds initially mainly to the less solvent accessible native Zn site. Similar behavior was observed previously for E2Co2SOD1 in which paramagnetic Co2+ ion is bound to the native Zn site.12 In the case of Ni2+, however, unlike Co2+, the steady increase in the relaxivities with increasing amount of Ni2+, even at low ratios of Ni2+ to protein (Figure 6) indicates that Ni2+ is not completely isolated from the solvent, presumably due to partial Ni2+ binding to the solvent accessible native Cu site (see below; I–III).
Scheme 1.


The instantaneous formation of Ag2Ni2SOD1 upon addition of 2 equivalents of Ag+ to new Ni2-SOD1 (Figure 3C) also suggests that most Ni2+ binds initially to the native Zn site, leaving the native Cu site empty for rapid Ag+ binding (IV). However, the lower molar absorptivity of the electronic spectrum compared with that of M(I)2Ni2SOD1 (Figure 2) indicates that only 70% of the Ni2+ is bound to the zinc site in new Ni2-SOD1 (I). The observation of ~30% relaxivity relative to the total relaxation (Figure 6) is consistent with the binding of ~30% Ni2+ into the solvent accessible Cu site and 70% Ni2+ into the solvent inaccessible Zn site. The observation of some residual signals after the addition of 2.0 equivalents Ag+ to new Ni2-SOD1 (Figure 3B) also suggests that Ni2+ is not bound exclusively to the zinc site under these conditions.
Ni2+ binding to the copper site
The second set of electronic absorption at 385 nm and 1H NMR signals a–d of new Ni2-SOD1, aged Ni2-SOD1, and Ni4-SOD1 in Figures 2 and 3 strongly resemble those of Ni2Zn2SOD1 and Ni2Cd2SOD10 and thus can be assigned to Ni2+ binding to the native Cu site(II). The detection of three solvent exchangeable hyperfine-shifted 1H NMR signals, a–c, in the spectra of new Ni2-SOD1 and Ni4-SOD1 (Figure 3) and in aged Ni2-SOD1 (Figure 5) indicates that there are at least three coordinated histidyl side chains in this site, i.e., His-44, 46, and 118 in the native Cu site (Figure 1).
We conclude that Ni2+ is bound to the Cu site in aged Ni2-SOD1, with His-46 and His-118 coordinated to the metal through the Nε nitrogen2 (Figure 1B) to afford four ortho-like broad imidazole C–H protons and two sharp meta-like imidazole N–H protons and His-44 coordinated through the Nδ nitrogen to yield one sharp meta-like Cδ–H signal (signal d), one sharp solvent exchangeable meta-like N–H signal, and one broad ortho-like C–H signal. The absence of a fourth His ring N–H signal in spectra of aged Ni2-SOD1 may be due to fast solvent exchangeability of this N–H proton or lack of coordination of His61 to the Ni2+ in the Cu site in this species.
Although Ni2+ is bound to the Cu site in Ni4-SOD1 and aged Ni2-SOD1, distinct differences in the spectral features are observed as compared to Ni2Zn2SOD,10 in which Ni2+ is also bound to the Cu site. For example: (a) The signals a–d in Ni4-SOD1 and aged Ni2-SOD1 are broader than the corresponding signals in Ni2Zn2SOD1 under the same conditions. (b) The T1 values of the signals b (9.5 ms) and d (9.1 ms) are shorter than those in Ni2Zn2SOD1 (17.8 and 21.6 ms, respectively) at 90 MHz. Four- and five-coordinate Ni2+ complexes usually have faster electronic relaxation rates than those of six-coordinate Ni2+ complexes due to the existence of low lying triplet excited states in the former case, which provides an efficient mechanism for electron relaxation, thus affording longer nuclear relaxation times.11 The shorter T1 of the signals a–d suggests that the Ni2+ in the Cu site in Ni4-SOD1 and aged Ni2-SOD1 may have an octahedral-like geometry, which could afford a more open coordination sphere (II); whereas the Ni2+ in Ni2Zn2SOD1 or in Ni2Cd2SOD, as previously shown,10 is more like to adopt a five-coordinate geometry analogous to that in the native enzyme (VII). High spin Ni2+ has a great tendency to acquire a square planar or an octahedral-like geometry due to a significant gain in ligand field stabilization energy. This geometric preference was shown in a series of crystallographic studies on different metal-substituted derivatives of carboxypeptidase A, in which Ni was shown to acquire a distorted square pyramidal geometry.13
Another difference between the Ni2+ in the Cu site of Ni2Zn2SOD, Ni2Cd2SOD, and Ni4-SOD1 is that His-61 serves as a bridging ligand in the first two derivatives (VII),10 but not in the third (III), since each of the six-imidazole NH signals from the six coordinated His side chains are observed (Figure 3B). A bridging His-61 does not contain an imidazole NH proton, which would result in five solvent exchangeable signals.
