

Abstract
The Biopharmaceutics Classification System (BCS) has found widespread utility in drug discovery, product development and drug product regulatory sciences. The classification scheme captures the two most significant factors influencing oral drug absorption; solubility and intestinal permeability and it has proven to be a very useful and a widely accepted starting point for drug product development and drug product regulation. The mechanistic base of the BCS approach has, no doubt, contributed to its wide spread acceptance and utility. Nevertheless, underneath the simplicity of BCS are many detailed complexities, both in vitro and in vivo which must be evaluated and investigated for any given drug and drug product. In this manuscript we propose a simple extension of the BCS classes to include subspecification of acid (a), base (b) and neutral (c) for classes II and IV. Sub-classification for Classes I and III (high solubility drugs as currently defined) is generally not needed except perhaps in border line solubility cases. It is well known that the , pKa physical property of a drug (API) has a significant impact on the aqueous solubility dissolution of drug from the drug product both in vitro and in vivo for BCS Class II and IV acids and bases, and is the basis, we propose for a sub-classification extension of the original BCS classification.
This BCS sub-classification is particularly important for in vivo predictive dissolution methodology development due to the complex and variable in vivo environment in the gastrointestinal tract, with its changing pH, buffer capacity, luminal volume, surfactant luminal conditions, permeability profile along the gastrointestinal tract and variable transit and fasted and fed states. We believe this sub-classification is a step toward developing a more science-based mechanistic in vivo predictive dissolution (IPD) methodology. Such a dissolution methodology can be used by development scientists to assess the likelihood of a formulation and dosage form functioning as desired in humans, can be optimized along with parallel human pharmacokinetic studies to set a dissolution methodology for Quality by Design (QbD) and in vitro–in vivo correlations (IVIVC) and ultimately can be used as a basis for a dissolution standard that will ensure continued in vivo product performance.
Keywords: BCS Class II, Sub-classification, In vivo predictive dissolution, Simulation, GastroPlus™
1. Introduction
The Biopharmaceutics Classification System (BCS) and the corresponding guidance issued by the FDA categorize drug substances into 4 groups based on aqueous solubility and intestinal membrane permeability (Amidon et al., 1995). The FDA BCS guidance allows biowaivers for BCS Class I drugs based on drug product in vitro dissolution for immediate-release (IR) solid oral dosage forms (CDER/FDA, 2000). The criterion for high solubility, high permeability and product dissolution is likely conservative relative to the current in vivo bioequivalence (BE) (80–125%) standard for BCS Class I drugs. However, as noted by Butler (Butler and Dress-man, 2010) it does not capture the most significant physicochemical differences that are critical to dosage form design and performance for BCS Class II and IV drugs/drug products. Drugs and drug products within BCS Class II, for example, low solubility and high permeable drugs, would be absorbed completely, if in solution. Thus in vivo dissolution is the critical determinant of in vivo absorption, bioavailability (BA) and the subsequent bio-equivalence (BE) determinations. Clearly the in vitro dissolution methodology needs further development, relative to the in vivo conditions, in order to be predictive of in vivo oral performance.
BCS Class II and IV drug product dissolution in vivo and in vitro is highly dependent on the acidic or basic nature of the drug, the drug solubility and formulation factors, in addition to the in vivo luminal environment. While these factors can be complex, in this manuscript we propose a BCS Class II and IV sub-classification with a, b, and c subclasses dependent on the acidic (a), basic (b), or neutral (c) characteristics of the drug in the physiological pH range (~pH < 7.5). Further, we propose developing a corresponding drug product in vivo predictive dissolution (IPD) methodology, based on these classes and subclasses that will be predictive of in vivo performance.
