

Abstract
Proteinuria is a major health-care problem that affects several hundred million people worldwide. Proteinuria is a cardinal sign and a prognostic marker of kidney disease, and also an independent risk factor for cardiovascular morbidity and mortality. Microalbuminuria is the earliest cue of renal complications of diabetes, obesity, and the metabolic syndrome. It can often progress to overt proteinuria that in 10–50% of patients is associated with the development of chronic kidney disease, ultimately requiring dialysis or transplantation. Therefore, reduction or prevention of proteinuria is highly desirable. Here we review recent novel insights into the pathogenesis and treatment of proteinuria, with a special emphasis on the emerging concept that proteinuria can result from enzymatic cleavage of essential regulators of podocyte actin dynamics by cytosolic cathepsin L (CatL), resulting in a motile podocyte phenotype. Finally, we describe signaling pathways controlling the podocyte actin cytoskeleton and motility and how these pathways can be manipulated for therapeutic benefit.
Keywords: albuminuria, aldosterone, cyclosporine, podocyte
TYPES OF PROTEINURIA
In a healthy person, urinary protein excretion is less than 150 mg/day and consists mainly of filtered plasma proteins (60%) and tubular Tamm-Horsfall proteins (40%). The main plasma protein in the urine is albumin, constituting about 20% of daily protein excretion. In healthy subjects, the daily amount of urinary albumin is less than 20 mg (13.8 mg/min).1 Proteinuria usually reflects an increase in glomerular permeability for albumin and other plasma macromolecules.1 A 24-h urine collection containing more than 150 mg of protein is considered pathological. There are several basic types of proteinuria; for example, glomerular, tubular, overflow, and exercise-induced (Table 1). Glomerular proteinuria is the most common form (around 90%). Low molecular weight molecules, such as β2-microglobulin, amino acids, and immunoglobulin light chains, have a molecular weight of about 25 kDa (albumin is 69 kDa). These smaller proteins are readily filtered across the glomerular filtration barrier and then fully reabsorbed by the proximal tubule. A variety of diseases that affect tubular and interstitial cell integrity impair the tubular reabsorption of these molecules.2 Some forms of glomerular diseases are also accompanied by tubular injury and tubular proteinuria.3 Pathological processes, such as multiple myeloma with a production of paraproteins, can result in increased excretion of low molecular weight proteins into the urine, a process termed overflow proteinuria. In this scenario, proteinuria results from the amount of filtered proteins exceeding the reabsorptive capacity of the proximal tubule.4 Dynamic exercise can also result in increased urinary excretion of proteins, predominantly of plasma origin, during and following physical exercise. A number of terms have been used to describe this phenomenon—post-exercise proteinuria, athletic pseudonephritis, exercise proteinuria, or exercise-induced proteinuria.5 Maximal rates of proteinuria occur approximately 30 min after exercise, with a resolution toward resting levels within 24–48 h. The magnitude of proteinuria varies from near normal to heavy (>7 g/day), with the greatest levels up to 100 times that of rest observed after high-intensity exercise, such as a marathon. It is noteworthy that post-exercise proteinuria is transient in nature and not associated with any particular renal disease, raising the intriguing possibility that at least some forms of proteinuria (e.g., post-exercise, post-prandial, infection-associated) may reflect a normal, physiological response of the human body (Table 1). In this review, we focus on the emerging concept that proteinuria can result from enzymatic cleavage of essential regulators of podocyte actin dynamics by cytosolic cathepsin L (CatL). Because of space limitations, genetic6 and other causes of proteinuria7 will not be discussed in detail.
Table 1.
Types of proteinuria
| Types | Characteristics |
|---|---|
| Glomerular | Most common form, up to 90% |
| Feature of chronic kidney disease | |
| Loss of albumin and higher molecular weight proteins | |
| Tubular | Low molecular weight proteins, such as β2-microglobulin |
| Overflow | Increased production, that is, light chains in multiple myeloma |
| Post-exercise | Transient benign |
| Can be up to 10 g/day | |
| Post-prandial | Transient physiological proteinuria |
| Possibly through insulin action in podocytes | |
| Infection-associated | Physiological response |
| Mediated by toll-receptors | |
| Possibly involved in clearing pathogens from the circulation |
CONSTITUENTS OF THE KIDNEY FILTRATION BARRIER
The kidney glomerulus (Figure 1a) is a highly specialized vascular bed that ensures the selective ultrafiltration of plasma so that the essential proteins are retained in the blood.8-10 The glomerular basement membrane (GBM) provides the primary structural support for the glomerular tuft. The basic unit of the glomerular tuft is a single capillary. The fenestrated glomerular endothelial cells and mesangial cells are located inside the GBM, whereas podocytes are attached to the outer aspect of the GBM (Figure 1a). The glomerular capillaries function as the filtration barrier.9,11 The filtration barrier is characterized by distinct charge and size selectivity, thereby ensuring that albumin and other plasma proteins are retained in the circulation.9,11 Proteinuria occurs when the permeability of the glomerular barrier is increased.11 Direct proof for this concept came from human monogenetic studies showing that mutations affecting podocyte proteins, including α-actinin-4,12 CD2AP,13 nephrin,14 PLCE1,15 podocin,16 and TRPC6,17,18 lead to renal disease owing to disruption of the filtration barrier and rearrangement of the podocyte actin cytoskeleton.6,7 Additional proteins regulating the podocyte actin cytoskeleton, such as Rho GDIa,19,20 podocalyxin,21 FAT1,22 Nck1/223,24 and synaptopodin,25,26 are also of critical importance for sustained function of the glomerular filtration barrier.7 Some years ago, using two-photon microscopy, Russo et al.27 claimed that the normal kidney filters nephrotic levels of albumin retrieved by proximal tubule cells. However, several recent studies convincingly refuted this alternative concept of a ‘leaky’ glomerular barrier.28-30 Using two-photon microscopy as well, Tanner28 and Peti-Peterdi30 independently and directly confirmed the classical view that the glomerular filter is the primary barrier for albumin and that the glomerular sieving coefficient for albumin is extremely low.28-30 The values determined by Tanner28 and Peti-Peterdi30 are somewhat higher than most values determined by kidney micropuncture, but they are still an order of magnitude less than the values reported by Russo et al.27 Tanner28 and Peti-Peterdi30 also demonstrated how the abnormally high glomerular sieving coefficient postulated by Russo et al. most likely resulted from technical limitations of the experimental approach.28,30 In conclusion, the classic view of a highly size- and charge-selective glomerular barrier still holds.28,31-33
Figure 1. Podocyte structure in health and disease.