We conclude that the coordination geometry of Ni2+ in the Cu site expands from a five-coordinate geometry to a six-coordinate geometry for the Ni2+ coordination sphere in the Cu site when His61 does not form an imidazolate bridge between Ni2+ and a metal ion in the Zn site. Thus Ni2+ in the Cu site appears to adopt an octahedral like geometry either when the Zn site is empty or contains Ni2+ (II, III; see below), whereas it adopts a more native-like five-coordinate geometry when either Zn or Cd is in the Zn site and the imidazolate bridge is intact (III).10 If His-61 does not serve as a ligand of Ni2+ in the Cu site of Ni4-SOD1 and aged Ni2-SOD1, an open coordination space in the copper site would be generated which may account for its different anion binding properties from those of Ni2Zn2SOD1 (see below).
Migration of Ni2+ions
The spectral features of new Ni2-SOD1 are consistent with Ni2+ binding in 70% of the native Zn sites and 30% of the native Cu sites (I), but those of aged Ni2-SOD1 are consistent with all of the Ni2+ bound to the native Cu site (II). We conclude that initial Ni2+ binding occurs rapidly and predominantly to the native zinc site, but that this initial product is kinetically rather than thermodynamically determined. This initial binding is followed by a slow rearrangement in which the Ni2+ migrates from the Zn site entirely to the Cu site (from I to become II).
Reversible metal migration after alteration of the solution conditions was previously observed for a couple of metal-substituted derivatives of Cu2Zn2SOD, including pH- and phosphate-dependent Co2+ migration from the Zn site to the empty Cu site in the derivative E2Co2SOD14 and pH-dependent migration of the Cu2+ back and forth between the Cu site and the empty Zn site in Cu2E2SOD.15 In those cases, the migration of the metal ions resulted in the formation of a binuclear center that was confirmed with optical, NMR, and EPR spectroscopies, whereas the Ni2+ in the Zn site of Ni2-SOD1 migrates completely into the Cu site in the current study. The different Ni2+ binding preferences toward the two sites in apo SOD1 are also reflected in the preparation of the several Ni2+ derivatives.9,10 For example, while Ni2+ binding to Ag2E2SOD1 and Cu2E2SOD1 to give the corresponding M2Ni2SOD1 derivative (VI) occurs within a day, the formation of the derivatives Ni2Zn2SOD1 and Ni2Co2SOD1 (VII) upon introduction of Ni2+ to the corresponding E2M’2SOD1 takes several days to reach completion.9,10 The fast Ni2+ binding to the tetrahedral native Zn site may be due to an electrostatic attraction of the metal ion to the negative Asp81 side chain (Figure 1B) combined with the relative flexibility of the Zn site relative to the Cu site in apo SOD1.16 The thermodynamic preference for Ni2+ binding to the Cu site can be attributed to the more favorable distorted square pyramidal coordination sphere with larger ligand field stabilization energy.