In the WHO and European Medicines Agency BE standards, as well as recommendations from several scientific workshops, certain BCS Class II drugs (e.g. BCS Class IIa, see below) have even been recommended as, or are candidates for a biowaiver. BCS Class IIa drugs, typically carboxylic acids with a pKa in the range of 4 to 5, are insoluble at typical, fasted, gastric pHs but soluble at intestinal pHs and, hence, are classified as BCS Class II or IV depending on intestinal jejunal permeability at pH = 6.5 or fraction dose absorbed determination in humans. With intestinal pH of ~6.5, the solubility and hence, likely the dissolution rate, of the acidic drugs (pKa ~ 4–5) would increase up to ~100 fold or more on entering the small intestine. Thus dissolution would likely be significantly faster than the average gastric emptying rate (half-time ~15 min) depending on dose and intrinsic solubility. Thus, as with BCS Class I drug products with rapid dissolution, the plasma levels for a BCS Class IIa drug would likely reflect gastric emptying and luminal pH differences, and not product differences. The objectives of this investigation are to use simulation methods to illustrate the oral absorption profiles of BCS Class II drugs. To illustrate the utility of this sub-classification we have chosen characteristic parameters based on the following example drugs: for Class IIa ibuprofen and ketoprofen (weak acids), for IIb; carvedilol and ketoconazole (weak bases) and for IIc; fenofibrate and danazol (neutral) to illustrate the general features of the absorption profiles to be expected from the proposed BCS sub-classes.
It has been reported that many BCS Class II drugs such as flurbiprofen (Davies, 1995), naproxen (Davies and Anderson, 1997; Faassen and Vromans, 2004), ketoprofen (Faassen and Vromans, 2004), rifampicin (Agrawal and Panchagnula, 2005; Becker et al., 2009; Panchagnula and Agrawal, 2004), and carbamazepine (Kovacevic et al., 2009; Wilson et al., 2005) exhibit good oral absorption, even though they are poorly soluble at pH conditions. Some BCS Class IIa weak acids, particularly the small molecule nonsteroidal anti-inflammatory drugs (NSAIDs), are reported to dissolve quickly and behave like BCS Class I drugs in the small intestine, even though they exhibit low solubility at acidic gastric pH (Yazdanian et al., 2004). They are thus classified as low solubility BCS Class II drugs since the FDA BCS classification uses the lowest solubility over the physiological pH range (pH = 1–7.5) as the reference classification solubility. Alternatively BCS Class IIb weak bases exhibit high solubility and (likely) dissolution rates at acidic pH in the stomach and may precipitate (in a very complex poorly understood manner) in the small intestine (Tubic-Grozdanis et al., 2008). The intestinal precipitation of a Class IIb drug will depend on numerous formulation and GI physiological factors (luminal composition) and environment at the time of dosing. For example, excipients may alter this precipitation (and thus solution concentration e.g. super saturation) process and further complicate the in vivo prediction. While BCS Class II weak acids and bases demonstrate complimentary solubility profiles due to the pH variation of the GI tract, the solubility of BCS Class IIc drugs would not be affected by this in vivo pH change. However, for BCS Class IIc drug products, the in vivo environment e.g. surfactants and lipids, play a significant but difficult to predict, role in drug dissolution. This solubilization can be estimated with more physiologically based media (Vertzoni et al., 2010) though these media may only partially represent the variation in the actual complex and time dependent in vivo processes. Thus, the BCS Class II drugs have divergent biopharmaceutical properties that have an enormous impact on their formulation, gastrointestinal solubility, dissolution rate and hence in vivo oral absorption and bioavailability profiles. Thus a BCS sub-classification based on these biopharmaceutical characteristics will aid in setting a predictive dissolution methodology, which we term an in vivo predictive dissolution (IPD) methodology, that will provide the best assurance of in vivo performance i.e. in vivo BE.
In this study we illustrate the oral absorption profiles of BCS Class II drugs using the simulation software GastroPlus™, and support this sub-classification of BCS Class II drugs into: weak acid (a), weak base (b), (drugs within the physiological pH range), and neutral (c). To illustrate this sub-classification we use; ibuprofen and ketoprofen (weakly acidic, pKa 4.9 and pKa 4.5), carvedilol and ketoconazole (weakly basic, pKa 7.8 and pKa 2.9/6.5) and fenofibrate and danazol (neutral) as model drugs. The oral drug absorption, the maximum concentration (Cmax), the time after administration of a drug when the maximum concentration is reached (Tmax), area under the concentration curve (AUC0-inf), fraction dose absorbed (Fa %), confidence intervals (CI) and absorption profiles are predicted in silico based on incorporating the effects of hypothetical in vivo dissolution kinetics. Additional significant factors complicating systemic availability prediction (Fsys), such as nonlinear, carrier mediated intestinal or hepatic absorption and secretion and, intestinal and hepatic metabolism are not considered. Further, factors influencing the detailed dissolution processes, in vitro and in vivo e.g. solubilization, buffer and buffer capacity are not considered in detail. In this report we focus on absorption and empirical (simulated) in vivo dissolution, and the resulting expected dissolution and absorption profiles based on this BCS sub-classification. We expect this BCS sub-classification to be a useful base for establishing mechanistic dissolution methodologies for in vitro–in vivo correlations (IVIVC) and Quality by Design (QbD) performance measures (Dickinson et al., 2008; Takano et al., 2012; Yu, 2008).