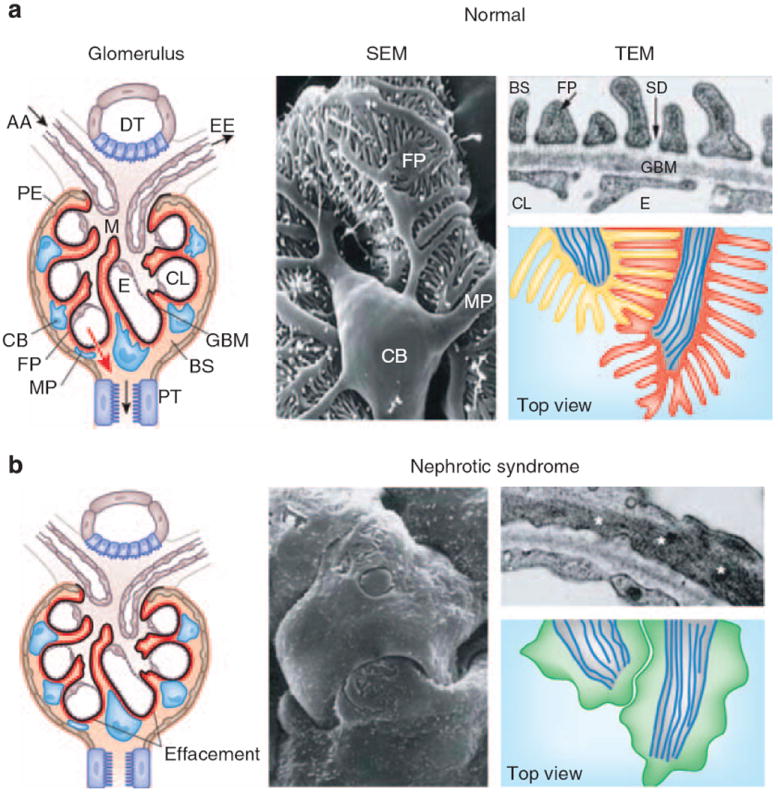
(a) Left: the glomerulus contains a capillary tuft that receives primary structural support from the glomerular basement membrane (GBM). Glomerular endothelial cells (E) embracing the capillary lumen (CL), and mesangial cells (M) are located on the blood side of the GBM, whereas podocyte foot processes (FP) cover the outer aspect of the GBM. Podocyte cell bodies (CB) and major processes (MT) are floating in primary urine in Bowman’s space (BS). Along its route from the blood space to the urine space (red arrow), the plasma ultrafiltrate passes sequentially through the fenestrated glomerular capillary endothelium, the GBM, and the filtration slits between neighboring podocyte foot processes with the interposed slit diaphragms. AA, afferent arteriole; DT, distal tubule; EE, efferent arteriole; PT, proximal tubule. Middle: scanning electron microscopy (SEM) illustrates the complexity of podocyte morphology. Looking from Bowman’s space, a cell body (CB) is seen that is linked to the capillaries by major processes (MP). Podocyte foot processes (FP) arise from MP and form the signature interdigitating pattern with FPs of neighboring podocytes, leaving in between the filtration slits. Only FPs are in direct contact with the GBM. Right: transmission electron microscopy (TEM) view (top) of the glomerular filtration barrier consisting of fenestrated endothelium (E), GBM, and podocyte FP with the interposed slit diaphragm (SD) covering the filtration slits. Top view (bottom) of normal FPs. In healthy podocytes, FPs regularly interdigitate. The highly organized parallel contractile actin bundles of interdigitating FPs from two adjacent podocytes are shown in red and yellow. Microtubules of MPs are shown in blue. (b) Left: in nephrotic syndrome with proteinuria, FPs lose their normal interdigitating pattern and show effacement instead. Middle: the loss of the normal cytoarchitecture and the development of meandering cell borders can be best viewed by SEM. Right: effaced FPs develop a continuous band of cytoplasm containing a dense band of short branched actin filaments (*). Top view (bottom) of effaced podocyte FPs showing the loss of the replacement of the regular interdigitating pattern by a simplified meandering cell border between effaced FPs. A continuous sheet of cytoplasm develops that is filled with reorganized, short, branched actin filaments (green).
PODOCYTES ARE PERICYTE-LIKE CELLS WITH AN ACTIN-BASED CONTRACTILE APPARATUS
Differentiated podocytes are mesenchymal-like cells that arise from epithelial precursors during renal development.34 Similar to pericytes, podocytes never embrace a capillary in total.10 Podocytes consist of three morphologically and functionally different segments: a cell body, major processes, and foot processes (FPs).10 From the cell body, major processes arise that split into FP (Figure 1a). FPs contain an actin-based cytoskeleton that is linked to the GBM.7 Podocyte FPs form a highly branched interdigitating network with FPs of neighboring podocytes connected by the slit diaphragm (SD) (Figure 1a). The SD is a modified adherens junction35 that covers the 30–50 nm wide filtration slits (Figure 1a), thereby establishing the final barrier to urinary protein loss.8 The extracellular portion of the SD is made up of rod-like units that are connected in the center to a linear bar, forming a zipper-like pattern, with pores about the same size as or smaller than albumin.10 The function of podocytes is largely based on their complex cell architecture, in particular on the maintenance of the normal FP structure with their highly ordered parallel contractile actin filament bundles7,36 (Figure 1a). FPs are functionally defined by three membrane domains: the apical membrane domain, the SD, and the basal membrane domain or sole plate that is associated with the GBM.7,37 All three domains are physically and functionally linked to the FP actin cytoskeleton. Proteins regulating the plasticity of the podocyte actin cytoskeleton are therefore of critical importance for sustained function of the glomerular filter.7
SIGNAL TRANSDUCTION AT THE SD REGULATES PODOCYTE ACTIN DYNAMICS
At the SD, multiple membrane proteins are present that are connected to the actin cytoskeleton through a variety of adaptor and effector proteins that may function as a key sensor and regulator of the permanent changes in FP shape and length.7 Changes in podocyte FP dynamics need to be precisely coordinated with FPs of neighboring podocytes, thereby preserving the integrity of the filtration barrier during FP movements, with functional coupling of opposing FPs and signaling cascades on both sides of the SD.7 Mutations in the NPHS1 gene encoding for the SD protein nephrin have been identified as the cause of congenital nephrotic syndrome of the Finnish type.14 It is noteworthy that nephrin is connected to the actin cytoskeleton through several adapter proteins and has a pivotal part in the regulation of podocyte actin dynamics.7,38 Among others (reviewed in detail by Faul et al.7), a recently discovered signaling pathway couples nephrin to the actin cytoskeleton through the adaptor protein Nck.23,24 After nephrin phosphorylation by Fyn,39 Nck binds to phospho-nephrin and Nck binds to N-WASP.23,24 This in turn leads to the activation of the Arp2/3 complex, a major regulator of actin dynamics.7,23,24,38
PODOCYTE DYSFUNCTION IS THE COMMON THREAD IN PROTEINURIC DISEASES
Podocytes can be injured in many forms of human and experimental glomerular disease, including minimal change disease (MCD), focal segmental glomerulosclerosis (FSGS), membranous glomerulopathy, diabetic nephropathy, and lupus nephritis.8,37 Characteristic changes are actin cytoskeleton reorganization of the involved FP, which typically leads to FP effacement and SD disruption12,40 (Figure 1b). Interference with any of the three FP domains changes the actin cytoskeleton from parallel contractile bundles36 into a dense network with FP effacement (reflected by the simplification of the FP structure and loss of the normal interdigitating pattern) (Figure 1b) and proteinuria.37 Causes of FP effacement and proteinuria include the following: (i) changes in SD structure or function,39,41,42 (ii) interference with the GBM or the podocyte–GBM interaction,43-49 (iii) dysfunction of the podocyte actin cytoskeleton,12,20,24,38,50-52 (iv) modulation of the negative surface charge of podocytes,53-55 and (v) activation of CatL-mediated proteolysis56-60 (see below). In addition, disturbances in the transcriptional regulation of podocyte function,61 modulation of vascular endothelial growth factor,62,63 transforming growth factor-β,64 adiponectin,65 notch,66,67 or aPKClamda68,69 signaling can also contribute to the pathogenesis of FP effacement and proteinuria. The early structural changes in podocyte morphology, such as FP effacement and SD disruption, are fully reversible (Figure 2). From a clinical point of view it is important to recognize that persistent podocyte injury harbors great risk to severe and progressive glomerular damage70,71 (Figure 2). The persistence of podocyte injury can cause podocyte cell death (through apoptosis or necrosis) or podocyte detachment from the GBM.72 Through a series of ensuing changes that have been reviewed in detail elsewhere,73 the loss of podocyte ultimately leads to glomerulosclerosis and end-stage renal failure.73 The contribution of proteinuria itself to the progression of end-stage renal failure is a matter of debate.74 Some studies have suggested that proteinuria causes podocyte75 or tubulointerstitial inflammation and progressive injury.76 On the other hand, severe experimental protein loss across the glomerular filter caused by repeated injection to rats with anti-nephrin antibody 5-1-6 did not lead to progressive renal failure.77 More importantly, patients with MCD or membranous glomerulopathy can present over years with nephrotic-range proteinuria without progressing to end-stage renal failure (Figure 2). Thus, the role of proteinuria in the progression of kidney failure probably depends on the type and the route of protein loss; that is, protein loss across the filtration barrier versus misdirected filtration into the periglomerular interstitium.73,78
Figure 2. Consequences of podocyte injury.
