

Abstract
The matrix 1 (M1) protein is a multifunctional protein in the life cycle of influenza virus. It plays an important role in virus budding and intracellular trafficking of viral ribonucleoproteins (vRNPs). The M1 protein consists of three domains based on the structure: N-terminal domain, Middle domain, and C-terminal domain. However, the functions of different domains of the M1 protein remain largely unclear. In this study, using bimolecular fluorescence complementation assays (BIFC) we demonstrated that swine importin α1 interacts with the M1 protein and transports it to the nucleus. Interestingly, M1 with mutated nuclear localization signal (NLS; 101-RKLKR-105 to 101-AALAA-105) still interacts with swine importin α1 and is localized in the nucleus, suggesting that the NLS located at residues 101–105 is not the only NLS within M1 recombinant protein containing 1–160 residues of M1 with mutated nuclear localization signal is able to interact with swine importin α1, but M1/60-252 domains cannot bind importin α1. Further mapping showed that the deletion of residues 1–20 impaired the interaction between N terminus of M1 and importin α1. Collectively, our data suggested that the N-terminal domain of M1 protein is critical for binding swine importin α1 and for nuclear localization.
The influenza virus matrix protein 1 (M1), the most abundant protein in virus particles, plays a critical role in virus morphology, replication, virus assembly and virus budding [1–4]. The M1 layer lies beneath the viral envelope interacting with both surface proteins and viral ribonucleoproteins (vRNPs). Furthermore, there is substantial evidence that the M1 protein is critical for virion morphology [1, 5–8], and that certain residues in the M1 protein are able to determine the ability of the virus to form filamentous or spherical virions. For instance, the combination of residues at positions 41, 95, and 218 of the M1 protein influence the virus filamentous morphology in the background of A/Victoria/3/75 influenza virus [4]. A/WSN/33 virus with its mutations in the six helical domains of the M1 displays a wide variety of morphological phenotypes [9]. Position 24 of the M1 can also influence the morphology of the influenza C virus [10]. Moreover, the M1 protein also plays an important role in virus budding. Although a single M1 is incapable of budding in a plasmid-transfected system [2], the lack of an inherent membrane targeting signal has been reported to be responsible for the failure of the M1 protein to bud into virus-like particles [11]. In the virion, M1 interacts with surface proteins HA, NA, and M2 by cross-linking their cytoplasmic tails, which may mediate recruiting of M1 proteins and vRNPs into the virion [12–14]. Cytoplasmic tail mutations of HA and NA result in a significant reduction of M1 in the virion [15, 16]. The M2 protein cytoplasmic tail has also been shown to interact with the M1 protein and to incorporate the M1 protein during virus assembly [12].
M1 is synthesized in the late stage of infection, and it plays an important role in the nuclear export of newly synthesized vRNPs. M1 proteins are synthesized in the cytoplasm and then actively imported into the nucleus, where M1 binds to vRNPs and the nuclear export protein (NEP). Furthermore, the NEP contains the nuclear export signal that is essential for the transportation of the vRNPs to the cytoplasm [17, 18]. One nuclear localization signal (NLS) was found in the M1 protein, located within AA positions 101–105 (101-RKLKR-105) of M1 that mediates recruit the M1 protein to the nucleus by interacting with importin α from host cells [19]. The M1 protein consists of 252 amino acids that have been divided into three domains: N-terminal domain, Middle domain, and C-terminal domain [20]. The Middle domain was found to mediate the binding to nucleoprotein (NP) and association with vRNPs [21]. The N terminus (2–164 residues) has been analyzed by X-ray crystallography and has been shown to contain nine α-helices linked by eight loops, with four helices formed at the N-terminal, and the other four helices located in the Middle domain of the M1 protein [22]. However, the C-terminal globular domain (165–252) has not yet been characterized by X-ray crystallography. It is predicted that the C-terminal of M1 is disordered [23]. Until now, the functions of the different domains of M1 still remain largely unclear.