Anion binding properties
Addition of azide to Cu2Ni2SOD1 was previously shown to cause spectral shifts due to changes in the coordination site of Cu2+ only, with no evidence of any interaction of azide with Ni2+ when it was bound in the Zn site,9 Similarly, addition of azide to M2Ni2SOD, M = Cu+ or Ag+ was shown to leave the spectra of those derivatives unaffected.10 By contrast, addition of azide to new Ni2-SOD1, in which Ni2+ likewise resides initially in the Zn site, caused immediate changes in the 1H NMR spectra, with the spectral features characteristic of Ni2+ in the Zn site disappearing entirely and while those of Ni2+ in the Cu site were retained (Figure 7C; V, Scheme 1), consistent with rapid, azide-assisted redistribution of Ni2+ from the Zn site to the Cu site. Anion-assisted metal redistribution was observed previously for E2Co2SOD, in which Co2+ in the Zn site at elevated pH migrates into the Cu site in the presence of phosphate, yielding one-half amount of the protein subunits fully occupied by two Co ions and leaving the remaining subunits in the apo form.14 In the current case, addition of azide anion, without changing the pH, causes Ni2+ in the Zn site to migrate completely into the Cu site. In combination with the observation of the time-dependent Ni2+ migration from the Zn site to the Cu site as Ni2-SOD1 is aged, this observation further indicates that the binding of Ni2+ to the zinc site is labile and kinetically favorable; while binding of Ni2+ to the Cu site is thermodynamically preferred. This conclusion also helps to explain results of earlier experiments in which Ni2M'2SOD1 retained its bound Ni2+ in the Cu site throughout extensive ultra-filtration to remove the bound azide in the copper site, whereas the Ni2+ in the Zn site of Cu2Ni2SOD1 was partially removed by a similar process.9,10
While the hyperfine-shifted 1H NMR signals in Ni2Zn2SOD1 shift gradually toward the diamagnetic region upon azide titration and become diamagnetic upon introduction of 2.0 equivalents of cyanide,10 the isotropically shifted 1H NMR signals of new Ni2-SOD1 are not moved toward the diamagnetic region and not eliminated by 2.0 equivalents of cyanide (Figure 7). Since the isotropically shifted features of Ni2-SOD1 are clearly affected by anion binding, the Ni2+ in the Cu site must have an open coordination sphere for anion binding. The 1H NMR spectrum of Ni2-SOD1 upon azide binding is also different from that of the azide-bonded Ni2Zn2SOD, indicating that the mode of binding of azide is different for the two derivatives. Since His-61 is not a bridging ligand in Ni2-SOD1, azide presumably can bind to Ni2+ in the Cu site in place of His-61 and adopt a coordination geometry similar to that of Ni2+ in Ni2Zn2SOD.
The three sharper signals in the cyanide adduct of Ni2-SOD1 in D2O (marked in Figure 7E) are consistent with the CεH protons of the three Nδ-coordinated histidines in the Zn site (Figure 1), while the broader signal at 42 ppm is similar to signal d (Figures 3 and 5) in new and aged Ni2-SOD1 and in Ni4-SOD1, which is consistent with Ni2+ in the Cu site. This result indicates that Ni2+ in the Cu and Zn sites in Ni2-SOD1 retains their geometries and paramagnetism upon cyanide binding (V). By contrast, Ni2+ in Ni2Zn2SOD1 acquires a square planar geometry and becomes diamagnetic upon cyanide binding.10 Similar to the case of azide binding to Ni2-SOD1 described herein, the differences in cyanide binding may be due to the more open metal binding coordination sphere in Ni2-SOD1 without a bridging His-61. Moreover, the disappearance of the three marked signals (Figure 7E) upon addition of excess cyanide may mean either that cyanide assists the migration of Ni2+ from the Zn site to the Cu site as azide does or that Ni2+ is removed from the Zn site by CN–.