2. Methods
Drug dissolution and fraction absorbed from the gastrointestinal tract were predicted using physicochemical and pharmacokinetic properties approximating those of the selected drugs and input into GastroPlus™ (version 7.0). First-pass metabolism and systemic availability were not considered at this stage. Such considerations are essential for optimal product development and systemic availability prediction. Our goal in this study is to illustrate the general expected oral absorption curves for BCS II subclasses simulated using approximate ‘real’ drug properties. We did not attempt to provide a detailed absorption and systemic availability analysis of the chosen drugs. Virtual trials with a 250-ml dose volume for an immediate release (IR) dosage form were performed to predict the oral absorption (Cmax, AUC0-inf, and Fa %) of 6 BCS Class II drugs, two in each BCS subclass, with the consequently varying dissolution in the GI tract. The predictions of oral absorption for BCS Class II drugs revealed absorption patterns, as expected, that depend on their biopharmaceutical (physicochemical) properties. The results demonstrate that the dissolution profiles of BCS Class II drugs along with pH changes in the GI tract would have intestinal absorption patterns and dissolution rates, which will vary depending on the luminal environment and drug product composition. This would control the overall rate of oral absorption for BCS Class II drugs, though systemic availability may be further reduced and complicated by intestinal or hepatic first-pass metabolism and excretion (including subsequent reabsorption). These BCS Class II absorption patterns are clearly shown to be dependent on the sub-classification, as expected. We believe the proposed BCS sub-classification can be used to set improved predictive dissolution methodology and standards for BCS Class II drug products and will provide better assurance of in vivo bioperformance.
2.1. Computer hardware and software
GastroPlus™ version 7.0 (SimulationPlus, Inc., Lancaster, CA) was run using an IBM computer with Intel Core™ 2 Duo processors. The computational software allows the input of various dissolution profiles and computes the corresponding pharmacokinetic profiles and parameters.
2.2. Simulation design
Oral drug absorption was computed based on (approximating) the physicochemical, pharmacokinetic, and drug dissolution properties of the six selected BCS Class II drugs using the previously reported simulation method (Tsume and Amidon, 2010). The gastric emptying time (15 min) as a mean value was adopted and the mean with coefficients of variation was used for these virtual trials. Since the AUC0-inf of drugs is calculated by extrapolation of the last two data points by GastroPlus™, the virtual trial over 24 h resulted in some variation in AUC0-inf for longer half-life drugs. These virtual trials were performed for 48 h. The variations in population physiology were defined as means with coefficients of variation (in log space) and individual trials were randomly selected within those ranges. A virtual trial with n = 500 was used as the IR reference for each drug product (dissolution profile).
