

Abstract
Background:
Phospholipidosis (PLD) is a lysosomal storage disorder induced by a class of cationic amphiphilic drugs. However, drug-induced PLD is reversible. Evidence of PLD from animal studies with some compounds has led to discontinuation of development. Regulatory authorities are likely to request additional studies when PLD is linked to toxicity.
Objective:
We conducted a trial to investigate urinary phenylacetylglycine (uPAG) as a biomarker for PLD.
Materials and methods:
Five groups of 12 male Wistar rats were dosed once with vehicle, 300 mg/kg or 1500 mg/kg of compound A (known to induce PLD), or 300 mg/kg or 1000 mg/kg of compound B (similar structure, but does not induce PLD) to achieve similar plasma exposures. Following dosing, urine and blood samples underwent nuclear magnetic resonance (NMR), proteomic, and biochemical analyses. Necropsies were performed at 48 and 168 h, organ histopathology evaluated, and gene expression in liver analyzed by microarray. Electron microscopic examination of peripheral lymphocytes was performed.
Results:
For compound A, uPAG increased with dose, correlating with lamellar inclusion bodies formation in peripheral lymphocytes. NMR analysis showed decreased tricarboxylic acid cycle intermediates, inferring mitochondrial toxicity. Mitochondrial dysfunction was suggested by uPAG increase, resulting from a switch to anaerobic metabolism or disruption of the urea cycle.
Discussion and conclusion:
uPAG shows utility as a noninvasive biomarker for mitochondrial toxicity associated with drug-induced PLD, providing a mechanistic hypothesis for toxicity associated with PLD likely resulting from combined direct and indirect mitochondrial toxicity via impairment of the proton motor force and alteration of fatty acid catabolism.
Keywords: biomarker, phospholipidosis, urinary phenylacetylglycine
Background
Phospholipidosis (PLD) is a storage disorder whereby excess phospholipids accumulate within the cell, particularly in the lysosome. PLD has been shown to be induced by a class of cationic amphiphilic drugs (CADs) which have a hydrophobic moiety with a charged cationic amine that interferes with phospholipid metabolism and turnover. Drug-induced PLD has been observed in vivo [Kodavanti and Mehendale, 1990; Lullmann-Rauch, 1979] and in vitro [Drenckhahn et al. 1976; Jagel and Lullmann-Rauch, 1984; McCloud et al. 1995; Ruben et al. 1991], and can affect many tissues. The first reports of drug-induced PLD were on alveolar macrophages [Franken et al. 1970; Reasor, 1981] which are particularly susceptible to the formation of multilamellar bodies. Affected macrophages are referred to as foamy macrophages as the cytoplasm appears ‘foamy’ when viewed with light microscopy.
PLD is thought to be an adaptive response to CAD exposure; the drug is sequestered in lamellar bodies to avoid potential toxicity to intracellular structures [Hostetler et al. 1985]. Subsequent accumulation of one or several types of lipid is thought to occur via inhibition of lysosomal phospholipases, most likely through binding of CADs to phospholipids [Drenckhahn et al. 1976; Lullmann et al. 1978]. Direct inhibition of lysosomal phospholipases [Kubo and Hostetler, 1985] or regulation of phospholipid synthesis may also contribute to lipid accumulation [Pappu and Hostetler, 1984].
Drug-induced PLD is reversible and does not necessarily cause toxicity, but is predictive of drug or metabolite accumulation in affected tissues and is a warning to investigate possible associated toxicities. The severity and reversibility of PLD depend on the dose, exposure, duration of treatment and pharmacology of the CAD. Evidence of PLD has previously led to discontinuation of development of some compounds. Fischer and colleagues have recently reported a new in-silico method for predicting the likelihood of compounds in causing PLD [Fischer et al. 2012]. However, evidence of PLD does not have to result in withdrawal of a molecule; PLD only presents a problem when linked to toxicity, and regulatory authorities are likely to require additional studies in these circumstances [Reasor, 2010]. Withdrawal of any compound is a serious issue for any pharmaceutical developer [Kaitin, 2008], and early recognition of characteristics that may cause a product to fail at later stages is preferable from both a human safety and economic perspective [Kola and Landis, 2004].
The phospholipidotic potential of a compound is most easily indicated by histopathological examination, such as the detection of foamy macrophages in various tissues, for example lungs, or vacuolation of epithelial cells. However, a definitive PLD diagnosis is based on ultrastructural changes detected by transmission electron microscopy (TEM). A metabonomic approach by Nicholls and colleagues to identify novel markers for PLD examined urinary levels of metabolites in rats following dosing with CAD [Nicholls et al. 2000]. Urinary levels of the tricarboxylic acid (TCA) cycle intermediates, citrate and 2-oxoglutarate were found to decrease, suggesting that the mitochondrial TCA cycle for the production of energy had been disrupted, and energy-yielding intermediates metabolized. In addition, the study highlighted that levels of urinary phenylacetylglycine (uPAG) increased, and thus PAG was proposed as a biomarker for PLD. A subsequent study of urinary metabolites in acclimatizing germ-free rats by Nicholls and colleagues demonstrated that uPAG levels were affected during the establishment of stable gut microflora [Nicholls et al. 2003]. However, the alterations in uPAG caused by microbes were not as substantial as those caused by metabolic dysfunction. uPAG has also been shown to be affected by parasitic infection [Garcia-Perez et al. 2010] and dietary intake [O’Sullivan et al. 2011]. Several subsequent studies of urinary metabolites in rats with PLD induced by amio- darone reported increases in uPAG [Delaney et al. 2004; Dieterle et al. 2006; Hasegawa et al. 2007]. PAG has therefore been suggested as a surrogate marker for PLD. PAG is formed metabolically by the conjugation of phenylacetyl coenzyme A (CoA) with glycine [Jones, 1982]. Phenylacetate, the precursor for phenylacetyl CoA, can be synthesized by oxidation of phenyl-containing fatty acids, or produced by the degradation of phenylalanine to phenylethylamine and phenylacetaldehyde, or via phenylpyruvate.
To support the hypothesis of PAG as a biomarker for PLD, a single-dose toxicology study was performed in a rat model. The effects of two compounds (A and B) with similar structures were assessed by clinical chemistry, histopathology, electron microscopy, metabolite and protein composition in urine, and gene expression in liver. The structure of these compounds mainly differs by a piperazine against a morpholine moiety in compound A, leading to a more basic pKa and greater potential to induce PLD compared with compound B, based on amphiphilicity. It was hypothesized that dosing with compound A, but not compound B, would induce an increase in PAG based on known histopathology findings of PLD with compound A only.