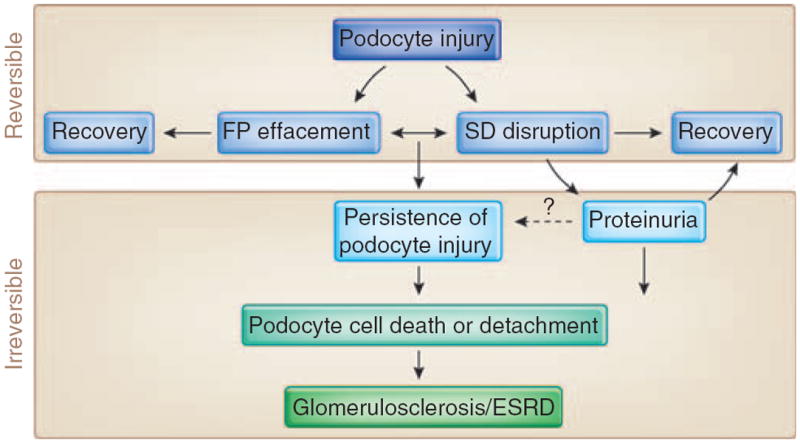
Podocytes can be injured in many human and experimental glomerular diseases, leading to structural changes, such as foot processes (FP) effacement and slit diaphragm (SD) disruption that are reversible.124 Persistence of podocyte injury can cause cell death or detachment of podocytes from the glomerular basement membrane (GBM).72 The resulting loss of podocyte will ultimately lead to irreversible glomerulosclerosis and end-stage renal failure (ESRD).73 The role of proteinuria in the progression of ESRD is a matter of debate. In some patients, nephrotic-range proteinuria can persist over years without progression to ESRD.
INCREASED FP MOTILITY AND THE ONSET OF PROTEINURIA
It has long been know that the podocyte FP actin cytoskeleton is highly dynamic, although the underlying mechanisms remained ill defined. Testaments to a dynamic FP regulation are experiments that used perfusion of rat kidneys with the polycation protamine sulfate (PS). This treatment causes FP effacement and SD disruption within 15 min79 and tyrosine phosphorylation of nephrin.24 The reperfusion with heparin for another 15 min can reverse PS-induced FP effacement80 and nephrin phosphorylation.24 PS-induced FP effacement involves the active reorganization of actin filaments,21,54 and disruption of the actin cytoskeleton by cytochalasin can prevent PS-induced FP effacement.81
THE ROLE OF CYTOSOLIC CatL AND B7-1 IN THE PATHOGENESIS OF PROTEINURIA
CatL is a member of the cathepsin family of cysteine proteases, which are involved primarily in protein breakdown in the lysosome.82 Two recent studies described a function for extralysosomal CatL in the nucleus.83,84 Regarding the role of CatL in kidney filter function, previous studies showed that a CatL inhibitor can reduce experimental proteinuria,85 but the underlying molecular mechanism remained unclear. In 2004, we reported that the onset of experimental proteinuria is associated with the induction of CatL expression and activity in podocytes.58 This study introduced the concept that the onset of proteinuria represents a migratory event in podocyte FP that is caused by the activation of CatL58 (Figure 3). Subsequently, we found that a cytoplasmic variant of CatL in podocytes is required for the development of proteinuria in mice through a mechanism that involves the cleavage of the large GTPase dynamin60 and synaptopodin57 (Figure 4). The clinical relevance of these findings was underscored by the observation that increased podocyte CatL expression was found in a variety of human proteinuric kidney diseases, including MCD, membranous glomerulopathy, FSGS, and diabetic nephropathy.60 Together these results support the notion that CatL-mediated proteolysis may have a key function in the development of many forms of proteinuria.60 The lipopolysaccharide (LPS) model of proteinuria also helped identifying an unanticipated role for costimulatory molecule B7-1 in podocytes as an inducible modifier of glomerular permselectivity86 (Figure 4). It is noteworthy that the expression of B7-1 in podocytes is correlated with the severity of human lupus nephritis, and mice lacking B7-1 are protected from LPS-induced proteinuria, suggesting a functional link between podocyte B7-1 expression and proteinuria.86 Functionally, LPS signaling through Toll-like receptor-4 reorganized the actin cytoskeleton of cultured podocytes.86 These findings also suggest a function for B7-1 in danger signaling by podocytes.86,87 LPS causes proteinuria by selectively targeting podocytes because podocyte-specific overexpression of CatL-resistant dynamin60 or synaptopodin57 (see below) is sufficient to safeguard against proteinuria. Key effectors of the LPS-induced proteinuria have been detected in podocytes in vivo in animals and in biopsies from patients with proteinuric kidneys diseases, including B7-1,86 CatL,60 and urokinase plasminogen activator receptor (uPAR).42 Although there is no report about cytosolic variant of cathepsin L in the proximal tubule, CatL is highly expressed in the tubular lysosomes. On the basis of published data, we do not think that there is experimental evidence or need for a role of tubular CatL in the pathogenesis of albuminuria, because the podocyte-specific interference with calcineurin–CatL signaling is sufficient to cause induction of albuminuria, or protection thereof, respectively.42,57,58,60
Figure 3. Induction of cathepsin L in podocytes precedes FP effacement and proteinuria.
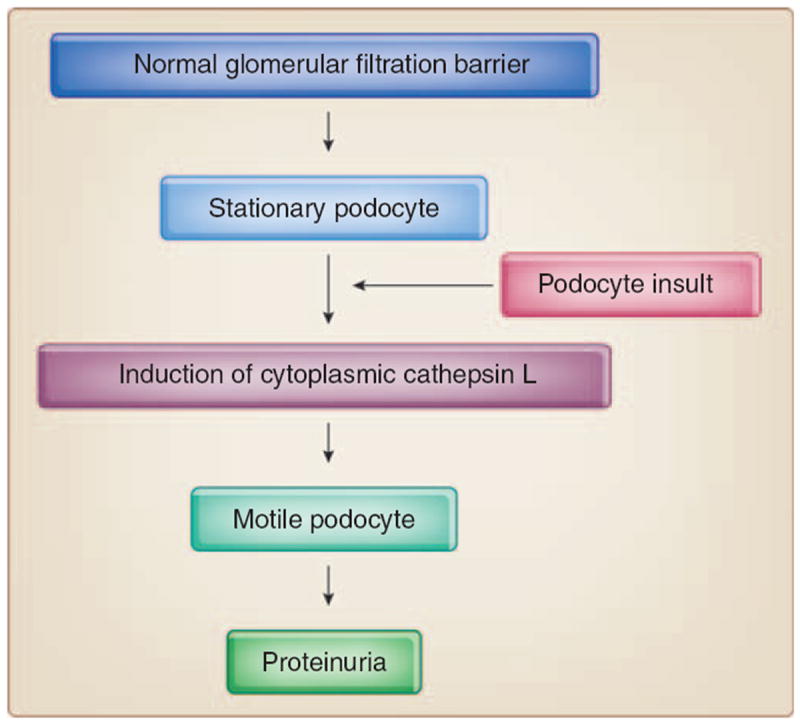
Upon an insult, stationary podocytes upregulate cytoplasm cytosolic cathepsin L expression and activity and develop motile podocyte foot processes (FPs). This migratory response leads to FP effacement, slit diaphragm remodeling, and proteinuria.58
Figure 4. Integrative model for the regulation of podocyte actin dynamics in health and disease.