2.3. Input parameters for pharmacokinetic simulations
The physicochemical and biopharmaceutical properties of ibuprofen, ketoprofen, carvedilol, ketoconazole, danazol, and fenofibrate used in the GastroPlus™ simulations, as well as the chemical, physiological, and pharmacological parameters for drugs cited in the literature are presented in Table 1 (Abernethy and Greenblatt, 1985; Avdeef et al., 1998; Baxter et al., 1986; Beetge et al., 2000; Cordero et al., 1997; Domanska et al., 2009; Dressman and Lennernäs, 2000; Fei et al., 2013; Geisslinger et al., 1995; GlaxoSmithKline, 2009; Hooper et al., 1991; Huang et al., 1986; Ige et al., 2013; Kasim et al., 2004; Keating and Croom, 2007; Loftsson et al., 2008; Mannisto et al., 1982; Oliary et al., 1992; Peeters et al., 2008; Perez de la Cruz Moreno et al., 2006; Planinsek et al., 2011; Prajapati et al., 2012; Vertzoni et al., 2010; Yun et al., 2006). Since the size of drug particles can significantly affect drug dissolution rate, the relatively larger particle size, a radius of 25 μm, to minimize the particle effects for dissolution was set as the mean particle size with a coefficient of variation (Le et al., 2006; Lin et al., 1982; Mutalik et al., 2008; Sheng et al., 2008; Takano et al., 2008; Tsume and Amidon, 2010). It is hard to predict the mean precipitation time because it depends on the formulation and physicochemical properties of drug compound. The range of precipitation time, 15–75 min, has been calculated from the various experiments (Carlert et al., 2010; Kambayashi and Dressman, 2013; Lindfors et al., 2008). The mean precipitation time of 900 s (15 min) was chosen and was used it with a coefficient of variation for virtual trials. Virtual trials were performed using the GastroPlus™ standard physiological conditions: Human Physiological-Fasted, which condition simplifies the drug solubility in this set of simulation because of minimal effects of bile components on drug dissolution, and Opt LogD Model SA/V 6.1. In those virtual trials, dose, dose volume, molecular weight, logP, pKa, particle density, and diffusion coefficient of drug compounds were fixed. All other parameters for virtual trials such as effective human permeability, intestinal transit time and pharmacokinetic clearance were defined as variables, which were randomly selected within a 5–20% (coefficient of variation) log–normal distribution based on their mean values. The physiological compartment parameters were adjusted such that the BCS Class II drugs would be absorbed in the entire small and large intestine with no absorption in the stomach.
Table 1.
Chemical/physiological/pharmacological parameters of BCS Class II drugs for GastroPlus™ simulation.
| Ibuprofen Acid |
Ketoprofen Acid |
Carvedilol Base |
Ketoconazole Base |
Fenofibrate Neutral |
Danazol Neutral |
||
|---|---|---|---|---|---|---|---|
| MW | 206.3 | 254.3 | 406.5 | 531.4 | 360.8 | 337.5 | |
| Dose | mg | 800a | 50f | 25j | 200m | 100r | 100v |
| Dose number | 1792A | 197A | 835A | 61A | 1429A | 23A | |
| Dose volume | ml | 250 | 250 | 250 | 250 | 250 | 250 |
| Solubility | mg/ml | 9.0 × 10−2b | 5.1 × 10−2g | 5.8 × 10−4k | 1.8 × 10−2n | 2.8 × 10−4s | 1.8 × 10−2w |
| logP | 3.6c | 2.8g | 3.8l | 4.3o | 5.3t | 4.5x | |
| pKa | 4.9d | 4.5h | 7.8l | 2.9/6.5p | NA | NA | |
| Mean precipitation time | s | 900 | 900 | 900 | 900 | 900 | 900 |
| Human Peff | ×10−4 cm2/s | 4.10B | 8.70i | 4.04B | 1.37B | 2.83B | 1.21B |
| Body weight | kg | 70 | 70 | 70 | 70 | 70 | 70 |
| Vc | l/kg | 0.2e | 0.4f | 1.6j | 0.7q | 0.4u | 118.8v |
| Total clearance | l/h/kg | 0.10a | 0.06f | 0.51j | 0.38m | 0.02r | 9.90v |
Vc; Volume of Central Compartment.
Calculated by GastroPlus™ 7.0.
Calculated by ADMET Predictor.
3. Results
The oral absorption of the BCS Class II reference drug product was predicted using GastroPlus™ virtual trials. The mean (reference) Cmax, AUC0-inf and Fa % ± standard deviation (SD), were obtained with an n = 500 virtual trial for an IR dosage.
3.1. BCS Class IIa drugs; pKa ≤ 5 (weak acid drugs; ibuprofen and ketoprofen)
The predicted oral absorption results, mean Cmax, AUC0-inf and Fa %, for ibuprofen and ketoprofen are shown in Table 1. The percentages of dissolved and absorbed ibuprofen and ketoprofen with an IR dosage were calculated by a GastroPlus™ simulation and plotted as a function of time for the first 10 h (Fig. 1a and b). Ibuprofen and ketoprofen exhibited similar Tmax (ibuprofen 4.6 h and ketoprofen 4.0 h) and Fa % values, and also exhibited nearly complete oral absorption (97.4% and 97.1%), respectively (Table 2). The percentages of dissolved and absorbed ibuprofen and ketoprofen with an IR dosage were also calculated and plotted as a function of time for the first 10 h in Figs. 1 and 2. Fig. 2 illustrates that the nearly complete dissolution of ibuprofen and ketoprofen is predicted to occur by 8 h after oral administration. The percentages of absorbed ibuprofen and ketoprofen were divided by the average transit time of each GI segment and the results were plotted as a function of those segments (Fig. 3). In Fig. 3, ibuprofen and ketoprofen are predicted to be absorbed more in the distal small intestine than in the proximal small intestine due to the environmental pH in the GI tract and their pH-dependent solubility/dissolution. The absorption pattern of ibuprofen suggests a higher fraction absorbed in the ileum than the jejunum in these simulations.