Importin α protein is a sub-family of karyopherin proteins, which is involved in the import of cargo proteins into the cell nucleus. Importin α can recognize the NLS of the cargo protein, and transport the protein into the nucleus with help of importin β. Previous studies showed that importin α isoforms pay a critical role in the host range of influenza viruses [24]. Human importin α1 and α7 can up regulate polymerase activity of human- but not avian-like influenza viruses [25]. Swine are important intermediate hosts in the cycle of influenza viruses, and also are considered as a “mixing vessel” of both avian and mammalian influenza viruses. However, how the swine importin α1 interact with influenza virus proteins remains unclear. In this study, the interaction between M1 and importin α1 from swine was assessed by bimolecular fluorescence complementation assays (BIFC). It was found that the N-terminal 1–160 residues of the M1 protein play an important role in interaction with swine importin α1, which binds to classical nuclear localization signal (cNLS)-containing proteins, and facilitates their translocation to the nucleus. Briefly, for the construction of BIFC plasmids, nucleotide sequences encoding the carboxyl [residues 155–238 (VC)] or amino-terminal [residues 1–173 (VN)], fragments of Venus fluorescence protein were fused to the N terminus of M1 of A/California/04/2009 (2009 pandemic H1N1) (designated as VC-M1) or swine importin α1 (designated as VN-IP1). Recombinent fragments were sub-cloned into the eukaryotic expression vector pPRE under the control of both the cytomegalovirus (CMV) immediate-early promoter and the bovine growth hormone polyadenylation signal. Pig kidney cells (PK-15) cultured in 96-well-plate were co-transfected with VC-M1 and VN-IP1 (400 ng each plasmid). An interaction between VC-M1 with VN-IP1 will result in bringing the C- and N-terminal domains of the Venus fluorescence protein together which can be recorded by fluorescence microscopy. Twenty-four hours post transfection, the fluorescence signal was observed and located in the nucleus of the PK-15 cell (Fig. 1a), indicating that swine importin α1 bound to the M1 protein and transferred it to the nucleus. Interestingly, after mutating 101-RKLKR-105 residues (NLS) of M1 to 101-AALAA-105 (VC-M1/nls-mut), the mutant protein still interacted with swine importin α1 and emitted a strong fluorescence signal from the nucleus, as shown in Fig. 1a. This suggests that 101-RKLKR-105 (NLS) are not the only residues responsible for nuclear transport of the M1 protein., i.e., another region of M1 may facilitate the M1 binding to swine importin α1. In summary, our results suggest that swine importin α1 can transport M1 and M1/nls-mut to the nucleus, and that this process can be independent of the previously identified NLS located on position of 101–105 in the M1.
Fig. 1.

Localization patterns of M1 and swine importin α1 proteins by BIFC (a). Relative fluorescence intensity of BIFC (b). VN-IP1 plasmid was transfected into PK-15 cells together with VC-M1 or VC-M1/nls-mut plasmids (400 ng for each plasmid). Individual construct was transfected in PK-15 as negative control. Twenty-four hours post transfection, fluorescence signal was observed under fluorescence microscope. VC-M1 expressed intact M1 protein of pandemic H1N1 virus; VC-M1/nls-mut expressed whole M1 protein containing nuclear localization signal mutation on 101–105 (101-AALAA-105)
To map the regions of the M1/nls-mut protein that are involved in the interactions with swine importin, different truncated M1/nls-mut genes (containing either 1–160 or 60–252 residues) of pandemic H1N1 (pH1N1) virus were cloned into VC vector (designated as VC-M1/1-160/nls-mut or VC-M1/60-252/nls-mut). Each truncated M1 plasmid was co-transfected with VN-IP1 into PK-15 cells. Twenty-four hours post transfection, the fluorescence signals were analyzed. The results showed that only the N-terminal part (1–160 residues) of M1 was critical for interacting with swine importin α1, since signal was emitted by a complex of VC-M1/1-160/nls-mut and VN-IP1 in the nucleus (Fig. 2a, b). In contrast, VC-M1/60-252/nls-mut did not produce a signal, indicating that there was no interaction of this mutant protein with swine importin α1 (Fig. 2a, b).
Fig. 2.