Methods
This study was performed in a laboratory approved by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC) and the Swiss Federal Act on Animal Protection (Swiss APA 1978) in accordance with Swiss Animal Protection law.
Five groups each of 12 male specific pathogen-free Wistar rats (HanBrl:WIST) were dosed once with vehicle, or 300 mg/kg or 1500 mg/kg of compound A, or 300 mg/kg or 1000 mg/kg of compound B. Blood and urine samples were collected in nonfasted stage. Samples for blood chemistry were collected approximately 24, 48 and 168 h after dosing. Blood samples for toxicokinetics were drawn from the retro-orbital plexus under light isoflurane anaesthesia before, and then 1, 3, 5, 8, 24, 48 and 168 h after dosing. Urine samples were collected in metabolism cages at 0–4°C automatically refrigerated by a Tecniplast sampling/cooling unit into labelled sample tubes containing 1 ml of an aqueous Na-azide (1%) solution. Before aliquoting urine, volumes were determined. The metabolism cages were cleaned with bidistilled water on days –3, 0, 2, 4 and 6 directly at the end of the sampling period and prior to the next. On days –1, 1, 3, 5 and 7 the collection units of the metabolism cages were exchanged. Urine samples were collected at 12 timepoints (four predose: –144, –40, –16 and 0 h; and eight postdose: 8, 24, 48, 72, 96, 120, 144 and 168 h) for nuclear magnetic resonance (NMR) spectroscopy, urinalysis, proteomics and liquid chromatography–mass spectrometry (LC-MS) [Schlotterbeck et al. 2006].
Necropsies were performed on five animals from each group at 48 h and 168 h after dosing, and the remaining two animals from each group were observed for toxicokinetic bleeding only. At necropsy, blood samples were collected from all animals for electron microscopic examination of peripheral lymphocytes, and major organs including liver, kidneys, lungs, mesenteric lymph nodes and spleen underwent subsequent histopathologic evaluation. Tissues were embedded in paraffin, cut at a nominal thickness of 2–4 mm, stained with haematoxylin and eosin, and examined under a light microscope. Buffy coats were prepared from a sample of at least 2 ml terminal blood and processed for electron microscopy following standard protocols.
Blood chemistry parameters were measured by routinely used procedures; samples for serum analysis were allowed to clot naturally at room temperature for no longer than 10 min and then cooled and centrifuged at 1000 g for 10 min at 4°C. Standard assays were performed on a Hitachi 917 spectrometer for the following parameters: creatinine (enzymatic colorimetric assay), phospholipids (phospholipase–cholinoxidase–peroxidase assay), albumin (bromocresol green assay), globulin (calculated as total protein minus albumin), and total protein (Biuret assay). Tyrosine was assayed by treatment with two volumes of phenylalanine lyase, pH 8.75 for 30 min at 37°C, followed by separation on a Waters Oasis HLB column (1 ml; Elstree, UK) and analysis by high-performance liquid chromatography.
Pharmacokinetic parameters were calculated using WinNonlin Pro v.3 (Pharsight, Inc., 1998, Cary, NC, USA). The maximum plasma concentration (C max) was determined directly from plasma concentration–time profiles. The apparent terminal half lives were derived from the equation t½ = ln 2/λz. λz was calculated by least squares linear regression of the terminal portion of the log-transformed plasma concentration curve.
Urine was processed for NMR analysis or proteomics as described previously in the literature [Keun et al. 2002]. Urine samples from high-dose groups collected at 48 h and 168 h postdosing were also subjected to proteomics analysis by two-dimensional polyacrylamide gel electrophoresis followed by staining, gel spot excision and matrix-assisted laser desorption ionization time-of-flight mass spectrometry as described elsewhere [Roessler et al. 2006].
Gene expression analysis was performed on RNA from the livers of all animals collected in RNALater (Applied Biosystems; Carlsbad, CA, USA) and determined using RG U34a GeneChip microarrays (Affymetrix; Santa Clara, CA, USA) following the provider’s instructions. Expression levels of phenylalanine hydroxylase (PAH) and carnitine palmitoyltransferase 1A (CPT1a) were determined by signal intensity with the M12337_at. To classify individual gene expression profiles, previous liver gene expression profiles from male Wistar rats were used to generate a predictive support vector machine (SVM)-based predictive model for the following hepatotoxic categories: control/nontoxic, direct acting, peroxisome proliferation, cholestasis and steatosis [Lee, 1995]. Subsequently, gene expression profiles from the livers of control and treated rats were compared with the SVM Hepatotox_SVM_OVA_Versionl [Steiner et al. 2004], and classified according to confidence.
Results
A similar level of plasma exposure was reached with both compounds. Maximum plasma concentrations were reached at 5–8 h postdose with compound A, and at 2–3 h with compound B. The maximum plasma concentration (C max) for compound A was 3.4 µg/ml with 300 mg/kg and 4.8 µg/ml with 1500 mg/kg, and for compound B was 2.8 µg/ml with 300 mg/kg and 2.3 µg/ml with 1000 mg/kg, suggesting saturation in the absorption of both compounds at higher doses. Terminal half lives were 32 and 34 h for the low- and high-dose groups with compound A, and 31 and 35 h for the low- and high-dose groups with compound B.
Body mean weight gain on day 2 increased by (+)0.8% and (+)0.5% in animals on control or on compound B 1000 mg/kg while mean body weight decreased by (–)3% and (–)2.7% for animals on compound A (300 and 1500 mg/kg respectively) and by (–)0.5% for animals on compound B 300 mg/kg.
Compounds A and B decreased liver function, as indicated by a reduction in total protein in blood samples taken at 168 h (Table 1). However, animals dosed with compound A had a severe alteration in liver function with a significant reduction in levels of creatinine, phospholipids, albumin, globulin and tyrosine in blood samples taken at 24 h and 168 h. These effects were not seen in animals dosed with compound B.
Table 1.