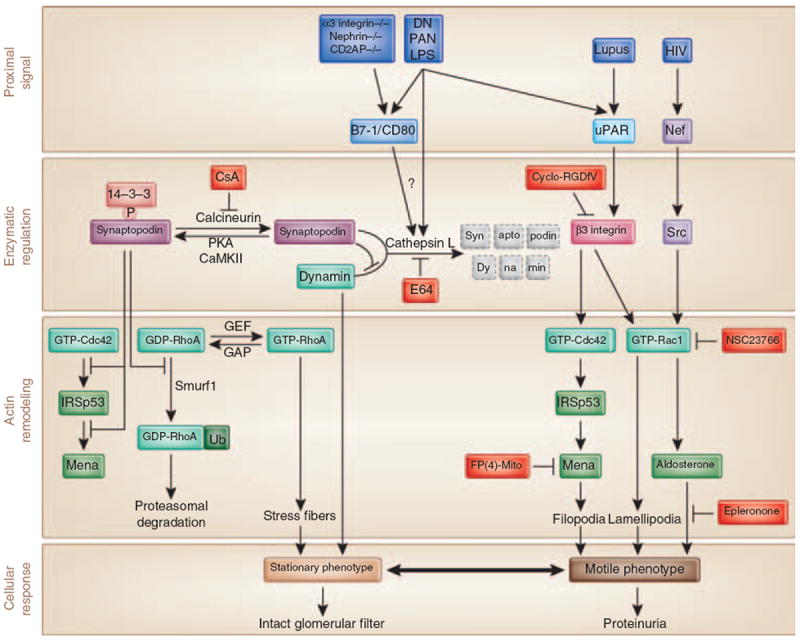
Induction of cytoplasmic cytosolic cathepsin L (CatL) enzyme is a common downstream effector in many glomerular diseases. Lipopolysaccharide (LPS) or various other proximal signals induce the expression of B7-186 and CatL58,60 in podocytes, which cause proteinuria through the increased degradation of synaptopodin57 and dynamin.60 Phosphorylation of synaptopodin by PKA or CaMKII promotes 14-3-3 binding, which protects synaptopodin against CatL-mediated cleavage, thereby stabilizing synaptopodin steady-state levels.57 Synaptopodin suppresses IRSp53–Mena-mediated filopodia by blocking the binding of Cdc42 and Mena to IRSp5396 and induces stress fibers by competitive blocking the Smurf-1-mediated ubiquitination of RhoA.26 Synaptopodin also prevents the CatL-mediated degradation of dynamin.57 Dephosphorylation of synaptopodin by calcineurin abrogates the interaction with 14-3-3. This renders the CatL cleavage sites of synaptopodin accessible and promotes the degradation of synaptopodin.57 LPS or other proximal signals can also activate Cdc42 and Rac1 through uPAR–β3-integrin signaling,42 through the loss of synaptopodin-mediated inhibition of Cdc42 signaling,96 or through Nef–Src-mediated activation of Rac1.52 As a consequence, the podocyte actin cytoskeleton shifts from a stationary to a motile phenotype, thereby causing foot process effacement and proteinuria. CsA and the CatL inhibitor E64 safeguard against proteinuria by stabilizing synaptopodin and dynamin steady-state protein levels in podocytes, FP(4)-Mito by blocking Cdcd42–IRSp53–Mena signaling,96 cycloRGDfV by blocking uPAR–β3-intergrin signaling,42 NSC23766 by blocking Rac1, and epleronone by blocking aldosterone signaling.20
ACTIVATION OF PROMIGRATORY Cdc42 AND Rac1 IN PODOCYTES CAUSES FP EFFACEMENT AND PROTEINURIA
The Rho family of small GTPases (RhoA, Rac1, and Cdc42) controls signal-transduction pathways that influence many aspects of cell behavior, including actin dynamics.88-90 At the leading edge, Rac1 and Cdc42 promote cell motility through the formation of lamellipodia and filopodia, respectively. On the contrary, RhoA promotes the formation of contractile actin–myosin containing stress fibers in the cell body and at the rear.88,89,91 We will now discuss how the onset of proteinuria can be caused by the activation of Cdc42 and Rac1 signaling and concomitant decrease of RhoA signaling in podocytes.7 uPAR is a glycosylphosphatidylinositol-anchored protein that has been shown to be a proteinase receptor for urokinase, but has also been involved in non-proteolytic pathways, mainly through its ability to form signaling complexes with other transmembrane proteins, such as integrins, caveolin, and G-protein-coupled receptors.92 uPAR is induced and activated in podocytes in response to proteinuric stimuli, such as LPS or PAN (Figure 4), leading to increased podocyte motility in vitro and FP effacement and proteinuria in vivo.42 Lipid raft-associated uPAR forms a complex with β3-integrin, thereby causing the activation of β3-integrin. This in turn promotes Cdc42 and Rac1 signaling, thereby causing podocyte FP effacement and proteinuria.42 Proteinuria caused by uPAR–β3-integrin signaling can be prevented and reduced by cycloRGDfV,42 a selective inhibitor of αvβ3-integrin.93 The activation of Rac1 signaling in podocytes also contributes to proteinuria in human immunodeficiency virus-associated nephropathy52 and RhoGDIalpha knockout mice.20 Rac1-induced proteinuria can be blocked by the inhibition of Rac1 or blocking its downstream target aldosterone20 (Figure 4). Thus, the activation of promigratory Cdc42 and Rac1 signaling in podocytes is a widespread cause of FP effacement and proteinuria.
SYNAPTOPODIN IS A KEY STABILIZER OF THE PODOCYTE ACTIN CYTOSKELETON
The actin-binding protein, synaptopodin, which is highly expressed in podocytes,94 exists in three isoforms: neuronal Synpo-short, renal Synpo-long, and Synpo-T.25 Synaptopodin mutant mice lacking Synpo-short and Synpo-long upregulate Synpo-T protein expression in podocytes, thereby rescuing kidney filter function during development.25 They do, however, display impaired recovery from PS-induced FP effacement and LPS-induced proteinuria.25 Bigenic heterozygosity for synaptopodin and CD2AP results in proteinuria and FSGS-like glomerular damage,95 thereby underscoring the importance of synaptopodin and CD2AP for sustained kidney filter function. Mechanistically, synaptopodin induces stress fibers by stabilizing RhoA26 and suppresses filopodia by disrupting Cdc42–IRSp53–Mena signaling complexes96 (Figure 4). The Mena inhibitor, FP(4)-Mito, suppresses aberrant filopodia formation in synaptopodin knockdown podocytes in vitro, and when delivered into mice protects against LPS-induced proteinuria.96 Thus, synaptopodin is an antagonistic regulator of RhoA and Cdc42 signaling that stabilizes the kidney filter by blocking the reorganization of the podocyte actin cytoskeleton into a migratory phenotype (Figure 4).
CURRENT CLINICAL THERAPIES TARGETING PROTEINURIA
When considering antiproteinuric therapy, one has to distinguish between acute new-onset proteinuria and proteinuria in the setting of chronic kidney disease. It is widely accepted that proteinuria reduction is an imperative therapeutic goal in chronic proteinuric kidney disease.97 On the basis of large randomized clinical trials, angiotensin-converting enzyme inhibitor (ACEI) and angiotensin II (Ang II) type 1-receptor blockers (ARB) therapy have developed into the most important antiproteinuric and renoprotective interventions.97 Many of the beneficial effects of these types of agents toward proteinuria can be explained by the blood pressure-lowering effect that leads also to a lowered glomerular filtration pressure. However, there are also numerous non-blood pressure-related effects of ACEI and ARB that can act directly on podocyte through targeting a local renin–angiotensin system.97,98 The inhibition of the renin–angiotensin system is also associated with restoration of nephrin expression in both experimental and human diabetic nephropathy.99 The expression of nephrin was retained at the SD when rats with experimental membranous nephropathy were treated with an ACE inhibitor or with an Ang II receptor antagonist. The resulting well-known effect of these drugs—limiting proteinuria in animal models of progressive nephropathy—therefore may depend on the capacity of Ang II blockers to preserve podocyte function.100 In a rat model of spontaneous proteinuria, the development of proteinuria is associated with the relocation of zonula occludens-1 from the SD to the podocyte cell body.101 The ACE inhibitor, lisinopril, not only prevented proteinuria but also redistributed zonula occludens-1 to the SD.101 Most direct support for a role of Ang signaling in podocytes as a cause of proteinuria and FSGS comes from a study with transgenic rats overexpressing an AT1 receptor selectively in podocytes.102 Hence, the renoprotective effects of ACEI and ARBs are associated with the preservation of podocyte function. Other examples where ACEI and ARB act benefitial on the glomerular filtration barrier are the modulation of podocyte function through modification of extracellular matrix, proinflammatory cytokine production, and blockade of the deleterious actions of radical oxygen species.97 There are numerous other interventions (Table 2) that have been shown to be antiproteinuric and renoprotective.76 Unfortunately, testing each of these antiproteinuric therapies in randomized clinical trials is presently not feasible. Interestingly, the inhibition of aldosterone by specific inhibitors can reduce proteinuria and Rac1 signaling independent of blood pressure, as shown in normotensive RhoGDIα knockout mice20 (Figure 4).
Table 2.