Fig. 1.

Amount of BCS Class II drugs dissolved and absorbed calculated in silico: (a) ibuprofen, (b) ketoprofen, (c) carvedilol, (d) ketoconazole, (e) fenofibrate, (f) danazol. Solid lines represent dissolved drugs and dot lines represent absorbed drugs.
Table 2.
Simulated Cmax, AUC, and Fa of selected BCS Class II drugs with an IR dosage.
| Cmax(×10−2 μg/ml) | AUC(×10−1 ng/ml h) | Predicted Fa (%) |
|
|---|---|---|---|
| Ibuprofen | 1582.0 ± 160.2 | 1159.3 ± 51.7 | 97.4 ± 1.2 |
| Ketoprofen | 102.7 ± 13.9 | 111.1 ± 8.3 | 97.1 ± 2.1 |
| Carvedilol | 2.6 ± 0.5 | 2.3 ± 0.4 | 45.9 ± 1.0 |
| Ketoconazole | 44.6 ± 8.9 | 63.0 ± 4.0 | 47.0 ± 6.9 |
| Fenofibrate | 93.3 ± 17.4 | 433.9 ± 80.2 | 47.8 ± 6.7 |
| Danazol | 3.5 ± 0.7 | 42.0 ± 1.2 | 36.4 ± 4.3 |
Data reported as mean ± SD.
Fig. 2.
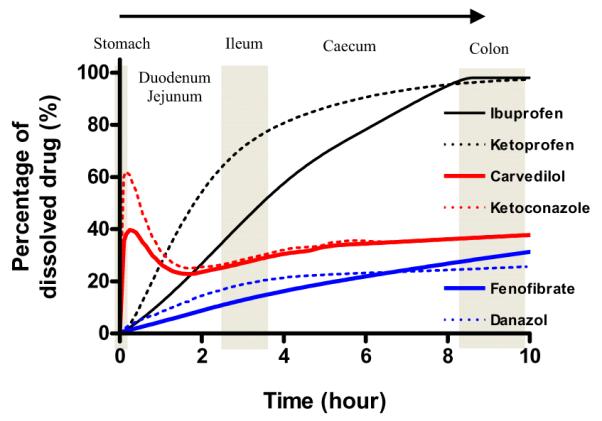
Percentage of amount dissolved with an IR dosage. Black solid and dot lines represent BCS Class II weak acids, Red solid and dot lines represent BCS class weak bases and blue solid and dot lines represent BCS class neutrals. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.