Blood protein and metabolite levels in samples taken from animals dosed with compound A, compound B or control, at 24 h and 168 h postdose.
| Time after dosing (h) | Group 1 (control) | Group 2 |
Group 3 |
Group 4 |
Group 5 |
Statistical test | |
|---|---|---|---|---|---|---|---|
| Compound A |
Compound B |
||||||
| 300 mg/kg | 1500 mg/kg A | 300 mg/kg B | 1000 mg/kg B | ||||
| Total protein (g/liter) | 24 | 60.321 (2.153) | 59.313 (1.422) | 58.957 (1.571) | 60.006 (1.907) | 59.552 (1.177) | ANOVA |
| 168 | 62.702 (0.782) | 61.576 (1.504) | 56.752** (2.273) | 60.762 (1.344) | 58.874** (2.388) | ANOVA and Dunnett | |
| Creatinine (µmol/liter) | 24 | 25.86 (1.76) | 23.31** (1.93) | 23.41* (1.59) | 26.45 (1.77) | 25.63 (1.65) | ANOVA and Dunnett |
| 168 | 29.58 (1.54) | 30.14 (1.92) | 26.77 (3.37) | 28.76 (1.82) | 29.02 (1.36) | ANOVA | |
| Phospholipids (mmol/liter) | 24 | 1.639 (0.124) | 1.351** (0.172) | 1.359** (0.200) | 1.543 (0.189) | 1.583 (0.221) | ANOVA and Dunnett |
| 168 | 1.626 (0.115) | 1.662 (0.215) | 1.232* (0.210) | 1.662 (0.163) | 1.612 (0.148) | ANOVA and Dunnett | |
| Albumin (g/liter) | 24 | 43.607 (1.342) | 41.361 (1.344) | 41.422 (1.550) | 42.179 (1.198) | 41.849 (0.759) | ANOVA |
| 168 | 44.264 (0.945) | 41.052* (1.977) | 30.270** (2.776) | 41.988) (1.356) | 41.484 (1.297) | ANOVA and Dunnett | |
| Globulin (g/liter) | 24 | 17.714 (1.241) | 17.952 (1.141) | 17.535 (1.307) | 17.827 (1.088) | 17.703 (1.012) | ANOVA |
| 168 | 18.438 (0.898) | 20.524 (1.183) | 26.483* (3.570) | 18.774 (0.365) | 17.390 (1.243) | Kruskal–Wallace and Dunn | |
| Tyrosine | 24 | 138 (19.6) | - | 113* (14.3) | – | 133 (19.6) | Two-sided Dunnett’s test |
| 48 | 125 (16.0) | - | 98.7* (5.96) | – | 119 (17.0) | Two-sided Dunnett’s test | |
Note: The bold text corresponds to parameters that are statistically significant (also see asterisk).
p < 0.05, **p < 0.01.
n = 10 for each at 24 h, n = 5 for each at 168 h (except group 3, where n = 4).
ANOVA, analysis of variance.
Gene expression analyses confirmed that compound A had clear hepatotoxic potential and showed evidence of steatosis (Table 2). At 48 h, all animals dosed with compound A (both high and low dose) were classified as steatotic compared with no animals dosed with compound B. Liver steatosis is an expected finding in lipid storage disorders.
Table 2.
Classification of individual gene expression profiles by a support vector machine (SVM)-based predictive model.
| Compound | Dose | Classification (n animals) | Classification status (% animals) | |
|---|---|---|---|---|
| Group 1 | Control | – | Control (5) | Unique (60%) |
| Fits into no class (40%) | ||||
| Group 2 | Compound A | 300 mg/kg | Steatotic (5) | Unique (60%) |
| Ambiguous (40%) | ||||
| Group 3 | Compound A | 1500 mg/kg | Steatotic (5) | Unique (100%) |
| Group 4 | Compound B | 300 mg/kg | Control (2) | Fits into no class (100%) |
| Cholestatic (1) | Fits into no class (100%) | |||
| Steatotic (2) | Fits into no class (100%) | |||
| Group 5 | Compound B | 1000 mg/kg | Control (4) | Unique (75%) |
| Fits into no class (25%) | ||||
| Cholestatic (1) | Fits into no class (100%) |
Gene expression profiles from the livers of control and treated rats were classified using the SVM Hepatotox_SVM_OVA_Version l. These were compared with predictive SVM-based models created from our database of liver gene expression profiles for the following hepatotoxic categories: control/nontoxic, direct acting, peroxisome proliferation, cholestasis, and steatosis.
Histopathology findings for compound A, but not compound B, were characteristic for PLD, and were more severe with the higher dose. The extent and timing of pathologic response differed according to tissue type; vacuolated macrophages in lungs, mesenteric lymph nodes and spleen, and vacuolated Kupffer cells and hepatocytes were affected by compound A only (Table 3). In addition to PLD histopathology findings, organ toxicity was observed in liver, kidney, spleen and mesenteric lymph nodes, particularly in animals administered with the higher dose of compound A, and consisted of single cell, focal or multifocal necrosis predominantly 168 h postdose.
Table 3.
Pathologic findings for animals necropsied at 168 h postdose.
| Group 1 | Group 2 |
Group 3 |
Group 4 |
Group 5 |
|
|---|---|---|---|---|---|
| Control | Compound A |
Compound B |
|||
| 300 mg/kg | 1500 mg/kg | 300 mg/kg | 1000 mg/kg | ||
| Kidneys | |||||
| Single cell necrosis of tubular epithelium | – | – | 4 (1.5) | – | – |
| Liver | |||||
| Glycogen increase | 5 (2.0) | 2 (1.0) | – | 5 (1.6) | 5 (1.8) |
| Haematopoiesis | 3 (1.0) | 4 (1.3) | – | 3 (1.0) | 4 (1.0) |
| Necrosis | – | 1 (2.0) | 2 (3.0) | – | |
| Vacuolated Kupffer cells | – | – | 3 (3.0) | – | – |
| Lung | |||||
| Alveolar histiocytosis | – | 5 (2.8) | 5 (3.6) | – | – |
| Mesenteric lymph nodes | |||||
| Histiocytosis | – | – | 5 (3.4) | – | – |
| Lymphoid depletion | – | – | 3 (4.0) | – | – |
| Inflammation | – | – | 5 (2.6) | – | – |
| Necrosis | – | – | 4 (2.8) | – | – |
| Spleen | |||||
| Haematopoiesis | 5 (2.2) | 4 (1.3) | – | 5 (2.2) | 5 (1.6) |
| Histiocytosis | – | – | 5 (2.6) | – | – |
| Lymphoid depletion | – | – | 3 (3.0) | – | – |
| Inflammation | – | – | 1 (2.0) | – | – |
For each group, five animals were necropsied and kidney, liver, lung, mesenteric lymph node and spleen pathology was graded as follows: grade 1 = minimal/very few/very small; grade 2 = slight/few/small; grade 3 = moderate/moderate number/moderate size; grade 4 = marked/many/large; grade 5 = finding unilateral in paired organs. Numbers are expressed as number of animals (average grade).