The simple complex Ni2+-His presumably has a quite flexible and open coordination sphere since His has only 3 coordinating groups. Cyanide binding to Ni2+-His does not yield a diamagnetic square-planar complex until more than 2 equivalents of cyanide is present, based on hyperfine-shifted 1H NMR and 13C NMR spectra.10 Likewise, Ni2-SOD1 remains paramagnetic upon binding of <6 equivalents of cyanide as demonstrated by the presence of hyperfine-shifted 1H NMR signals (Figure 7E), which may also be attributed to a more open coordination sphere (V). The effect of cyanide binding on the hyperfine-shifted 1H NMR signals due to the Ni2+ in the Zn site (marked signals in Figure 7E) suggests a possible interaction of this Ni2+ with anion; whereas the disappearance of the signals indicates either that this Ni2+ becomes diamagnetic or has migrated to the Cu site (V). By contrast, the Ni2+ site in Ni2Zn2SOD1 (VII) becomes diamagnetic upon binding two equivalents of cyanide, yielding a square planar Ni2+ coordination (VIII).10 The disappearance of the signals due to Ni2+ in the zinc site in the presence of excess amounts of cyanide and azide (Figure 7) also implies that Ni2+ binds to the Cu site with a higher affinity than to the Zn site.
Similarities between the coordination chemistry of Cu2+ and Ni2+ when bound to SOD1
Previous studies of Cu2Zn2SOD1 and Cu2E2SOD1 demonstrated that the four His ligands are coordinated to Cu2+ in both derivatives, but that the coordination geometry, which has a significant degree of rhombic character in the former, has relaxed to a more tetragonal geometry in the latter and that this difference became even more pronounced as the pH was lowered.17 These differences are thus analogous to those between Ni2Zn2SOD1 and Ni2E2SOD, as described above, with Ni2+ in the former derivative adopting a five-coordinate geometry (VII) while the geometry of the latter appears octahedral like (II).
Analogous properties of Ni2+ and Cu2+ in SOD1 are also evident from comparisons of the derivatives Ni4-SOD1 and Cu2Cu2SOD. Both metal ions show a thermodynamic preference for binding to the native Cu site relative to the Zn site18 and a tendency for the His-bridge to break (i.e., lack of the bridging His61 in the former and breakage of the bridging His upon lyophilization 19 and upon azide anion binding in the latter20). These two derivatives show that binding of a non-native metal to the Zn site renders significant influence on anion binding properties of the metal ion in the Cu site.
Reevaluation of Structure-Function Relationships in Cu2 Zn2SOD
The picture that emerges from this study and from the many other studies of metal ion substitution in wild type Cu2Zn2SOD1 proteins is that this protein has a remarkably high degree of stability, an unusual ability to bind tightly a wide range of metal ions with different geometric preferences, and several unusual examples of kinetically controlled metal ion migration reactions between different metal binding sites. Until recently, researchers in the SOD1 field, including us, have tended to assume that these unusual properties were related in some way to optimization of enzymatic activity. However, recent studies of the biophysical properties of ALS-mutant human SOD1 proteins demonstrate that this assumption about the reasons for the unusual metal binding properties of wild type SOD1 must be reevaluated.