Antiproteinuric strategies (modified from Wilmer et al.76)
| Intervention | Goal/comment |
|---|---|
| Level 1 | |
| Control blood pressure (BP) | The greater the proteinuria, the greater the benefit of lowering BP |
| Angiotensin-converting enzyme inhibitor (ACEI) therapy | Use ACEI even if normotensive |
| Angiotensin II type 1-receptor blocker (ARB) therapy | Proven antiproteinuric and renoprotective therapy |
| Combination ACEI and ARB therapies | Adding ARB to maximum ACEI appears to reduce proteinuria further |
| Avoid dihydropyridine calcium-channel blockers (DHCCBs) unless needed for BP control | DHCCBs are excellent antihypertensive agents, but they are not antiproteinuric and may promote kidney disease progression; ARB therapy may mitigate these effects |
| β-Blocker therapy | β-Blocker therapy is antiproteinuric compared with DHCCB therapy |
| Control protein intake | Goal is 0.7–0.8 g/kg/day. Effect on proteinuria is nearly the same as that of the low BP goal |
| Level 2 | |
| Restrict NaCl intake | Goal is 80–120 mmol/day (≈2.0 to 3.0 g Na). |
| Control fluid intake | Goal is urine volume <2.0 l/day unless higher fluid intake is needed for specific reasons |
| Nondihydropyridine calcium-channel blocker therapy | This CCB class is considered antiproteinuric |
| Control blood lipids | Statins are considered antiproteinuric and renoprotective |
| Aldosterone antagonist therapy | Spironolactone is antiproteinuric in humans and in animal models independent of BP control |
| Smoking cessation | Cigarette smoking in humans increases proteinuria/albuminuria and is associated with faster kidney disease progression |
| Avoid estrogen/progestin replacement therapy in postmenopausal women with kidney disease | Estrogens may have renoprotective effects that explain slower progression of kidney disease in premenopausal women compared with men of the same age but may have adverse effects in postmenopausal women |
| Supine/recumbent posture when feasible. | Nephrotic-range proteinuria decreases by as much as 50% during recumbency. |
| Avoid severe exertion | Severe exercise may increase proteinuria substantially |
| Reduce obesity | Obesity apparently causes glomerulomegaly and proteinuria |
| Level 3 | |
| Decrease elevated homocysteine | Hyperhomocystinuria is associated with microalbuminuria and increased cardiovascular risks. |
| Folic acid, B6, and B12 may lower homocysteine levels | |
| Antioxidant therapies | Antioxidant therapies of several types reduce proteinuria in both experimental models and in patients with diabetic nephropathy |
| Sodium bicarbonate therapy to correct metabolic acidosis | NaHCO3 therapy is not antiproteinuric; however, it blocks complement activation in the tubular compartment and, therefore, may block tubular injury caused by proteinuria |
| NSAID therapy in severe untreatable nephrotic syndrome | NSAIDs (both COX 2 and nonspecific COX inhibitors) are antiproteinuric but are also nephrotoxic. Thus, NSAID use should be reserved for severe untreatable nephrotic syndrome |
| Other therapies based on animal studies | Avoid excessive caffeine, iron overload. Allopurinol, pentoxifylline, mycophenolate therapy |
In contrast to chronic forms of proteinuria, treatment of new-onset proteinuria often includes immunomodulatory drugs, such as glucocorticoids, cyclosporine A (CsA), FK506, mycophenolate mofetil, and Rituximab.103 What is the rationale for using immune-suppressing agents in treating acute proteinuria? Historically, some rationale derived from the ‘soluble mediator hypothesis’ that MCD is a disorder of the immune system in which release of T-cell-derived circulating factor(s) causes proteinuria.104 Despite the lack of convincing experimental support, this hypothesis has remained the dominant paradigm for the treatment of nephrotic syndrome, and the antiproteinuric effect of glucocorticoid therapy was attributed to its T-cell-repressing action.103 However, recent studies have shown that glucocorticoid receptors are present on podocytes and translocate to the podocyte nucleus upon dexamethasone treatment.105 Glucocorticoids can also increase the stability of actin filaments, increase actin polymerization, and activate cytoskeleton-associated kinases in podocytes.105 These findings suggest that the antiproteinuric effect of glucocorticoids is, at least in part, mediated by a direct effect on the podocyte actin cytoskeleton independent of immunosuppression.
THE ANTIPROTEINURIC EFFECT OF CsA RESULT FROM THE STABILIZATION OF SYNAPTOPODIN IN PODOCYTES
Calcineurin is a ubiquitously expressed serine/threonine phosphatase.106 The best-characterized function of calcineurin is the regulation of nuclear factor of activated T cells (NFAT) signaling. The immunosuppressive action of the calcineurin inhibitor, CsA, stems from the inhibition of NFAT signaling in T cells.107 CsA can also induce a remission of proteinuria caused by diseases, including MCD and FSGS.103 T-cell dysfunction is associated with some forms of proteinuria, including a subset of MCD in children. Therefore, it is has been assumed that the antiproteinuric effect of CsA results from the inhibition of NFAT signaling in T cells.103 CsA can also reduce proteinuria in human108 and experimental109 Alport’s syndrome, a non-immunological disease, raising doubts about the above hypothesis. Moreover, LPS-induced proteinuria can develop independent of T cells,86 and mice lacking synaptopodin display impaired recovery from LPS-induced proteinuria.25 Therefore, we reasoned that podocytes are a direct target of CsA, independent of NFAT inhibition in T cells and found that CsA blocks the calcineurin-mediated dephosphorylation of synaptopodin, thereby preserving the phosphorylation-dependent synaptopodin-14-3-3β interaction (Figure 4).57 Preservation of this interaction, in turn, protects synaptopodin from CatL-mediated degradation and preserves a stable filtration barrier (Figure 4). The inducible expression of CatL-resistant synaptopodin in podocytes can prevent not only the LPS-induced degradation of synaptopodin and proteinuria but also the degradation of the other CatL target dynamin (Figure 4) and zonula occludens-1, a vital component of the SD.101 Moreover, the inducible expression of dominant active calcineurin in podocytes is sufficient to cause the degradation of synaptopodin, thereby inducing proteinuria.57 These data unveiled a calcineurin signaling pathway, which is operative in podocytes and contributes to the maintenance of kidney filter function (Figure 4). In contrast to most other calcineurin-controlled signaling events,106,107,110,111 the antiproteinuric effect of CsA does not result from the inhibition of NFAT signaling. Altogether, the antiproteinuric effect of CsA results, at least in part, from the maintenance of synaptopodin protein levels in podocytes, which safeguard against proteinuria by maintaining a stationary podocyte phenotype (Figure 4).57
OUTLOOK
Intense research efforts aimed at deciphering the pathogenesis of proteinuria are starting to pay off and novel therapies are in sight. A CatL-mediated enzymatic disease process in podocytes suits well to explain the common pathological feature of podocyte FP effacement that is observed in most cases of proteinuria (Figure 4). The growing list of targets that are cleaved by cytosolic CatL harbors potential for renal protection by stabilizing these substrates. Motility of FP is directly affected by CatL-mediated proteolysis or by signaling pathways that change the migratory behavior of podocytes into a motile phenotype (Figure 4); for example, by activating Cdc4242,96 and Rac1 signaling.20,42,52 The relevance of these changes is underscored by the findings that inhibition of Cdc42 signaling with FP(4)-Mito96 or Rac1 signaling with NSC23766 or epleronone20 can prevent proteinuria. The antiangiogenic and antitumor agent, cycloRGDfV, a selective inhibitor of αvβ3 integrin,93 is currently being tested in clinical trials against a number of human cancers, including recurrent glioblastoma multiforme,112 pediatric brain tumors,113 and prostate cancer.114 CycloRDGfV also holds substantial promise for the development of novel antiproteinuric therapeutics by blocking activated uPAR–β3-integrin signaling and podocyte FP hypermotility.42 The findings that the antiproteinuric effect of CsA results from a direct effect on podocytes suggests that nephrologists have been using the right approach for the wrong reason, and raises the exciting possibility that other proteinuric diseases, such as diabetic nephropathy, may also be treated by agents that stabilize the podocyte cytoskeleton.115 Clearly, this notion will be of more practical value if the antiproteinuric effect of CsA can be achieved by lower, non-immunosuppressive doses or if selective agents can be developed that avoid the serious side effects of long-term CsA treatment.116 Looking ahead, it will be important to understand the crosstalk between the calcineurin–CatL pathways and other proteins and emerging signaling pathways that govern podocyte actin dynamics and cell migration,7 including Kibra,117 ROS, Ang II,102,118 transforming growth factor-β,119 TRPC6,120,121 myosins,122,123 or nephrin–nck signaling.23,24,38 Integrating these other mechanism with the calcineurin–Catl pathway and identifying the calcineurin–Catl pathway-dependent versus independent pathways, leading to proteinuria and/or progressive kidney disease, are necessary work lying ahead. In conclusion, based on the consideration discussed above, novel calcineurin (synaptopodin) and CatL substrates (dynamin, synaptopodin), as well as the inhibition of cytosolic CatL, itself provide promising starting points for the development of selective, antiproteinuric, and podocyte-protective drugs.