Electron microscopy of peripheral lymphocytes demonstrated the presence of intracytoplasmic inclusions for compound A, with timings coinciding with histopathologic changes (Figure 1). Lamellar inclusions were more prevalent with the higher dose than the lower dose of compound A for animals necropsied 48 h following administration. Animals necropsied 168 h following administration with the lower dose of compound A exhibited decreased or no lymphocyte response compared with 48 h following administration, indicating partial or complete recovery. At the high dose there were no indication of recovery within 168 h following administration.
Figure 1.
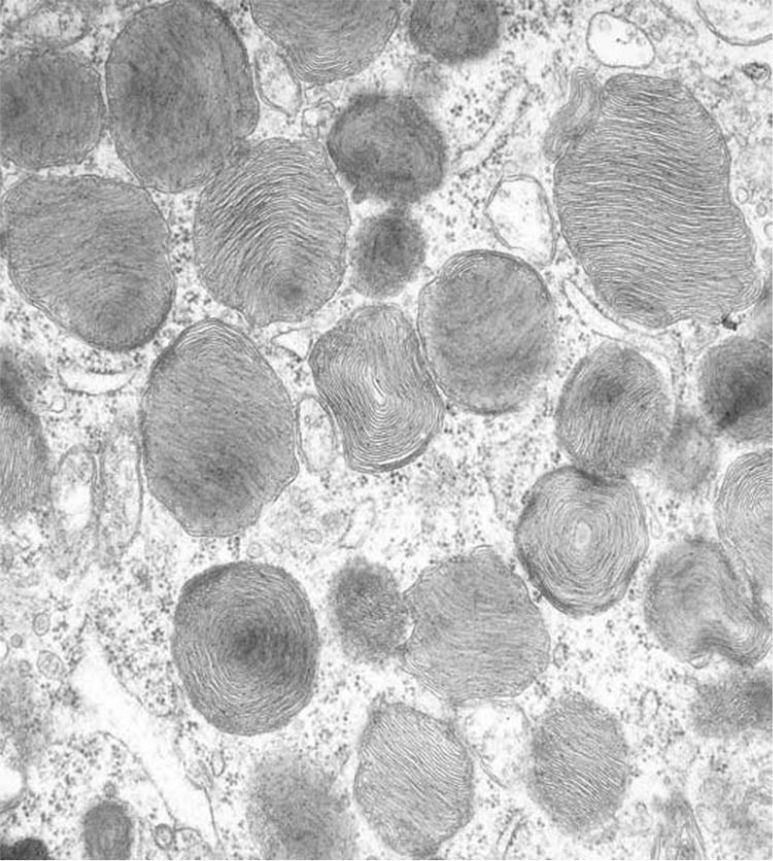
Electron microscopy of peripheral lymphocytes demonstrating the presence of intracytoplasmic inclusions for compound A.
NMR analysis of urine samples indicated that compound A caused a disruption to the TCA cycle at the higher dose (Figure 2). Levels of TCA cycle intermediates, citrate, isocitrate, 2-oxoglutarate, succinate, fumarate and malate were decreased compared with the control, following the higher dose of compound A; however, levels were not affected following dosing with compound B. Changes are summarized in Table 4 and indicated schematically in Figure 3 by arrows. uPAG was seen to increase by two- to fourfold with higher doses of compound A (Figure 4). The lower dose of compound A also produced a twofold increase in uPAG at 24 h, whereas neither dose of compound B affected uPAG levels. Proteomic analysis revealed no clear differences between any of the urine samples (data not shown).
Figure 2.

Levels of tricarboxylic acid cycle intermediates by nuclear magnetic resonance analysis, following dosing and normalized to time-matched control animals. Levels of urinary citrate and 2-oxoglutarate are shown for animals dosed with high and low doses of compound A (left) and compound B (right).
Table 4.
Summary of metabolic changes upon dosing with compounds A and B.
| Drug | Blood | Urine | Histopathology | Liver toxicogenomics |
|---|---|---|---|---|
| Compound A | Creatinine ↓ (L, H)
Phospholipids ↓ (L, H) ALP, total protein, globulin. ↓ (H) Albumin ↓ ( H) Albumin/globulin ↓ (L, H) ASAT, ALAT ↑(H) Tyrosine ↓ (H)* |
Biochemistry
pH ↓ (L, H) Creatinine ↓ (H) Phospholipids ↓ (L, H) Ca++, K+ ↓ (L, H) Inorganic PO4 ↑ (L, H) NMR analysis PAG ↑ (2× L, 2-4x H) Citrate, 2OG, fumarate, malate, isocitrate, succinate ↓ (H) Taurine ↓ (H) Carnitine, creatine ↑ (H) Proteomics Clear sign of nephrotoxicity (H)* |
Vacuolated foamy macrophages lungs, mesenteric lymph nodes (L, H), spleen (H)
Vacuolated hepatocytes (L,H). Associated toxicity (necrosis) in liver, kidney, spleen, mesenteric lymph nodes (predominantly H). |
Call of hepatotoxicity, steatosis/cholestasis.
Partly reversible at 168 h PAH ↓ FA-metabolism ↑ FA synthase ↓ Stearoyl CoA desaturase ↓ ATP citrate lyase ↓ Fumarate hydratase ↑ General stress response |
| Compound B | Total protein ↓ (H) |
NMR analysis
No significant drug-induced changes |
Single cell liver necrosis (L, H, 48 h, postdose) | Individual animals with borderline cholestasis-like evidence (L, H)
Slight PAH ↓ Slight FA synthase ↓ Slight ATP citrate lyase ↓ Slight fumarate hydratase ↑ |
Tested only with high doses.
2OG, 2-oxoglutarate; ALAT, alanine aminotransferase; ALP, alkaline phosphatase; ATP, adenosine triphosphate; ASAT, aminotransferase; FA, fatty acid; H, high dose; L, low dose; PAG, phenylacetylglycine; PAH, phenylalanine hydroxylase.
Figure 3.

Schematic diagram showing metabolic changes upon dosing with compound A. Phenylacetylglycine (PAG) is formed metabolically from phenylalanine (PA) via phenylacetyl coenzyme A (CoA) and phenylpyruvate. The metabolism of PA is linked to the mitochondrial tricarboxylic acid (TCA) cycle via acetyl CoA. Dosing with compound A but not compound B caused an increase in urinary PAG (as indicated schematically by yellow arrows), and the higher dose of compound A caused disruption to the TCA cycle as indicated by a decrease in TCA cycle intermediates compared with the control. Downregulation of phenylalanine hydroxylase (PAH) by compound A after 48 h correlated with a decrease in tyrosine levels. An increase in urinary inorganic phosphate also indicated disruption to oxidative phosphorylation with compound A. (Adapted from the Krebs cycle and Kaufman [1999].) AA, amino acid; ADP, adenosine diphosphate; ATP, adenosine triphosphate; CPT1, carnitine palmitoyltransferase 1; DOPA, dihydroxyphenylalanine; FADH2, reduced form of flavin adenine dinucleotide; GTP, guanine triphosphate; NADH, nicotinamide adenine dinucleotide plus hydrogen.