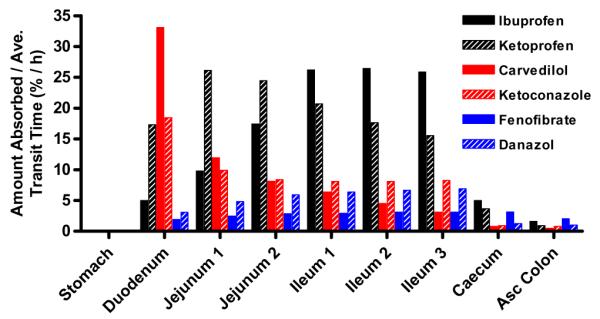
The absorption rates of BCS Class II drugs in each GI segment. Percentages of amount absorbed after oral administration of an IR dosage are divided by the average transit time and are plotted as a function of each GI segment. Black bars represent BCS Class II weak acids, Red bars represent BCS class weak bases and blue bars represent BCS class neutrals. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. BCS Class IIb drugs; pKa ≥ 6 (weak base drugs; carvedilol and ketoconazole)
The prediction results of mean Cmax, AUC0-inf, and Fa % ± SD for carvedilol and ketoconazole are shown in Table 2. Carvedilol and ketoconazole exhibited predicted oral absorption of 45.9% and 47.0%, respectively (Table 2). The percentages of dissolved and absorbed carvedilol and ketoconazole with an IR dosage were calculated by a GastroPlus™ simulation and plotted as a function of time for the first 10 h (Fig. 1c and d). The percentages of absorbed carvedilol and ketoconazole were divided by the average transit time of each GI segment and plotted as a function of those segments (Fig. 3). Figs. 1 and 2 illustrate the quick dissolution of carvedilol and ketoconazole due to the acidic pH in stomach. The precipitation of carvedilol and ketoconazole was calculated to be 30–60 min after oral administration. Thus, the majority of the absorption for carvedilol and ketoconazole (68.8–77.5%) was predicted at the lower pH environment in the duodenum and proximal jejunum (jejunum 1) due to their higher solubility and their high permeability (Fig. 3) in this region of the intestine. Contrary to weak acids, carvedilol and ketoconazole were absorbed more in the proximal small intestine than in the distal small intestine. Particularly, the absorption pattern of carvedilol clearly revealed more absorption at the duodenum and proximal jejunum than at the ileum.
3.3. BCS Class IIc drugs; No pKa (neutral drugs; fenofibrate and danazol)
The prediction results for the oral absorption of fenofibrate and danazol are shown in Table 2. Fenofibrate and danazol exhibited oral absorption of 47.8% and 36.4%, respectively (Table 2). The percentages of dissolved and absorbed drug with an IR dosage were calculated by GastroPlus™ simulation and are plotted as a function of time for the first 10 h (Fig. 1e and f). The percentages of absorbed fenofibrate and danazol were divided by the average transit time of each GI segment and those results were plotted as a function of those segments (Fig. 3). Fig. 1e and f illustrate that the slow dissolution rate/solubility of fenofibrate and danazol is the rate-limiting step for its oral absorption and exhibited a relatively constant dissolution profile throughout the intestine in these simulations. These observations are due to the independence of drug solubility on gastrointestinal pH in these simulations. Both, fenofibrate and danazol were absorbed from entire small and large intestines independent of the environmental pH in GI tract (Fig. 3).
4. Discussion
The Biopharmaceutics Classification System (BCS) is a system classifying a drug substance (API) based on its minimum aqueous solubility in the pH range of 1–7.5, dose and human fraction absorbed or intestinal membrane permeability (CDER, 2000). This system categorizes drugs into four classes according to their permeability and solubility (CDER, 2000). It has been suggested that the regulatory criterion for a high soluble drug, whose highest dose (approved) strength is soluble in 250 ml aqueous media over the pH range of 1.0–7.5(6.8), is conservative for BCS Class I drugs and that further biowaivers for acidic drugs, BCS Class IIa, should be considered (Rinaki et al., 2004; Yazdanian et al., 2004). In this report we suggest a further BCS sub-classification based on drug biopharmaceutical properties that can be used as a beginning basis for setting an in vivo predictive dissolution (IPD) methodology.
It has been reported that some BCS Class II compounds, which have particular biopharmaceutical characteristics, such as weak acids, particularly the high permeability, low molecular weight non-steroidal anti-inflammatory drugs (NSAIDs), are rapidly and completely absorbed orally (Davies, 1995; Davies and Anderson, 1997; Faassen and Vromans, 2004). The scientific basis for this is that poorly soluble weak acids with pKa in the physiological range i.e. pKa values ≤5.0, such as ibuprofen and naproxen would have a solubility of ≥1 mg/ml at around pH 6.5, (average pH value of the fasted state in the jejunum), resulting in rapid (less than 0.5 h) and reliable dissolution (Dressman et al., 2001). On the other hand, poorly soluble weak bases with pKa values in the physiological range of 3–6 such as ketoconazole and carvedilol would have high solubility in the stomach, typically at pH 1–3, and subsequently may precipitate in the higher pH of the small intestine (the presence of food, the pH of the gastric contents and intestinal secretions are variable factors). For neutral drugs such as fenofibrate and danazol, the solubility would not be affected by regional pH changes, though luminal composition changes e.g. lipid and bile salt presence, digestion and/or absorption of luminal contents (particularly foods), as well as formulation factors can affect the in vivo dissolution in a complex time dependent manner (Sugano, 2010).