Acknowledgments
This work was supported in part by the US National Institutes of Health Grants DK057683, DK062472 (PM), and DK073495 (JR).
Footnotes
DISCLOSURE
The authors declared no competing interests.
References
- 1.Orth SR, Ritz E. The nephrotic syndrome. N Engl J Med. 1998;338:1202–1211. doi: 10.1056/NEJM199804233381707. [DOI] [PubMed] [Google Scholar]
- 2.Bergon E, Granados R, Fernandez-Segoviano P, et al. Classification of renal proteinuria: a simple algorithm. Clin Chem Lab Med. 2002;40:1143–1150. doi: 10.1515/CCLM.2002.201. [DOI] [PubMed] [Google Scholar]
- 3.de Zeeuw D, Lewis EJ, Remuzzi G, et al. Renoprotective effects of reninangiotensin-system inhibitors. Lancet. 2006;367:899–900. doi: 10.1016/S0140-6736(06)68374-8. [DOI] [PubMed] [Google Scholar]
- 4.Carroll MF, Temte JL. Proteinuria in adults: a diagnostic approach. Am Fam Physician. 2000;62:1333–1340. [PubMed] [Google Scholar]
- 5.Bellinghieri G, Savica V, Santoro D. Renal alterations during exercise. J Ren Nutr. 2008;18:158–164. doi: 10.1053/j.jrn.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 6.Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354:1387–1401. doi: 10.1056/NEJMra052131. [DOI] [PubMed] [Google Scholar]
- 7.Faul C, Asanuma K, Yanagida-Asanuma E, et al. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet. 2000;24:333–335. doi: 10.1038/74139. [DOI] [PubMed] [Google Scholar]
- 9.Farquhar MG. The primary glomerular filtration barrier—basement membrane or epithelial slits? Kidney Int. 1975;8:197–211. doi: 10.1038/ki.1975.103. [DOI] [PubMed] [Google Scholar]
- 10.Mundel P, Kriz W. Structure and function of podocytes: an update. Anat Embryol (Berl) 1995;192:385–397. doi: 10.1007/BF00240371. [DOI] [PubMed] [Google Scholar]
- 11.Farquhar MG. The glomerular basement membrane: not gone, just forgotten. J Clin Invest. 2006;116:2090–2093. doi: 10.1172/JCI29488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 13.Lowik MM, Groenen PJ, Pronk I, et al. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. 2007;72:1198–1203. doi: 10.1038/sj.ki.5002469. [DOI] [PubMed] [Google Scholar]
- 14.Kestila M, Lenkkeri U, Mannikko M, et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 15.Hinkes B, Wiggins RC, Gbadegesin R, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. 2006;38:1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 16.Boute N, Gribouval O, Roselli S, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 17.Winn MP, Conlon PJ, Lynn KL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 18.Reiser J, Polu KR, Moller CC, et al. TRPC6 is a glomerular slit diaphragmassociated channel required for normal renal function. Nat Genet. 2005;37:739–744. doi: 10.1038/ng1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Togawa A, Miyoshi J, Ishizaki H, et al. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene. 1999;18:5373–5380. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 20.Shibata S, Nagase M, Yoshida S, et al. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–1376. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 21.Schmieder S, Nagai M, Orlando RA, et al. Podocalyxin activates RhoA and induces actin reorganization through NHERF1 and Ezrin in MDCK cells. J Am Soc Nephrol. 2004;15:2289–2298. doi: 10.1097/01.ASN.0000135968.49899.E8. [DOI] [PubMed] [Google Scholar]
- 22.Moeller MJ, Soofi A, Braun GS, et al. Protocadherin FAT1 binds Ena/VASP proteins and is necessary for actin dynamics and cell polarization. EMBO J. 2004;23:3769–3779. doi: 10.1038/sj.emboj.7600380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones N, Blasutig IM, Eremina V, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–823. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- 24.Verma R, Kovari I, Soofi A, et al. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest. 2006;116:1346–1359. doi: 10.1172/JCI27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asanuma K, Kim K, Oh J, et al. Synaptopodin regulates the actinbundling activity of alpha-actinin in an isoform-specific manner. J Clin Invest. 2005;115:1188–1198. doi: 10.1172/JCI23371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asanuma K, Yanagida-Asanuma E, Faul C, et al. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- 27.Russo LM, Sandoval RM, McKee M, et al. The normal kidney filters nephrotic levels of albumin retrieved by proximal tubule cells: Retrieval is disrupted in nephrotic states. Kidney Int. 2007;71:504–513. doi: 10.1038/sj.ki.5002041. [DOI] [PubMed] [Google Scholar]
- 28.Tanner GA. Glomerular sieving coefficient of serum albumin in the rat: a two-photon microscopy study. Am J Physiol Renal Physiol. 2009;296:F1258–F1265. doi: 10.1152/ajprenal.90638.2008. [DOI] [PubMed] [Google Scholar]
- 29.Tanner GA, Rippe C, Shao Y, et al. Glomerular permeability to macromolecules in the Necturus kidney. Am J Physiol Renal Physiol. 2009;296:F1269–F1278. doi: 10.1152/ajprenal.00371.2007. [DOI] [PubMed] [Google Scholar]
- 30.Peti-Peterdi J. Independent two-photon measurements of albumin GSC give low values. Am J Physiol Renal Physiol. 2009;296:F1255–F1257. doi: 10.1152/ajprenal.00144.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haraldsson B, Jeansson M. Glomerular filtration barrier. Curr Opin Nephrol Hypertens. 2009;18:331–335. doi: 10.1097/MNH.0b013e32832c9dba. [DOI] [PubMed] [Google Scholar]
- 32.Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]
- 33.Navar LG. Glomerular permeability: a never-ending saga. Am J Physiol Renal Physiol. 2009;296:F1266–F1268. doi: 10.1152/ajprenal.00152.2009. [DOI] [PubMed] [Google Scholar]
- 34.Reeves W, Caulfield JP, Farquhar MG. Differentiation of epithelial foot processes and filtration slits: sequential appearance of occluding junctions, epithelial polyanion, and slit membranes in developing glomeruli. Lab Invest. 1978;39:90–100. [PubMed] [Google Scholar]
- 35.Reiser J, Kriz W, Kretzler M, et al. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol. 2000;11:1–8. doi: 10.1681/ASN.V1111. [DOI] [PubMed] [Google Scholar]
- 36.Drenckhahn D, Franke RP. Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab Invest. 1988;59:673–682. [PubMed] [Google Scholar]
- 37.Kerjaschki D. Caught flat-footed: podocyte damage and the molecular bases of focal glomerulosclerosis. J Clin Invest. 2001;108:1583–1587. doi: 10.1172/JCI14629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tryggvason K, Pikkarainen T, Patrakka J. Nck links nephrin to actin in kidney podocytes. Cell. 2006;125:221–224. doi: 10.1016/j.cell.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Verma R, Wharram B, Kovari I, et al. Fyn binds to and phosphorylates the kidney slit diaphragm component Nephrin. J Biol Chem. 2003;278:20716–20723. doi: 10.1074/jbc.M301689200. [DOI] [PubMed] [Google Scholar]
- 40.Tryggvason K, Wartiovaara J. Molecular basis of glomerular permselectivity. Curr Opin Nephrol Hypertens. 2001;10:543–549. doi: 10.1097/00041552-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 41.Simons M, Schwarz K, Kriz W, et al. Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am J Pathol. 2001;159:1069–1077. doi: 10.1016/S0002-9440(10)61782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei C, Moller CC, Altintas MM, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63. doi: 10.1038/nm1696. [DOI] [PubMed] [Google Scholar]
- 43.Regele HM, Fillipovic E, Langer B, et al. Glomerular expression of dystroglycans is reduced in minimal change nephrosis but not in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2000;11:403–412. doi: 10.1681/ASN.V113403. [DOI] [PubMed] [Google Scholar]
- 44.Raats CJ, Bakker MA, van den Born J, et al. Hydroxyl radicals depolymerize glomerular heparan sulfate in vitro and in experimental nephrotic syndrome. J Biol Chem. 1997;272:26734–26741. doi: 10.1074/jbc.272.42.26734. [DOI] [PubMed] [Google Scholar]