Figure 4.

Urinary phenylacetylglycine (PAG) from rats dosed with compound A, compound B or control. (a) Top panel shows an example of a 1H NMR spectrum of urine taken from a control rat. The two boxes indicate the aliphatic (left) and aromatic (right) signals from PAG. The latter is shown in more detail in the expansion panel, where 15 spectra at 144 h postdose are shown as a stacked plot. (b) and (c) Relative mean PAG concentrations for compounds A and B respectively, related to time-matched control samples.
Gene expression analysis indicated downregulation of phenylalanine hydroxylase (PAH) at mRNA level, and induction of CPT1a following dosing with both compounds, at all doses (Figure 5). The effect on PAH downregulation was greater with compound A; however, the upregulation of CPT1a was greater with compound B. For compound A, at 48 h, downregulation of PAH correlated with decreased tyrosine levels while compound B at 1000 mg/kg did not change the plasma concentrations of tyrosine.
Figure 5.

(a) Expression levels of phenylalanine hydroxylase (PAH) at 48 h postdose, represented by signal intensity with the M12337_at probe. The five treatment groups each containing five animals are plotted in separate columns. (b) Expression levels of carnitine palmitoyltransferase 1A (CPT1a) for each animal at 48 h postdose, represented by signal intensity with the L07736_at probe.
Discussion
PAG as a biomarker for phospholipidosis in association with mitochondrial toxicity
Diagnosis and screening for phospholipidosis involves examination of peripheral lymphocytes by electron microscopy and biochemical assay of tissue phospholipid content. However, these methods have limited throughput. Fluorescent staining and immunohistochemical approaches are useful in identifying PLD [Casartelli et al. 2003; Ulrich et al. 2009; Xia et al. 1997; Matsuzawa and Hostetler, 1980; Obert et al. 2007] but do not indicate whether PLD might result in associated toxicity. As drug-induced PLD is reversible and is not always associated with toxicity, a noninvasive biomarker that indicates PLD associated with mitochondrial toxicity is highly desirable.
PAG is published as a marker for phospholipidosis [Garcia-Perez et al. 2010; Espina et al. 2001; Robertson et al. 2010] even after single compound administration [Nicholls et al. 2000; Delaney et al. 2004; Hasegawa et al. 2007], but the predictivity as a marker is critically discussed within the expert field and it has not been linked to mitochondrial toxicity. In a model tested to detect phospholipidosis with metabonomics techniques after single compound administration using 20 non-phospholipidogenic and 17 phospholipidogenic compounds, Lienemann and colleagues concluded that the induction of phospholipidosis by the test compounds could not be predicted using NMR-based urine analysis or the previously published biomarker PAG [Lienemann et al. 2008].
The results of our trial investigating uPAG as a biomarker for PLD show that, following dosing with CAD, uPAG increases in a dose-dependent manner in line with a traditional marker for PLD, that is the formation of lamellar bodies in peripheral lymphocytes and in association with organ toxicity. We propose that uPAG (phenylacetylglycine in rodents, phenylacetylglutamine in primates and humans) is a suitable noninvasive biomarker for mitochondrial toxicity associated with drug-induced PLD. Subsequent sections provide the mechanistic hypothesis for considering uPAG as such a biomarker, based on biochemistry considerations.
PAG and phenylketonuria
In addition to drug-induced PLD, an increase in uPAG is seen in patients with phenylketonuria (PKU), a metabolic autosomal recessive disorder. PKU is associated with an increase in ketone bodies, including phenylpyruvate and phenylacetyl CoA, and the latter is conjugated with glutamine to form PAG. PKU is caused by a lack of phenylalanine hydroxylase, which catalyzes the production of tyrosine from phenylalanine [Berg et al. 2006]. The resulting accumulation of phenylalanine is thought to donate amino groups through amino transferase activity and deplete 2-oxoglutarate. The 2-oxoglutarate entity is an intermediate of the TCA cycle and in its absence the cycle cannot function to produce energy aerobically. In patients with PKU, mitochondrial toxicity is associated with abnormal brain development and mental retardation [King, 2012].
The biochemical changes observed in PKU are similar to the pattern of TCA cycle changes following dosing with CAD, that is, an increase in ketone bodies due to anaerobic metabolism which can lead in either situation to the generation of PAG.
Mechanism for mitochondrial dysfunction
Mitochondrial dysfunction may be induced directly or indirectly.
Direct mitochondrial dysfunction
Mechanisms for direct mitochondrial dysfunction potentially include uncoupling of nicotinamide adenine dinucleotide (NAD) – nicotinamide adenine dinucleotide with hydrogen (NADH) via CAD associated with protons on the exterior of mitochondria, passing through the membrane with the bound proton, and dissociating the proton on the interior of the mitochondrion. In the dysfunctional mitochondrion, the electron transport generates no adenosine triphosphate (ATP), since the translocated protons do not return to the interior through ATP synthase. Also when NADH production by the TCA cycle is disrupted, oxidative phosphorylation is affected, which is inferred by an increase in free inorganic phosphate in urine samples since NADH is required to efficiently synthesize ATP from inorganic phosphate and adenosine diphosphate. Mitochondrial uncoupling by a protonophoretic mechanism has been inferred for bupivacaine, an amphiphilic basic amine compound [Sun and Garlid, 1992; Irwin et al. 2002]. It is interesting to note that compound A is also an amphiphilic amine, whereas compound B is a nonbasic amine at physiological pH.
Another potential dysfunction is an increase in the ratio of acetyl CoA to oxaloacetate and a decrease in ATP levels may result in an excess of acetyl CoA and a switch to anaerobic respiration, leading to the production of ketones in the mitochondria.
A decrease in urinary levels of intermediates in the TCA cycle supports the hypothesis that direct mitochondrial toxicity is occurring. Our results show that compound A at higher dose levels induced a decrease in urinary levels of citrate and 2-oxoglutarate. A similar biochemical effect was reported following dosing of Han Wistar rats with CAD, when urinary levels of citrate and 2-oxoglutarate were also decreased [Nicholls et al. 2000].