Over 150 different mutations in the SOD1 gene have been linked to familial amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease characterized by motor neuron death. Biophysical characterization of ALS-mutant SOD1 proteins has revealed that the mutations can have very different effects on the properties of the protein, depending upon where the mutation occurs. Mutations that alter residues at or near the metal binding region of the proteins, the so-called “metal binding region mutations”, e.g. His46Arg or His48Gln (His46 and His48 are copper ligands in wild type SOD1), significantly alter both the metal ion binding properties and consequently the SOD1 activities of the mutant enzymes. But ALS-mutant SOD1 proteins in which the mutations occur in regions of the protein that are far removed from the metal binding region, the so-called “wild type-like mutations”, e.g., Ala4Val or Gly37Arg, can usually be isolated from in vivo expression systems properly metallated by copper and zinc ions, and these biologically metallated forms of the ALS-mutant SOD1 proteins have been found to differ very little from metallated wild type SOD1 with respect to SOD activities, structures, and stabilities.21
What about the stabilities and metal ion binding properties of the apoproteins derived from the ALS mutant SOD1 proteins? These apoproteins can be readily prepared from the as-isolated proteins by removal of any metal ions present. As expected, in the case of the metal binding region mutant proteins, because the metal binding regions have been significantly altered from those of the wild type protein, the proteins are frequently isolated with few if any bound metal ions, and the apoproteins show highly abnormal metal ion binding behaviors. Interestingly, the stabilities of the metal binding region ALS-mutant apoproteins are frequently very similar to that of the wild type apoprotein.22 In the case of the wild type-like mutant SOD1 proteins, which in their biologically metallated forms are very similar to the wild type protein, large effects of these ALS-causing mutations may only become evident after removal of the metal ions. Not only are the wild type-like ALS-mutant apoproteins often found to be severely destabilized relative to the wild type apoprotein, but their in vitro metal binding properties are often found to be significantly altered, particularly at the native Zn site.1a, 23 Moreover, wild type apo SOD1 under appropriate conditions of buffer and pH shows a high degree of specificity in binding Cu ions preferentially to the Cu sites and Zn ions preferentially to the Zn sites,24 whereas reconstitution of the ALS-mutant SOD1 apoproteins under the same conditions gives severely mismetallated proteins that do not spontaneously rearrange to the correctly metallated forms.25 For a few cases of wild type like ALS-mutant SOD1 proteins, e.g., Asp101Asn SOD1, the enzymatic activities and thermodynamic properties appear to be identical to those of wild type SOD1 even in the absence of metal ions. In these cases, it has been postulated that the disease-causing property may be linked to changes in the charges of the proteins,8d as when Asp is changed to Asn, but another possibility arises from the observation that a group of diverse ALS-mutant SOD1 proteins, including Asp101Asn SOD1, have impaired folding kinetics relative to wild type SOD1.26
The observation that the ALS-causing mutations in the wild type-like ALS-mutant SOD1 proteins have drastically altered the stabilities of their apoproteins and the kinetics and thermodynamics of their in vitro metal ion binding reactions is in sharp contrast to the observation that these same mutant proteins, when properly metallated by Cu and Zn in vivo, have SOD activities, structures, and stabilities very similar to those of wild type SOD1 indicates that the remarkable stabilities and in vitro metal binding abilities of the wild type SOD1 apoprotein are not required for optimization of the SOD activity.27 Instead it appears that these unique properties evolved to optimize the free-energy landscape of folding, disulfide bond formation, and efficient in vivo metallation of the immature disulfide-reduced nascent SOD1 apoprotein. Particularly relevant in this regard are recent demonstrations of the strong influences of metal ions on the kinetics of SOD1 folding.26,28 In addition, Oliveberg and his group have recently demonstrated that Zn2+ catalyzes the folding of apo SOD1 by transient docking at the native Cu site followed by a slower migration to the native Zn site after that latter site is formed.29 This type of metal ion migration from one metal binding site to another in SOD1 is reminiscent of the kinetically controlled metal ions migrations of non-native metal ions between metal binding sites such as are described in the current report.
Conclusion
Cu2Zn2SOD contains two metal binding sites of very different geometries, ligand types, labilities, and stabilities and thus can serve as a unique system for the study of metal-protein binding. Although Ni2+ usually has a high preference to form octahedral or square planar complexes, the rigidity of protein framework may force Ni2+ to adopt other geometries in proteins where it can have very different spectroscopic properties, with the result that this metal ion can serve as a particularly valuable probe of the metal binding sites to which it binds. We report here that Ni2+ can bind to either or both metal binding sites of apo SOD1 and that the binding properties and spectroscopic features of Ni2+ in the Cu and the Zn sites of apo SOD are similar to those of the Ni2+ in Ni2Zn2SOD and in M(I)2Ni2SOD, respectively.9,10 Comparison of anion binding to these several Ni2+-substituted derivatives reveals some significant differences, which has been attributed to the absence of a bridging His-61 in the Ni2+-only derivatives reported herein. Thus we present here another example of the unique in vitro non-native metal binding and migration behaviors of wild type SOD1 proteins. Although such behaviors were originally discovered long before SOD1 was linked to ALS, it is appropriate now to ask how these properties are altered by the ALS-causing mutations in SOD1. What is revealed by a comparison of these properties with those of the ALS-mutant SOD1 proteins is that, even when in cases where the properly metallated ALS-mutant SOD1 protein behaves just like wild type SOD1, the metal-binding characteristics are likely to be severely altered. While it is now generally agreed that SOD1-linked ALS is a disease of protein misfolding, it has been controversial whether or not alterations in metal ion binding properties of SOD1 play any role in the disease process. Based on the comparisons described above, we conclude that it is likely that the large alterations in metal binding behavior caused by the ALS-causing mutations are indeed relevant to disease causation.