- 45.Kreidberg JA, Donovan MJ, Goldstein SL, et al. Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- 46.Noakes PG, Miner JH, Gautam M, et al. The renal glomerulus of mice lacking s-laminin/laminin beta 2: nephrosis despite molecular compensation by laminin beta 1. Nat Genet. 1995;10:400–406. doi: 10.1038/ng0895-400. [DOI] [PubMed] [Google Scholar]
- 47.Kretzler M, Teixeira VP, Unschuld PG, et al. Integrin-linked kinase as a candidate downstream effector in proteinuria. FASEB J. 2001;15:1843–1845. doi: 10.1096/fj.00-0832fje. [DOI] [PubMed] [Google Scholar]
- 48.Lorenzen J, Shah R, Biser A, et al. The role of osteopontin in the development of albuminuria. J Am Soc Nephrol. 2008;19:884–890. doi: 10.1681/ASN.2007040486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sachs N, Kreft M, van den Bergh Weerman MA, et al. Kidney failure in mice lacking the tetraspanin CD151. J Cell Biol. 2006;175:33–39. doi: 10.1083/jcb.200603073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smoyer WE, Mundel P. Regulation of podocyte structure during the development of nephrotic syndrome. J Mol Med. 1998;76:172–183. doi: 10.1007/s001090050206. [DOI] [PubMed] [Google Scholar]
- 51.Kos CH, Le TC, Sinha S, et al. Mice deficient in alpha-actinin-4 have severe glomerular disease. J Clin Invest. 2003;111:1683–1690. doi: 10.1172/JCI17988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu TC, He JC, Wang ZH, et al. HIV-1 Nef disrupts the podocyte actin cytoskeleton by interacting with diaphanous interacting protein. J Biol Chem. 2008;283:8173–8182. doi: 10.1074/jbc.M708920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orlando RA, Takeda T, Zak B, et al. The glomerular epithelial cell antiadhesin podocalyxin associates with the actin cytoskeleton through interactions with ezrin. J Am Soc Nephrol. 2001;12:1589–1598. doi: 10.1681/ASN.V1281589. [DOI] [PubMed] [Google Scholar]
- 54.Takeda T, McQuistan T, Orlando RA, et al. Loss of glomerular foot processes is associated with uncoupling of podocalyxin from the actin cytoskeleton. J Clin Invest. 2001;108:289–301. doi: 10.1172/JCI12539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Galeano B, Klootwijk R, Manoli I, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117:1585–1594. doi: 10.1172/JCI30954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asanuma K, Shirato I, Ishidoh K, et al. Selective modulation of the secretion of proteinases and their inhibitors by growth factors in cultured differentiated podocytes. Kidney Int. 2002;62:822–831. doi: 10.1046/j.1523-1755.2002.00539.x. [DOI] [PubMed] [Google Scholar]
- 57.Faul C, Donnelly M, Merscher-Gomez S, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–938. doi: 10.1038/nm.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reiser J, Oh J, Shirato I, et al. Podocyte migration during nephrotic syndrome requires a coordinated interplay between cathepsin L and alpha3 integrin. J Biol Chem. 2004;279:34827–34832. doi: 10.1074/jbc.M401973200. [DOI] [PubMed] [Google Scholar]
- 59.Ronco P. Proteinuria: is it all in the foot? J Clin Invest. 2007;117:2079–2082. doi: 10.1172/JCI32966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sever S, Altintas MM, Nankoe SR, et al. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest. 2007;117:2095–2104. doi: 10.1172/JCI32022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quaggin SE. Transcriptional regulation of podocyte specification and differentiation. Microsc Res Tech. 2002;57:208–211. doi: 10.1002/jemt.10076. [DOI] [PubMed] [Google Scholar]
- 62.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bottinger EP, Bitzer M. TGF-ss Signaling in Renal Disease. J Am Soc Nephrol. 2002;13:2600–2610. doi: 10.1097/01.asn.0000033611.79556.ae. [DOI] [PubMed] [Google Scholar]
- 65.Sharma K, Ramachandrarao S, Qiu G, et al. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest. 2008;118:1645–1656. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Waters AM, Wu MY, Onay T, et al. Ectopic notch activation in developing podocytes causes glomerulosclerosis. J Am Soc Nephrol. 2008;19:1139–1157. doi: 10.1681/ASN.2007050596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Niranjan T, Bielesz B, Gruenwald A, et al. The Notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14:290–298. doi: 10.1038/nm1731. [DOI] [PubMed] [Google Scholar]
- 68.Huber TB, Hartleben B, Winkelmann K, et al. Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J Am Soc Nephrol. 2009;20:798–806. doi: 10.1681/ASN.2008080871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hirose T, Satoh D, Kurihara H, et al. An essential role of the universal polarity protein, aPKClambda, on the maintenance of podocyte slit diaphragms. PLoS ONE. 2009;4:e4194. doi: 10.1371/journal.pone.0004194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 1998;54:687–697. doi: 10.1046/j.1523-1755.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- 71.Le Hir M, Keller C, Eschmann V, et al. Podocyte bridges between the tuft and Bowman’s capsule: an early event in experimental crescentic glomerulonephritis. J Am Soc Nephrol. 2001;12:2060–2071. doi: 10.1681/ASN.V12102060. [DOI] [PubMed] [Google Scholar]
- 72.Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol. 2002;13:3005–3015. doi: 10.1097/01.asn.0000039661.06947.fd. [DOI] [PubMed] [Google Scholar]
- 73.Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int. 2005;67:404–419. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 74.Zandi-Nejad K, Eddy AA, Glassock RJ. Why is proteinuria an ominous biomarker of progressive kidney disease? Kidney Int Suppl. 2004:S76–S89. doi: 10.1111/j.1523-1755.2004.09220.x. [DOI] [PubMed] [Google Scholar]
- 75.Morigi M, Buelli S, Angioletti S, et al. In response to protein load podocytes reorganize cytoskeleton and modulate endothelin-1 gene: implication for permselective dysfunction of chronic nephropathies. Am J Pathol. 2005;166:1309–1320. doi: 10.1016/S0002-9440(10)62350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilmer WA, Rovin BH, Hebert CJ, et al. Management of glomerular proteinuria: a commentary. J Am Soc Nephrol. 2003;14:3217–3232. doi: 10.1097/01.asn.0000100145.27188.33. [DOI] [PubMed] [Google Scholar]
- 77.Kikuchi H, Kawachi H, Ito Y, et al. Severe proteinuria, sustained for 6 months, induces tubular epithelial cell injury and cell infiltration in rats but not progressive interstitial fibrosis. Nephrol Dial Transplant. 2000;15:799–810. doi: 10.1093/ndt/15.6.799. [DOI] [PubMed] [Google Scholar]
- 78.Gross ML, Hanke W, Koch A, et al. Intraperitoneal protein injection in the axolotl: the amphibian kidney as a novel model to study tubulointerstitial activation. Kidney Int. 2002;62:51–59. doi: 10.1046/j.1523-1755.2002.00402.x. [DOI] [PubMed] [Google Scholar]
- 79.Seiler MW, Venkatachalam MA, Cotran RS. Glomerular epithelium: structural alterations induced by polycations. Science. 1975;189:390–393. doi: 10.1126/science.1145209. [DOI] [PubMed] [Google Scholar]
- 80.Seiler MW, Rennke HG, Venkatachalam MA, et al. Pathogenesis of polycation-induced alterations (‘fusion’) of glomerular epithelium. Lab Invest. 1977;36:48–61. [PubMed] [Google Scholar]
- 81.Kerjaschki D. Polycation-induced dislocation of slit diaphragms and formation of cell junctions in rat kindey glomeruli. The effects of low temperature, divalent cations, colchicine, and cytochalasin B. Lab Invest. 1978;39:430–440. [PubMed] [Google Scholar]
- 82.Honey K, Rudensky AY. Lysosomal cysteine proteases regulate antigen presentation. Nat Rev Immunol. 2003;3:472–482. doi: 10.1038/nri1110. [DOI] [PubMed] [Google Scholar]
- 83.Goulet B, Baruch A, Moon NS, et al. A cathepsin L isoform that is devoid of a signal peptide localizes to the nucleus in S phase and processes the CDP/Cux transcription factor. Mol Cell. 2004;14:207–219. doi: 10.1016/s1097-2765(04)00209-6. [DOI] [PubMed] [Google Scholar]