Indirect mitochondrial dysfunction
Indirect mitochondrial toxicity is indicated by altered transport of fatty acids into mitochondria. Levels of carnitine, required for the transport of fatty acids from the cytosol for the catabolism of lipids, are increased in rat’s urine following dosing with compound A which may indicate a fatty acid oxidation disorder. This disorder may be caused by complex formation between drug and phospholipid, leading to a decrease in phospholipase activity and decreased transport of fatty acid CoA to the mitochondria. Amiodarone is an example of a drug reported to be associated with a mitochondrial effect [Fromenty et al. 1990; Varbiro et al. 2003] and is a powerful inhibitor of phospholipases [Jacobson et al. 1997; Shaikh et al. 1987]. Amiodarone is also a potent inducer of PLD.
The transport of long-chain fatty acids across the mitochondrial membrane involves carnitine, which if not acylated to form acylcarnitine is not metabolized and is excreted in the urine; levels of urinary carnitine were increased in our study. Urine carnitine level increase may also indicate an increased export of short chain fatty acyl out of the mitochondria and the removal of toxic acyl CoA from cells and tissues. A concomitant decrease in TCA cycle intermediates results in anaerobic metabolism, leading to ketone bodies and PAG formation. In addition, CPT1a, which mediates the transport of long-chain fatty acids across the membrane by binding these to carnitine, was upregulated with compound A; however, CPT1a was also upregulated by compound B, thus this effect might not be connected with the toxicity of the compounds with regard to removal of excess fatty acids. Liver histopathological findings were indicative of PLD for compound A but not for compound B.
In addition to disruption of the TCA cycle, these findings suggest that mitochondrial toxicity is also associated with disruption to the urea cycle. The urea cycle is closely linked biochemically with the TCA cycle via α ketoglutarate. Fumarate, malate and 2-oxoglutarate were depleted following dosing with compound A. When the levels of 2-oxoglutarate are decreased this may indicate a disruption of the urea cycle and ammonia elimination; ammonia will then be eliminated via glutamate and PAG. The liver of humans and primates contains an enzyme that conjugates glutamine with phenylacetate (a by product of phenylalanine catabolism) to form phenylacetylglutamine (i.e. the human PAG), which is excreted in urine [James et al. 1972; Moldave and Meister, 1957].
Ketones bodies resulting from increased fatty acid oxidation in the liver may be used as a major fuel instead of glucose. Conversion of free ammonium and α ketoglutarate to glutamate and incorporation of ammonia into glutamate forming glutamine is part of nitrogen balance, taking into account that about 80% of the excreted nitrogen is in the form of urea via urea cycle or Krebs bi-cycle. The short-term regulation of urea cycle occurs principally at mitochondrial carbamoyl phosphate synthetase 1, which is relatively inactive in the absence of N-acetylglutamate generated from acetyl CoA and glutamate. The operation of the TCA and urea cycles is, therefore, dependent upon a common metabolite, that is, acetyl CoA. Acetyl CoA may be oxidized in TCA or used in the synthesis of ketone bodies.
Another link between the urea and TCA cycles is fumarate, an intermediate of the TCA cycle, derived from aspartate, phenylalanine and tyrosine for input to the Krebs cycle. Fumarate released from argininosuccinate is converted into malate and then to oxaloacetate by making use of the TCA cycle enzymes, fumarase and malate dehydrogenase respectively. Oxaloacetate then undergoes transamination with glutamate to regenerate aspartate, which is then reused by the urea cycle [Anand and Anand, 1993].
The formation of phenylacetylglutamine is used in the diagnosis of a number of inborn errors of the urea cycle enzymes that lead to the accumulation of toxic concentrations of ammonia in body fluids. In affected children treated with phenylacetate, nitrogen derived from protein catabolism is excreted in the form of phenylacetylglutamine rather than urea [Brusilow, 1991]. An inborn error in one of the enzymes of the urea cycle affects not only the operation of the urea cycle, but also the TCA cycle by siphoning of the α ketoglutarate by reductive amination [Devlin, 1997]. Increased demand in α ketoglutarate for production of glutamate/PAG is also seen in PKU and in PLD with low TCA intermediates.
In summary, the possible mitochondrial effects caused by similar biochemical changes in PKU and PLD are depicted in Table 5.
Table 5.
Biochemical changes caused by mitochondrial toxicity associated with drug-induced PLD and by phenylketonuria.
| Mitochondrial toxicity associated with drug-induced PLD | PKU | |
|---|---|---|
| PAH | Downregulated at mRNA level for drug A and B | Deficit |
| Blood tyrosine levels | Decreased after high dose of drug
A correlated with PAH down regulation at mRNA level |
Decreased |
| TCA intermediates/ | ||
| Fumarate is a link between TCA and urea cycles | Depletion in e.g. fumarate, malate, 2- oxoglutarate with high dose of drug A | Depletion in fumarate and 2-oxoglutarate |
| Urine | Low pH with drug A (dose dependent) | Ketone bodies |
| PAG | High with drug A (dose dependent) | High |
PAH: phenylalanine hydroxylase; PLD: phospholipidosis; PKU: phenylketonuria; TCA: tricarboxylic acid
Taken together, these implicate PAG as a biomarker for compounds inducing PLD associated with mitochondrial toxicities. It is likely mitochondrial toxicity results from a combined direct and indirect toxicity via impairment of the proton motor force and alteration of fatty acid catabolism. Reduction in TCA intermediates such as fumarate and α ketoglutarate will also impact the urea cycle, hence the elimination of nitrogen.
Conclusion
The risk of mitochondrial toxicity associated with PLD should be minimized during drug development using preclinical screening tools and detected early in clinical trials with CADs. Levels of uPAG increased in a dose-dependent manner following induction of mitochondrial toxicity associated with drug-induced PLD. We stipulate uPAG (phenylacetylglycine in rodents, phenylacetylglutamine in humans) is a suitable non-invasive biomarker to distinguish compounds inducing PLD with or without associated mitochondrial toxicity. Further mechanistic investigations should be performed to address whether organ toxicity associated with PLD-inducing compounds results from indirect and direct mitochondrial toxicity, including TEM examination of mitochondria.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: The authors declare no conflict of interest in preparing this article.
Contributor Information
Lucette Doessegger, Safety Risk Management/Licensing and Early Development, Building 682, Office 235, F. Hoffmann-La Roche AG, CH-4070, Basel, Switzerland.
Georg Schmitt, Non-Clinical Safety, F. Hoffmann-La Roche AG, Basel, Switzerland.
Barbara Lenz, Non-Clinical Safety, F. Hoffmann-La Roche AG, Basel, Switzerland.
Holger Fischer, Non-Clinical Safety, F. Hoffmann-La Roche AG, Basel, Switzerland.