Localization patterns of differently truncated M1/nls-mut and swine importin α1 proteins by BIFC (a). Relative fluorescence intensity of BIFC (b). Western blotting of differently truncated M1/nls-mut in VC vector (c). VN-IP1 plasmid was transfected into PK-15 cells together with different truncated VC-M1/nls-mut plasmids (400 ng each plasmid). Twenty-four hours post transfection, fluorescence signal was observed and measured, and cells were subjected to western blotting. VC-M1/1-160/nls-mut expressed 1–160 amino acids of M1 containing mutation on 101–105 (101-AALAA-105); VC-M1/60-252/nls-mut expressed 60–252 amino acids of M1 containing mutation on 101–105 (101-AALAA-105); VC-M1/20-160/nls-mut and VC-M1/1-140/nls-mut indicated expression of 20–160 or 1–140 amino acids of M1 with mutations on 101–105 (101-AALAA-105), respectively
In view of these experimental results, we assumed that there must be another region in the N terminus of M1 that might be responsible for interacting with importin α1 and nuclear importing. To locate the suspected novel region involved in the interaction with importin α1, two truncated M1/1-160/nls-mut proteins with deletions of residues 1–20 or 140–160 were generated and cloned into the BIFC system. Transfection of the VC-M1/20-160/nls-mut together with VN-IP1 did not produce any fluorescent signal. Also, no signal was observed in cells transfected with VC-M1/1-140/nls-mut and VN-IP1 either (Fig. 2a, b). To determine and quantify protein expression of different truncated M1 proteins cloned into the VC vector, PK-15 cells were transfected by different truncated VC-M1 plasmids, lysed, and subjected to western blotting. The VC vector has a HA tag fused to the carboxyl (residues 155–238 [VC]) fragment of the Venus fluorescence protein. A monoclonal antibody against HA tag (Invitrogen, USA) was used in western blotting. The results showed that VC-M1/1-160/nls-mut, VC-M1/60-252/nls-mut, and VC-M1/20-160/nls-mut express well in transfected PK-15 cells, whereas significantly lower level expression of the VC-M1/1-140/nls-mut protein was observed (Fig. 2c). To further confirm these results, all truncated M1 (M1/1-160/nls-mut, M1/60-252/nls-mut, M1/1-140/nls-mut, and M1/20-160/nls-mut) or the intact M1 protein (M1 and M1/nls-mut) genes were cloned into the PRE-3Flag vector. This allows the expression of genes in mammalian cell lines with the 3Flag tag fused to the N terminus of the expressed proteins. All plasmids were transfected separately into PK-15 cells cultured in 6-well plates (3 μg each plasmid) after verification of correct sequences by sequencing. Forty-eight hours post transfection, PK-15 cells were collected, lysed, and submitted to western blotting to quantify the protein expression levels using the monoclonal mouse anti-flag antibody (Sigma, USA). The western blotting result showed that M1, M1/nls-mut, and M1/1-160/nls-mut were expressed at similar levels in PK-15 cells, suggesting that the RKLKR/AALAA mutation and C-terminal truncation did not affect the expression of the M1 protein (Fig. 3). The M1/60-252/nls-mut and M1/20-160/nls-mut were also expressed in PK-15 cells, although at slightly lower levels when compared to the other M1 proteins. In contrast, no detectable protein expression of the M1/1-140/nls-mut was observed (Fig. 3). Although both VC-M1/20-160/nls-mut and VC-M1/1-160/nls-mut expressed well, only the VC-M1/1-160/nls-mut interacted with VN-IP1, while VC-M1/20-160/nls-mut did not, suggesting that residues 1–20 are critical for the interaction between the N terminus of M1 and importin α1 protein.
Fig. 3.
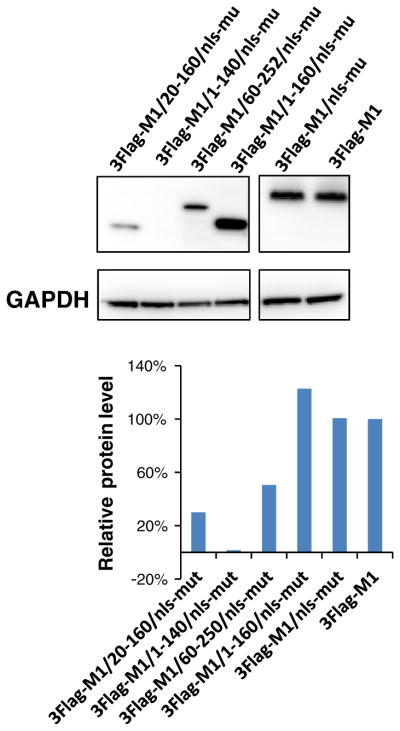
Western blotting. Transfected PK-15 cells were harvested and lysed at 48 h after transfection and analyzed by Western blotting using monoclonal antibodies against Flag. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. Relative protein amounts were calculated and normalized using ratios to GAPDH
Although M1/60-252/nls-mut and M1/20-160/nls-mut did not interact with swine importin α1, the localization of those proteins in cells was unclear. Immunofluorescence Assay (IFA) was performed on the PK-15 cells transfected with 3Flag-M1/1-160/nls-mut, 3Flag-M1/60-252/nls-mut, 3Flag-M1/20-160/nls-mut and 3Flag-M1/1-140/nls-mut plasmids, respectively. The results showed all truncated M1 expressed well except M1/1-140/nls-mut (Fig. 4). The M1/1-160/nls-mut was found in both nucleus and cytoplasm. In contrast, the M1/60-252/nls-mut only located in the cytoplasm (Fig. 4), which confirmed that the importin α1 can bind and transfer M1/1-160/nls-mut to the nucleus, but not M1/60-252/nls-mut. Surprisingly, the M1/20-160/nls-mut protein was observed in both nucleus and cytoplasm, suggesting that M1/20-160/nls-mut protein might be present in the nucleus either by passive diffusion or through interaction with other importin α proteins of the swine importin α family.