Acknowledgments
This research was supported by grants from the National Institutes of Health (R01GM28222 and P01NS049134).
Footnotes
Dedicated to Professor Ivano Bertini for his inspiration, encouragement, and collaboration in our paramagnetic NMR pursuits of SOD1 and other metalloproteins
References
- 1.a Valentine JS, Doucette PA, Potter SZ. Ann Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]; b Valentine JS, Pantoliano MW. In: Copper Proteins, Metals in Biology Series. Spiro TG, editor. Vol. 3. Wiley; New York: 1981. pp. 292–358. Chapter 8. [Google Scholar]; c Bertini I, Mangani S, Viezzoli MS. Adv Inorg Chem. 1998;45:127–250. [Google Scholar]
- 2.a Tainer JA, Getzoff ED, Beem KM, Richardson JS, Richardson DC. J Mol Biol. 1982;160:181–217. doi: 10.1016/0022-2836(82)90174-7. [DOI] [PubMed] [Google Scholar]; b Perry JJP, Shin DS, Getzoff ED, Tainer JA. Biochim Biophys Acta. 2010;1804:245–262. doi: 10.1016/j.bbapap.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a Bertini I, Lanini G, Luchinat C, Messori L, Monnanni R, Scozzafava A. J Am Chem Soc. 1985;107:4391–4396. [Google Scholar]; b Bertini I, Luchinat C, Monnanni R. J Am Chem Soc. 1985;107:2178–2179. [Google Scholar]
- 4.a Bertini I, Banci L, Piccioli M, Luchinat C. Coord Chem Rev. 1990;100:67–103. [Google Scholar]; b Bertini I, Luchinat C, Piccioli M. Prog Nucl Magn Reson Spect. 1994;26:91–139. [Google Scholar]
- 5.McCord JM, Fridovich I. J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 6.a Beyer WF, Fridovich I, Mullenbach GT, Hallewell Robert. J Biol Chem. 1987;262:11182–11187. [PubMed] [Google Scholar]; b Banci L, Bertini I, Luchinat C, Hallewell RA. J Am Chem Soc. 1988;110:3629–3633. [Google Scholar]; c Getzoff ED, Cabelli DE, Fisher CL, Parge HE, Viezzoli MS, Banci L, Hallewell RA. Nature. 1992;358:347–351. doi: 10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- 7.a Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]; b Deng H-X, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung W-Y, Getzoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soriano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallewell RA, Pericak-Vance MA, Siddique T. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 8.a Lyons TJ, Gralla EB, Valentine JS. In: Metal Ions in Biological Systems. Sigel A, Sigel H, editors. Marcel Dekker; New York: 1999. pp. 125–177. [PubMed] [Google Scholar]; b Sheng YW, Chattopadhyay M, Whitelegge J, Valentine JS. Curr Top Med Chem. 2012;12:2560–2572. doi: 10.2174/1568026611212220010. [DOI] [PubMed] [Google Scholar]; c Chattopadhyay M, Valentine JS. Antiox Redox Sign. 2009;11:1603–1614. doi: 10.1089/ars.2009.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shaw BF, Valentine JS. Trends Biochem Sci. 2007;32:78–85. doi: 10.1016/j.tibs.2006.12.005. [DOI] [PubMed] [Google Scholar]; e Valentine JS, Hart PJ. Proc. Natl Acad Sci USA. 2003;100:3617–3622. doi: 10.1073/pnas.0730423100. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, Hart PJ. Nat Struc Biol. 2003;10:461–467. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]; g Potter SZ, Valentine JS. J Biol Inorg Chem. 2003;8:373–380. doi: 10.1007/s00775-003-0447-6. [DOI] [PubMed] [Google Scholar]