Götz Schlotterbeck, Fachhochschule Nordwestschweiz/Hochschule für Life Sciences, Institut für Chemie und Bioanalytik, Muttenz, Switzerland.
Elke-Astrid Atzpodien, Non-Clinical Safety, F. Hoffmann-La Roche AG, Basel, Switzerland.
Hans Senn, Discovery Technology, F. Hoffmann-La Roche AG, Basel, Switzerland.
Laura Suter, Fachhochschule Nordwestschweiz/Hochschule für Life Sciences, Institut für Chemie und Bioanalytik, Muttenz, Switzerland.
Miklos Csato, Non-Clinical Safety, F. Hoffmann-La Roche AG, Basel, Switzerland.
Stefan Evers, Roche Glycart AG, Schlieren, Switzerland.
Thomas Singer, Non-Clinical Safety, F. Hoffmann-La Roche AG, Basel, Switzerland.
References
- Anand U., Anand C. (1993) The energy cost of urea synthesis. Biochem Educ 21: 198–199 [Google Scholar]
- Berg J., Tymoczko J., Stryer L. (2006) Biochemistry. New York: W.H. Freeman [Google Scholar]
- Brusilow S. (1991) Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatr Res 29: 147–150 [DOI] [PubMed] [Google Scholar]
- Casartelli A., Bonato M., Cristofori P., Crivellente F., Dal Negro G., Masotto I., et al. (2003) A cell-based approach for the early assessment of the phospholipidogenic potential in pharmaceutical research and drug development. Cell Biol Toxicol 19: 161–176 [DOI] [PubMed] [Google Scholar]
- Delaney J., Neville W., Swain A., Miles A., Leonard M., Waterfield C. (2004) Phenylacetylglycine, a putative biomarker of phospholipidosis: its origins and relevance to phospholipid accumulation using amiodarone treated rats as a model. Biomarkers 9: 271–290 [DOI] [PubMed] [Google Scholar]
- Devlin T. (1997) Textbook of biochemistry with clinical correlations. 4th ed. New York: Wiley-Liss [Google Scholar]
- Dieterle F., Ross A., Schlotterbeck G., Senn H. (2006) Metabolite projection analysis for fast identification of metabolites in metabonomics. Application in an amiodarone study. Anal Chem 78: 3551–3561 [DOI] [PubMed] [Google Scholar]
- Drenckhahn D., Kleine L., Lullmann-Rauch R. (1976) Lysosomal alterations in cultured macrophages exposed to anorexigenic and psychotropic drugs. Lab Invest 35: 116–123 [PubMed] [Google Scholar]
- Espina J., John P., Shockcor J., Herron W., Car B., Contel N., et al. (2001) Detection of in vivo biomarkers of phospholipidosis using NMR-based metabonomic approaches. Magn Reson Chem 39: 559–565 [Google Scholar]
- Fischer H., Atzpodien E., Csato M., Doessegger L., Lenz B., Schmitt G., et al. (2012) In silico assay for assessing phospholipidosis potential of small druglike molecules: training, validation, and refinement using several data sets. J Med Chem 55: 126–139 [DOI] [PubMed] [Google Scholar]
- Franken G., Lullmann H., Siegfriedt A. (1970) The occurrence of huge cells in pulmonary alveoli of rats treated by an anorexic drug. Arzneimittelforschung 20: 417. [PubMed] [Google Scholar]
- Fromenty B., Fisch C., Labbe G., Degott C., Deschamps D., Berson A., et al. (1990) Amiodarone inhibits the mitochondrial beta-oxidation of fatty acids and produces microvesicular steatosis of the liver in mice. J Pharmacol Exp Ther 255: 1371–1376 [PubMed] [Google Scholar]
- Garcia-Perez I., Earll M., Angulo S., Barbas C., Legido-Quigley C. (2010) Chemometric analysis of urine fingerprints acquired by liquid chromatography-mass spectrometry and capillary electrophoresis: application to the schistosomiasis mouse model. Electropheresis 31: 2349–2355 [DOI] [PubMed] [Google Scholar]
- Hasegawa M., Takenaka S., Kuwamura M., Yamate J., Tsuyama S. (2007) Urinary metabolic fingerprinting for amiodarone-induced phospholipidosis in rats using FT-ICR MS. Exp Toxicol Pathol 59: 115–120 [DOI] [PubMed] [Google Scholar]
- Hostetler K., Reasor M., Yazaki P. (1985) Chloroquine-induced phospholipid fatty liver. J Biol Chem 260: 215–219 [PubMed] [Google Scholar]
- Irwin W., Fontaine E., Agnolucci L., Penzo D., Betto R., Bortolotto S., et al. (2002) Bupivacaine myotoxicity is mediated by mitochondria. J Biol Chem 277: 12221–12227 [DOI] [PubMed] [Google Scholar]
- Jacobson W., Stewart S., Gresham G., Goddard M. (1997) Effect of amiodarone on the lung shown by polarized light microscopy. Arch Pathol Lab Med 121: 1269–1271 [PubMed] [Google Scholar]
- Jagel M., Lullmann-Rauch R. (1984) Lipidosis-like alterations in cultured macrophages exposed to local anaesthetics. Arch Toxicol 55: 229–232 [DOI] [PubMed] [Google Scholar]
- James M., Smith R., Williams R., Reidenberg M. (1972) The conjugation of phenylacetic acid in man, sub-human primates and some non-primate species. Proc R Soc Lond B Biol Sci 182: 25–35 [DOI] [PubMed] [Google Scholar]
- Jones A. (1982) Some observations of the urinary excretions of glycine conjugates by laboratory animals. Xenobiotica 12: 387–395 [DOI] [PubMed] [Google Scholar]
- Kaitin K. (2008) Obstacles and opportunities in new drug development. Clin Pharmacol Ther 83: 210–212 [DOI] [PubMed] [Google Scholar]
- Kaufman S. (1999) A model of human phenylalanine metabolism in normal subjects and in phenylketonuric patients. Proc Natl Acad Sci U S A 96: 3160–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keun H., Ebbels T., Antti H., Bollard M., Beckonert O., Schlotterbeck G., et al. (2002) Analytical reproducibility in (1)H NMR-based metabonomic urinalysis. Chem Res Toxicol 15: 1380–1386 [DOI] [PubMed] [Google Scholar]
- King W. (2012) Phenylketonuria. Available at: http://themedicalbiochemistrypage.org/pku.php (accessed 14 June 2012).