Fig. 4.

Immunofluorescence assay. PK-15 cells were transfected by 3Flag-M1/60-250/nls-mut, 3Flag-M1/1-160/nls-mut, 3Flag-M1/1-140/nls-mut, and 3Flag-M1/20-160/nls-mut, respectively (400 ng per plasmid). Twenty-four hours post transfection, IFA was conducted by a monoclonal mouse anti-flag antibody (Sigma, USA)
Both IFA and western blotting results suggest that residues 140–160 play an important role in maintaining the stability or biological half-life of the N-terminal proportion of the M1 protein (Figs. 3, 4). Based on the crystal structure of the N-terminal region of the M1 protein (Fig. 5), residues 89–160 form a four-helix bundle subdomain. The helix formed by 140–160 residues is very close to the helix formed by 1–20 residues. Thus, both helixes may interact with each other that in turn stabilizes the functional structure of the M1 protein. This hypothetical model is based on the observation that the deletion of 140–160 residues resulted in the destruction of the N-terminal domain structure of the M1 protein (Fig. 5). In the virion, the M1 protein interacts with both the surface proteins (HA, NA, and M2) and with RNPs [26, 27] and also supports the virus shape, which requires a stable structure for the M1 protein. Cryo-electron tomography study also showed that M1 formed a helical net under the viral membrane, which provided structural support for virus morphology [9].
Fig. 5.

Structure of dimeric N-terminal domain (2–158 AA) of the M1 protein. Crystal Structure of the N-terminal domain (2–158 AA) of M1 protein is from influenza A virus (PDB = 3MD2). The structure was constructed using Pymol and the surface is shown as transparent. Residues of 2–20 are shown in red; Residues of 140–158 are shown in cyan. Numbers in parenthesis indicate the residue based on starting methionine of the M1 protein (Color figure online)
In the life cycle of influenza virus, M1 displays bidirectional traffic through the nuclear envelope. In the late stage of infection, the M1 protein is actively transported to the nucleus, then nuclear M1 associated with vRNPs is exported to the cytoplasm by interacting with the NEP protein. Furthermore, recent work has identified a leucine-rich nuclear export signal (NES) in the M1 protein [28]. Mutations in NES of M1 caused nuclear retention of the protein and impaired the efficiency of the nuclear export of vRNPs, thereby affecting the virus replication, indicating nuclear export without NEP. Since a classical NLS exists in the M1 protein, precisely how the M1 protein associats with vRNPs and prevents the re-importation into the nucleus remains unknown. One possibility is that interaction with NEP or vRNPs may block the domain of the M1 protein that is recognized by importin α1. It has also been reported that NEP binds a N-terminal domain between residues 89 and 164 of the M1 protein [17].
In this study, we have shown that the N terminus (especially residues 1–20) of M1 is critical for interacting with swine importin α1. The previously identified classical NLS located at residues 101–105 is not the only NLS within the N terminus of M1 protein. Our results suggested that a novel and functional NLS exists in M1’s N-terminal domain.
Acknowledgments
We thank Robert Kahn for proof-reading of this manuscript. This project was funded by the CEIRS program of the National Institute of Allergy and Infectious Disease, National Institute of Health under contract numbers HHSN266200700005C.
Contributor Information
Qinfang Liu, Department of Diagnostic Medicine/Pathobiology, Kansas State University, Manhattan, KS, USA.
Bhupinder Bawa, Department of Diagnostic Medicine/Pathobiology, Kansas State University, Manhattan, KS, USA.
Jingjiao Ma, Department of Diagnostic Medicine/Pathobiology, Kansas State University, Manhattan, KS, USA.
Feng Li, Department of Biology and Microbiology, South Dakota State University, Brookings, SD, USA.
Wenjun Ma, Department of Diagnostic Medicine/Pathobiology, Kansas State University, Manhattan, KS, USA.