- 84.Duncan EM, Muratore-Schroeder TL, Cook RG, et al. Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell. 2008;135:284–294. doi: 10.1016/j.cell.2008.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baricos WH, Cortez SL, Le QC, et al. Evidence suggesting a role for cathepsin L in an experimental model of glomerulonephritis. Arch Biochem Biophys. 1991;288:468–472. doi: 10.1016/0003-9861(91)90222-5. [DOI] [PubMed] [Google Scholar]
- 86.Reiser J, Von Gersdorff G, Loos M, et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. 2004;113:1390–1397. doi: 10.1172/JCI20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reiser J, Mundel P. Danger signaling by glomerular podocytes defines a novel function of inducible b7-1 in the pathogenesis of nephrotic syndrome. J Am Soc Nephrol. 2004;15:2246–2248. doi: 10.1097/01.ASN.0000136312.46464.33. [DOI] [PubMed] [Google Scholar]
- 88.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 89.Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 90.Jaffe AB, Hall A. RHO GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005 doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 91.Jaffe AB, Hall A. Cell biology. Smurfing at the leading edge. Science. 2003;302:1690–1691. doi: 10.1126/science.1092874. [DOI] [PubMed] [Google Scholar]
- 92.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 93.Stupp R, Ruegg C. Integrin inhibitors reaching the clinic. J Clin Oncol. 2007;25:1637–1638. doi: 10.1200/JCO.2006.09.8376. [DOI] [PubMed] [Google Scholar]
- 94.Mundel P, Heid HW, Mundel TM, et al. Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes. J Cell Biol. 1997;139:193–204. doi: 10.1083/jcb.139.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huber TB, Kwoh C, Wu H, et al. Bigenic mouse models of focal segmental glomerulosclerosis involving pairwise interaction of CD2AP, Fyn, and synaptopodin. J Clin Invest. 2006;116:1337–1345. doi: 10.1172/JCI27400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yanagida-Asanuma E, Asanuma K, Kim K, et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am J Pathol. 2007;171:415–427. doi: 10.2353/ajpath.2007.070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Reiser J, Mundel P. Dual effects of RAS blockade on blood pressure and podocyte function. Curr Hypertens Rep. 2007;9:403–408. doi: 10.1007/s11906-007-0074-7. [DOI] [PubMed] [Google Scholar]
- 98.Durvasula RV, Petermann AT, Hiromura K, et al. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–39. doi: 10.1111/j.1523-1755.2004.00362.x. [DOI] [PubMed] [Google Scholar]
- 99.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–1634. doi: 10.2337/diabetes.54.6.1626. [DOI] [PubMed] [Google Scholar]
- 100.Benigni A, Tomasoni S, Gagliardini E, et al. Blocking angiotensin II synthesis/activity preserves glomerular nephrin in rats with severe nephrosis. J Am Soc Nephrol. 2001;12:941–948. doi: 10.1681/ASN.V125941. [DOI] [PubMed] [Google Scholar]
- 101.Macconi D, Ghilardi M, Bonassi ME, et al. Effect of angiotensin-converting enzyme inhibition on glomerular basement membrane permeability and distribution of zonula occludens-1 in MWF rats. J Am Soc Nephrol. 2000;11:477–489. doi: 10.1681/ASN.V113477. [DOI] [PubMed] [Google Scholar]
- 102.Hoffmann S, Podlich D, Hahnel B, et al. Angiotensin II Type 1 Receptor Overexpression in Podocytes Induces Glomerulosclerosis in Transgenic Rats. J Am Soc Nephrol. 2004;15:1475–1487. doi: 10.1097/01.asn.0000127988.42710.a7. [DOI] [PubMed] [Google Scholar]
- 103.Meyrier A. Treatment of focal segmental glomerulosclerosis. Expert Opin Pharmacother. 2005;6:1539–1549. doi: 10.1517/14656566.6.9.1539. [DOI] [PubMed] [Google Scholar]
- 104.Shalhoub RJ. Pathogenesis of lipoid nephrosis: a disorder of T-cell function. Lancet. 1974;2:556–560. doi: 10.1016/s0140-6736(74)91880-7. [DOI] [PubMed] [Google Scholar]
- 105.Ransom RF, Lam NG, Hallett MA, et al. Glucocorticoids protect and enhance recovery of cultured murine podocytes via actin filament stabilization. Kidney Int. 2005;68:2473–2483. doi: 10.1111/j.1523-1755.2005.00723.x. [DOI] [PubMed] [Google Scholar]
- 106.Aramburu J, Heitman J, Crabtree GR. Calcineurin: a central controller of signalling in eukaryotes. EMBO Rep. 2004;5:343–348. doi: 10.1038/sj.embor.7400133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 108.Charbit M, Gubler MC, Dechaux M, et al. Cyclosporin therapy in patients with Alport syndrome. Pediatr Nephrol. 2007;22:57–63. doi: 10.1007/s00467-006-0227-y. [DOI] [PubMed] [Google Scholar]
- 109.Chen D, Jefferson B, Harvey SJ, et al. Cyclosporine a slows the progressive renal disease of alport syndrome (X-linked hereditary nephritis): results from a canine model. J Am Soc Nephrol. 2003;14:690–698. doi: 10.1097/01.asn.0000046964.15831.16. [DOI] [PubMed] [Google Scholar]
- 110.Heit JJ, Apelqvist AA, Gu X, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. 2006;443:345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- 111.Horsley V, Aliprantis AO, Polak L, et al. NFATc1 balances quiescence and proliferation of skin stem cells. Cell. 2008;132:299–310. doi: 10.1016/j.cell.2007.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reardon DA, Fink KL, Mikkelsen T, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26:5610–5617. doi: 10.1200/JCO.2008.16.7510. [DOI] [PubMed] [Google Scholar]
- 113.MacDonald TJ, Stewart CF, Kocak M, et al. Phase I clinical trial of cilengitide in children with refractory brain tumors: Pediatric Brain Tumor Consortium Study PBTC-012. J Clin Oncol. 2008;26:919–924. doi: 10.1200/JCO.2007.14.1812. [DOI] [PubMed] [Google Scholar]
- 114.Beekman KW, Colevas AD, Cooney K, et al. Phase II evaluations of cilengitide in asymptomatic patients with androgen-independent prostate cancer: scientific rationale and study design. Clin Genitourin Cancer. 2006;4:299–302. doi: 10.3816/CGC.2006.n.012. [DOI] [PubMed] [Google Scholar]
- 115.Mathieson PW. Proteinuria and immunity—an overstated relationship? N Engl J Med. 2008;359:2492–2494. doi: 10.1056/NEJMcibr0806881. [DOI] [PubMed] [Google Scholar]
- 116.Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715–2729. doi: 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 117.Duning K, Schurek EM, Schluter M, et al. KIBRA modulates directional migration of podocytes. J Am Soc Nephrol. 2008;19:1891–1903. doi: 10.1681/ASN.2007080916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hsu HH, Hoffmann S, Endlich N, et al. Mechanisms of angiotensin II signaling on cytoskeleton of podocytes. J Mol Med. 2008;86:1379–1394. doi: 10.1007/s00109-008-0399-y. [DOI] [PubMed] [Google Scholar]
- 119.Wu DT, Bitzer M, Ju W, et al. TGF-beta concentration specifies differential signaling profiles of growth arrest/differentiation and apoptosis in podocytes. J Am Soc Nephrol. 2005;16:3211–3221. doi: 10.1681/ASN.2004121055. [DOI] [PubMed] [Google Scholar]
- 120.Moller CC, Wei C, Altintas MM, et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29–36. doi: 10.1681/ASN.2006091010. [DOI] [PubMed] [Google Scholar]
- 121.Moller CC, Flesche J, Reiser J. Sensitizing the slit diaphragm with TRPC6 ion channels. J Am Soc Nephrol. 2009;20:950–953. doi: 10.1681/ASN.2008030329. [DOI] [PubMed] [Google Scholar]
- 122.Kopp JB, Smith MW, Nelson GW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet. 2008;40:1175–1184. doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Krendel M, Kim SV, Willinger T, et al. Disruption of Myosin 1e promotes podocyte injury. J Am Soc Nephrol. 2009;20:86–94. doi: 10.1681/ASN.2007111172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Oh J, Reiser J, Mundel P. Dynamic (re)organization of the podocyte actin cytoskeleton in the nephrotic syndrome. Pediatr Nephrol. 2004;19:130–137. doi: 10.1007/s00467-003-1367-y. [DOI] [PubMed] [Google Scholar]