- 9.a Ming L-J, Valentine JS. J Am Chem Soc. 1987;109:4426–4428. [Google Scholar]; b Ming L-J, Banci L, Luchinat C, Bertini I, Valentine JS. Inorg Chem. 1988;27:4458–4463. [Google Scholar]; c Bertini I, Luchinat C, Ming L-J, Piccioli M, Sola M, Valentine JS. Inorg Chem. 1992;28:4433–4435. [Google Scholar]
- 10.Ming L-J, Valentine JS. J Am Chem Soc. 1990;112:6374–6383. [Google Scholar]
- 11.a La Mar GN, Horrocks WD Jr, Holm RH, editors. NMR of Paramagnetic Molecules. Academic; New York: 1973. [Google Scholar]; b Bertini I, Luchinat C. NMR of Paramagnetic Molecules in Biological Systems. Benjamin/Cummings; Menlo Park, CA.: 1986. [Google Scholar]; c Ming L-J. Nuclear Magnetic Resonance of Paramagnetic Metal Centers in Proteins and Synthetic Complexes. In: Que L Jr, editor. Physical Methods in Bioinorganic Chemistry. University Science Books; Sausalito, CA: 1999. pp. 375–464. Chapter 8. [Google Scholar]
- 12.Ming L-J, Banci L, Luchinat C, Bertini I, Valentine JS. Inorg Chem. 1988;27:728–733. [Google Scholar]
- 13.Rees DC, Howard JB, Chakrabarti P, Yeates T, Hsu BT, Hardman KD, Libscomb WN. In: Zinc Enzymes. Bertini I, Luchinat C, Maret W, Zeppezauer M, editors. Birkhauser; Boston: 1986. Chapter 10. [Google Scholar]
- 14.Ming L-J, Valentine JS. J Am Chem Soc. 1990;112:4256–4264. [Google Scholar]
- 15.Pantoliano MW, McDonnell PJ, Valentine JS. J Am Chem Soc. 1979;101:6454–6455. [Google Scholar]
- 16.Banci L, Bertini I, Cramaro F, Del Conte R, Viezzoli MS. Biochemistry. 2003;32:9543–9553. doi: 10.1021/bi034324m. [DOI] [PubMed] [Google Scholar]
- 17.Pantoliano MW, Valentine JS, Nafie LA. J Am Chem Soc. 1982;104:6310–6317. [Google Scholar]
- 18.Valentine JS, Pantoliano MW, McDonnell PJ, Burger AR, Lippard SJ. Proc Natl Acad Sci USA. 1979;76:4245–4249. doi: 10.1073/pnas.76.9.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strothkampt KG, Lippard SJ. J Am Chem Soc. 1982;104:852–853. [Google Scholar]
- 20.Strothkamp KG, Lippard SJ. 1981;20:7488–7493. doi: 10.1021/bi00529a025. [DOI] [PubMed] [Google Scholar]
- 21.a Rodriguez JA, Valentine JS, Eggers DK, Roe JA, Tiwari A, Brown RH, Hayward LJ. J Biol Chem. 2002;277:15932–15937. doi: 10.1074/jbc.M112088200. [DOI] [PubMed] [Google Scholar]; b Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown RH. J Biol Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez JA, Shaw BF, Durazo A, Sohn SH, Doucette PA, Nersissian AM, Faull KF, Eggers DK, Tiwari A, Hayward LJ, Valentine JS. Proc Natl Acad Sci USA. 2005;102:10516–10521. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a Nishida CR, Gralla EB, Valentine JS. Proc Natl Acad Sci USA. 1994;91:9906–9910. doi: 10.1073/pnas.91.21.9906. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]; c Lyons TJ, Liu H, Goto JJ, Nersissian A, Roe JA, Graden JA, Cafe C, Ellerby LM, Bredesen DE, Gralla EB, Valentine JS. Proc Natl Acad Sci USA. 1996;93:12240–12244. doi: 10.1073/pnas.93.22.12240. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lyons TJ, Nersissian A, Huang HJ, Yeom H, Nishida CR, Graden JA, Gralla EB, Valentine JS. J Biol Inorg Chem. 2000;5:189–203. doi: 10.1007/s007750050363. [DOI] [PubMed] [Google Scholar]
- 24.a Beem KM, Rich WE, Rajagopalan KV. J Biol Chem. 1974;249:7298–7305. [PubMed] [Google Scholar]; b Beem KM, Richardson DC, Rajagopalan KV. Biochemistry. 1977;16:1930–1936. doi: 10.1021/bi00628a027. [DOI] [PubMed] [Google Scholar]
- 25.Goto JJ, Zhu H, Sanchez RJ, Nersissian A, Gralla EB, Valentine JS, Cabelli DE. J. Biol Chem. 2000;275:1007–1014. doi: 10.1074/jbc.275.2.1007. [DOI] [PubMed] [Google Scholar]
- 26.Bruns CK, Kopito RR. EMBO J. 2007;26:855–866. doi: 10.1038/sj.emboj.7601528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das A, Plotkin SS. Proc. Natl Acad Sci USA. 2013;110:3871–3876. doi: 10.1073/pnas.1216597110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.a Assfalg M, Banci L, Bertini I, Turano P, Vasos PR. J Mol Biol. 2003;330:145–158. doi: 10.1016/s0022-2836(03)00533-3. [DOI] [PubMed] [Google Scholar]; b Svensson AKE, Bilsel O, Kondrashkina E, Zitzewitz JA, Matthews CR. J Mol Biol. 2006;364:1084–1102. doi: 10.1016/j.jmb.2006.09.005. [DOI] [PubMed] [Google Scholar]; c Kayatekin C, Zitzewitz JA, Matthews CR. J Mol Biol. 2008;384:540–555. doi: 10.1016/j.jmb.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Teilum K, Smith MH, Schulz E, Christensen LC, Solomentsev G, Oliveberg M, Akke M. Proc Natl Acad Sci USA. 2009;106:18273–18278. doi: 10.1073/pnas.0907387106. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Nordlund A, Leinartaite L, Saraboji K, Aisenbrey C, Grobner G, Zetterstrom P, Danielsson J, Logan DT, Oliveberg M. Proc Natl Acad Sci USA. 2009;106:9667–9672. doi: 10.1073/pnas.0812046106. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Sanchez-Ruiz JM. Biphys Chem. 2010;148:1–15. doi: 10.1016/j.bpc.2010.02.004. [DOI] [PubMed] [Google Scholar]; g Lynch SM, Boswell SA, Colon W. Biochemistry. 2004;43:16525–16531. doi: 10.1021/bi048831v. [DOI] [PubMed] [Google Scholar]; h Lynch SM, Colon W. Biochem Biophys Res Commun. 2006;340:457–461. doi: 10.1016/j.bbrc.2005.12.024. [DOI] [PubMed] [Google Scholar]; i Rumfeldt JAO, Lepock JR, Meiering EM. J Mol Biol. 2009;385:278–298. doi: 10.1016/j.jmb.2008.10.003. [DOI] [PubMed] [Google Scholar]; j Doyle CM, Rumfeldt JA, Broom HR, Broom A, Stathopulos PB, Vassall KA, Almey JJ, Meiering EM. Arch Biochem Biophys. 2013;531:44–64. doi: 10.1016/j.abb.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Leinartaite L, Saraboji K, Nordlund A, Logan DT, Oliveberg M. J Am Chem Soc. 2010;132:13495–13504. doi: 10.1021/ja1057136. [DOI] [PubMed] [Google Scholar]