Jürgen A. Richt, Email: jricht@vet.k-state.edu, Department of Diagnostic Medicine/Pathobiology, Kansas State University, Manhattan, KS, USA
References
- 1.Roberts PC, Lamb RA, Compans RW. Virology. 1998;240(1):127–137. doi: 10.1006/viro.1997.8916. [DOI] [PubMed] [Google Scholar]
- 2.Rossman JS, Lamb RA. Virology. 2011;411(2):229–236. doi: 10.1016/j.virol.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hui EK, Yap EM, An DS, Chen IS, Nayak DP. J Gen Virol. 2004;85(Pt 7):1877–1884. doi: 10.1099/vir.0.79906-0. [DOI] [PubMed] [Google Scholar]
- 4.Elleman CJ, Barclay WS. Virology. 2004;321(1):144–153. doi: 10.1016/j.virol.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Smirnov Yu A, Kuznetsova MA, Kaverin NV. Arch Virol. 1991;118(3–4):279–284. doi: 10.1007/BF01314038. [DOI] [PubMed] [Google Scholar]
- 6.Bourmakina SV, Garcia-Sastre A. J Gen Virol. 2003;84(Pt 3):517–527. doi: 10.1099/vir.0.18803-0. [DOI] [PubMed] [Google Scholar]
- 7.Muraki Y, Murata T, Takashita E, Matsuzaki Y, Sugawara K, Hongo S. J Virol. 2007;81(16):8766–8773. doi: 10.1128/JVI.00075-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calder LJ, Wasilewski S, Berriman JA, Rosenthal PB. Proc Natl Acad Sci USA. 2010;107(23):10685–10690. doi: 10.1073/pnas.1002123107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burleigh LM, Calder LJ, Skehel JJ, Steinhauer DA. J Virol. 2005;79(2):1262–1270. doi: 10.1128/JVI.79.2.1262-1270.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muraki Y, Washioka H, Sugawara K, Matsuzaki Y, Takashita E, Hongo S. J Gen Virol. 2004;85(Pt 7):1885–1893. doi: 10.1099/vir.0.79937-0. [DOI] [PubMed] [Google Scholar]
- 11.Wang D, Harmon A, Jin J, et al. J Virol. 2010;84(9):4673–4681. doi: 10.1128/JVI.02306-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen BJ, Leser GP, Jackson D, Lamb RA. J Virol. 2008;82(20):10059–10070. doi: 10.1128/JVI.01184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen BJ, Takeda M, Lamb RA. J Virol. 2005;79(21):13673–13684. doi: 10.1128/JVI.79.21.13673-13684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ksenofontov AL, Fedorova NV, Badun GA, et al. Mol Biol (Mosk) 1999;33(5):881–886. [PubMed] [Google Scholar]
- 15.Jin H, Leser GP, Zhang J, Lamb RA. EMBO J. 1997;16(6):1236–1247. doi: 10.1093/emboj/16.6.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Leser GP, Pekosz A, Lamb RA. Virology. 2000;269(2):325–334. doi: 10.1006/viro.2000.0228. [DOI] [PubMed] [Google Scholar]
- 17.Akarsu H, Burmeister WP, Petosa C, et al. EMBO J. 2003;22(18):4646–4655. doi: 10.1093/emboj/cdg449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimizu T, Takizawa N, Watanabe K, Nagata K, Kobayashi N. FEBS Lett. 2010;585(1):41–46. doi: 10.1016/j.febslet.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 19.Ye Z, Robinson D, Wagner RR. J Virol. 1995;69(3):1964–1970. doi: 10.1128/jvi.69.3.1964-1970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh M, Hotta H. Nihon Rinsho. 1997;55(10):2581–2586. [PubMed] [Google Scholar]
- 21.Noton SL, Medcalf E, Fisher D, Mullin AE, Elton D, Digard P. J Gen Virol. 2007;88(Pt 8):2280–2290. doi: 10.1099/vir.0.82809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sha B, Luo M. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 4):458–460. doi: 10.1107/S0907444997000152. [DOI] [PubMed] [Google Scholar]
- 23.Ksenofontov AL, Dobrov EN, Fedorova NV, et al. Mol Biol (Mosk) 2011;45(4):689–696. [PubMed] [Google Scholar]
- 24.Gabriel G, Klingel K, Otte A, et al. Nat Commun. 2011;2:156. doi: 10.1038/ncomms1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hudjetz B, Gabriel G. PLoS Pathog. 2012;8(1):e1002488. doi: 10.1371/journal.ppat.1002488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bui M, Wills EG, Helenius A, Whittaker GR. J Virol. 2000;74(4):1781–1786. doi: 10.1128/jvi.74.4.1781-1786.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Lamb RA. Virology. 1996;225(2):255–266. doi: 10.1006/viro.1996.0599. [DOI] [PubMed] [Google Scholar]
- 28.Cao S, Liu X, Yu M, et al. J Virol. 2012;86(9):4883–4891. doi: 10.1128/JVI.06586-11. [DOI] [PMC free article] [PubMed] [Google Scholar]