- Kodavanti U., Mehendale H. (1990) Cationic amphiphilic drugs and phospholipid storage disorder. Pharmacol Rev 42: 327–354 [PubMed] [Google Scholar]
- Kola I., Landis J. (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3: 711–715 [DOI] [PubMed] [Google Scholar]
- Kubo M., Hostetler K. (1985) Mechanism of cationic amphiphilic drug inhibition of purified lysosomal phospholipase A1. Biochemistry 24: 6515–6520 [DOI] [PubMed] [Google Scholar]
- Lee W. (1995) Drug-induced hepatotoxicity N Engl J Med 333: 1118–1127 [DOI] [PubMed] [Google Scholar]
- Lienemann K., Ploetz T., Pestel S. (2008) NMR-based urine analysis in rats: prediction of proximal tubule kidney toxicity and phospholipidosis. J Pharmacol Toxicol Meth 58: 41–49 [DOI] [PubMed] [Google Scholar]
- Lullmann H., Lullmann-Rauch R., Wassermann O. (1978) Lipidosis induced by amphiphilic cationic drugs. Biochem Pharmacol 27: 1103–1108 [DOI] [PubMed] [Google Scholar]
- Lullmann-Rauch R. (1979) Drug-induced lysosomal storage disorders. Front Biol 48: 49–130 [PubMed] [Google Scholar]
- Matsuzawa Y., Hostetler K. (1980) Studies on drug-induced lipidosis: subcellular localization of phospholipid and cholesterol in the liver of rats treated with chloroquine or 4,4′-bis (diethylaminoethoxy)alpha, beta-diethyldiphenylethane. J Lipid Res 21: 202–214 [PubMed] [Google Scholar]
- McCloud C., Beard T., Kacew S., Reasor M. (1995) In vivo and in vitro reversibility of chlorphentermine-induced phospholipidosis in rat alveolar macrophages. Exp Mol Pathol 62: 12–21 [DOI] [PubMed] [Google Scholar]
- Moldave K., Meister A. (1957) Synthesis of phenylacetylglutamine by human tissue. J Biol Chem 229: 463–476 [PubMed] [Google Scholar]
- Nicholls A., Mortishire-Smith R., Nicholson J. (2003) NMR spectroscopic-based metabonomic studies of urinary metabolites in acclimatizing germ-free rats. Chem Res Toxicol 16: 1395–1404 [DOI] [PubMed] [Google Scholar]
- Nicholls A., Nicholson J., Haselden J., Waterfield C. (2000) A metabonomic approach to the investigation of drug-induced phospholipidosis: an NMR spectroscopy and pattern recognition study. Biomarkers 5: 410–423 [DOI] [PubMed] [Google Scholar]
- Obert L., Sobocinski G., Bobrowski W., Metz A., Rolsma M., Altrogge D., et al. (2007) An immunohistochemical approach to differentiate hepatic lipidosis from hepatic phospholipidosis in rats. Toxicol Pathol 35: 728–734 [DOI] [PubMed] [Google Scholar]
- O’Sullivan A., Gibney M., Brennan L. (2011) Dietary intake patterns are reflected in metabolomic profiles: potential role in dietary assessment studies. Am J Clin Nutr 93: 314–321 [DOI] [PubMed] [Google Scholar]
- Pappu A., Hostetler K. (1984) Effect of cationic amphiphilic drugs on the hydrolysis of acidic and neutral phospholipids by liver lysosomal phospholipase A. Biochem Pharmacol 33: 1639–1644 [DOI] [PubMed] [Google Scholar]
- Reasor M. (1981) Drug-induced lipidosis and the alveolar macrophage. Toxicology 20: 1–33 [DOI] [PubMed] [Google Scholar]
- Reasor M. (2010) Drug-Induced Phospholipidosis. FDA Center for Drug Evaluation and Research, Advisory Committee for Pharmaceutical Science and Clinical Pharmacology. Maryland, 14 April 2010: 229–259 [Google Scholar]
- Robertson D., Shipkova P., Aranibar N., Hnatyshyn S., Williams J., Gong L., et al. (2010) Urinary metabonomic assessment of drug-induced phospholipidosis (PLD) in the rat. Presented at 49th Society of Toxicology Annual Meeting, 7–11 March 2010, Salt Lake City, UT [Google Scholar]
- Roessler M., Mantovani-Endl L., Hagmann M., Palme S., Berndt P., Engel A., et al. (2006) Identification of PSME3 as a novel serum tumor marker for colorectal cancer by combining two-dimensional polyacrylamide gel electrophoresis with a strictly mass spectrometry-based approach for data analysis. Mol Cell Proteomics 5: 2092–2101 [DOI] [PubMed] [Google Scholar]
- Ruben Z., Anderson S., Kacew S. (1991) Changes in saccharide and phospholipid content associated with drug storage in cultured rabbit aorta muscle cells. Lab Invest 64: 574–584 [PubMed] [Google Scholar]
- Schlotterbeck G., Ross A., Dieterle F., Senn H. (2006) Metabolic profiling technologies for biomarker discovery in biomedicine and drug development. Pharmacogenomics 7: 1055–1075 [DOI] [PubMed] [Google Scholar]
- Shaikh N., Downar E., Butany J. (1987) Amiodarone – an inhibitor of phospholipase activity: a comparative study of the inhibitory effects of amiodarone, chloroquine and chlorpromazine. Mol Cell Biochem 76: 163–172 [DOI] [PubMed] [Google Scholar]
- Steiner G., Suter L., Boess F., Gasser R., de Vera M., Albertini S., et al. (2004) Discriminating different classes of toxicants by transcript profiling. Environ Health Perspect 112: 1236–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Garlid K. (1992) On the mechanism by which bupivacaine conducts protons across the membranes of mitochondria and liposomes. J Biol Chem 267: 19147–19154 [PubMed] [Google Scholar]
- Ulrich R., Kilgore K., Sun E., Cramer C., Ginsberg L. (2009) An in vitro fluorescent assay for the detection of drug-induced cytoplasmic lamellar bodies. Toxicol Methods 1: 89–105 [Google Scholar]
- Varbiro G., Toth A., Tapodi A., Veres B., Sumegi B., Gallyas F., Jr (2003) Concentration dependent mitochondrial effect of amiodarone. Biochem Pharmacol 65: 1115–1128 [DOI] [PubMed] [Google Scholar]
- Xia Z., Appelkvist E., DePierre J., Nassberger L. (1997) Tricyclic antidepressant-induced lipidosis in human peripheral monocytes in vitro, as well as in a monocyte-derived cell line, as monitored by spectrofluorimetry and flow cytometry after staining with Nile red. Biochem Pharmacol 53: 1521–1532 [DOI] [PubMed] [Google Scholar]