From these simulations it was also observed that the colonic absorption of BCS Class II drugs may have a significant impact on their overall oral absorption due to their longer residence times (and high permeability) in the large intestine (Fig. 3). It is generally assumed for BCS Class II drugs that their incomplete dissolution in the small intestine is due to the limited and variable residence time in the various intestinal segments and the potential small residual intestinal fluid volume (Fadda et al., 2010; Marciani et al., 2010; Schiller et al., 2005). In the fasted state, the small-intestinal transit time, approximately 3–4 h, is significantly longer than the average gastric residence time of approximately 15–60 min (Davis et al., 1986; Kaus and Fell, 1984; Kortejarvi et al., 2007; Oberle et al., 1990; Reddy et al., 1999). Also, it has been reported that the residence times in the caecum and colon are around 3–7 h and 12–24 h, respectively, which are also significantly longer than the average 3 h residence time reported for the small intestine (Ibekwe et al., 2008; Laird et al., 2010; Madsen and Krogsgaard, 1989). In these simulations, the default residence times used for the caecum, and ascending colon were 4.2 h, and 12.6 h, respectively.
It has been reported that the measured pH of the human intestine in the fasted state ranged 6.3–7.5 in duodenum, 6.2–6.7 in proximal small intestine, 6.3–7.3 in middle small intestine, 6.7–7.7 in distal small intestine, and 5.5–7.6 in ascending colon (Ibekwe et al., 2008; Perez de la Cruz Moreno et al., 2006). The pH values of the caecum and colon were set to 6.4 and 6.8, respectively, as mean pH values in these simulations. At those pH values, a weakly acidic drug such as ibuprofen (pKa 4.9) would exhibit high solubility in the jejunum, ileum, caecum, and colon and, therefore, have sufficient residence time in the small intestine for 3–4 h and in the caecum and colon for 15–31 h for complete absorption. As a result, it is highly likely that ibuprofen, for example, would behave similar to a BCS Class I drug in the intestine even though it is classified as a low solubility drug due to its poor solubility at gastric pH. Therefore, weak acids of BCS Class IIa may be completely absorbed due to their high permeability and high solubility and dissolution rate in the small and large intestine and sufficient residence time throughout the whole intestine. In our simulation, ibuprofen and ketoprofen exhibited essentially complete absorption with an IR dose, including a small amount of absorption from the caecum and colon. Indeed, it has been reported that weakly acidic BCS Class IIa drug products have been shown to be completely absorbed orally due to their high solubility at the pH range (pH 6.0–6.4) of the small intestine (Davies, 1998; Dressman et al., 2001). Thus, BCS Class IIa, drugs and drug products with acidic pKa values ≤5.0 in IR oral dosage forms would have sufficient time for complete dissolution and absorption in the small intestine and biowaivers are scientifically sound, if a relevant dissolution test standard is met.
Weakly basic drugs such as carvedilol (pKa 7.8) and ketoconazole (pKa 2.9/6.5) would exhibit low solubility in the distal small intestine, caecum and large intestine, but would exhibit high solubility in the stomach and proximal small intestine. Basic drugs with pKa values the range of 3–6 in IR oral dosage forms may have sufficient time for dissolution in the stomach and exhibit good absorption in the proximal intestinal region. The BCS Class IIb drugs that have biopharmaceutical properties similar to those of carvedilol and ketoconazole will have a relatively short residence time in the duodenum and proximal intestine of 0.5–1 h (Holtmann et al., 1997; Mudie et al., 2010). These BCS Class IIb drugs may then precipitate as the drug moves from the favorable pH conditions in the stomach to the less favorable pH environment in the distal small intestine. This phenomenon is predicted by our in silico simulation (Fig. 2). Thus, the gastric emptying rate, the intestinal permeability and the precipitation time would be crucial factors in determining the absorption of weakly basic IR oral dosage forms for BCS Class IIb Drugs. Under fasted state conditions, in vitro dissolution studies suggest that a high degree of super-saturation for weakly basic drugs may occur in vivo before precipitation (Kostewicz et al., 2004) and this would drive a higher than expected absorption rate in the upper small intestine. It has been suggested that the actual measured times for drug precipitation tend to be much longer than the predicted times by mathematical calculations (Muller et al., 1991). Since the drug concentration in solution (super saturated or not) at the absorption sites is the key driving force for absorption, the absorptive rate of weakly basic compounds of BCS Class IIb drugs at the small intestine may be significantly higher than suggested by these simulations. As a result, the super-saturation of those weak bases and the delay of drug precipitation may give BCS Class IIb, weak bases, enough time for complete absorption before reaching the ileum. This is one of the most difficult factors to predict in vivo and reproduce by in vitro methods which requires significantly further study. Newer dissolution methods, such as the artificial stomach-duodenum model (Carino et al., 2010), are beginning to address this in vivo dissolution question.
For neutral drugs such as fenofibrate and danazol, the drug solubility would not be affected by pH changes in the GI tract, though disintegration and dissolution could be altered by the gastric environment. The drug solubility and dissolution rate would be significantly affected by their particle size and size distribution, and especially formulations factors (Anby et al., 2012; Williams et al., 2012b), as well as the in vivo luminal environment (Jinno et al., 2008; Sheng et al., 2008; Willmann et al., 2010). The BCS Class IIc drugs that have biopharmaceutical properties similar to those of fenofibrate will have longer residence times throughout small and large intestines i.e. slower absorption (residence time; 18–35 h) and the absorption of those drugs would be determined by their (in vivo) drug solubility, dissolution rates and permeability along the GI Tract. Neutral drugs would have dissolution rates depending on the in vivo environment, and can exhibit high oral extent of drug absorption from the caecum and colon (5–13% and 13–25%, respectively) due to more drug actually reaching those portions of the intestine.
4.1. Absorption profiles
Figs. 1 and 3 summarize the absorption rate profiles and intestinal segmental absorption profiles from the simulation results for the drugs from each BCS Class II sub-class. While the time scales for the oral absorption processes from IR products are often in the early portions of a usual pharmacokinetic profile, we note the expected profiles below. Particularly for BCS Class IIb and IIc, drugs in IR dosage forms, the absorption profile can continue throughout transit through the intestine. From these profiles we can draw some general conclusions regarding absorption from the BCS subclasses.
BCS Class IIa: There will be an initial and variable lag in absorption due to slow gastric disintegration and limited dissolution in the stomach and hence appearance in the systemic circulation. This lag and variability will be dependent on dosing time relative to the gastric motility phase (Oberle and Amidon, 1987; Oberle et al., 1990). The subsequent absorption rate and rate profile will depend mainly on the gastric emptying and dissolution rate. In these simulations the absorption rate increases as the drug transits along the intestine due to the increasing pH, and corresponding increases in dissolution rate. For these BCS Class IIa drugs. Variation in intestinal motility, fluid secretions and absorption (not included in these simulations) as well as permeability variation along the intestine will lead to further variation and time dependent absorption that will depend on the drug and formulation factors.
BCS Class IIb: The absorption rate of drugs in this class will depend initially on the disintegration and dissolution in the gastric environment and gastric emptying, with IR drug products generally showing a relatively rapid initial dissolution and gastric emptying rate limited absorption from the duodenum, relative to motility phase and duodenal environment for Class IIb drug products. Subsequent precipitation, solubilization, dissolution and absorption generally decrease the absorption rate after the initial rapid phase. In these simulations, absorption rate decreases along the gastrointestinal tract due to increasing pH and lower solubility. In general these processes will be complex due to drug, formulation and the in vivo variable intestinal contents. The fraction absorbed may be less than 90% for a BCS Class IIb drug product depending on dose, formulation and in vivo solubility in addition to the variable GI luminal conditions (motility, fluid, and solubilization).
BCS Class IIc: These low solubility drugs in drug products will generally show slow and prolonged absorption depending on the formulation (Williams et al., 2013). The fraction absorbed (Fabs) and systemic availability (Fsys) will be dependent on the in vivo solubilization, dissolution as well as motility (transit). These drugs will likely exhibit the most direct dissolution rate controlled absorption rate correlation with an in vivo predictive dissolution methodology (Williams et al., 2012a).
Analogous generalizations can be made for BCS Class IV (not explored in this study), with intestinal permeability and permeability variation along the GI tract playing a larger role. The role of non-sink conditions (confinement effect) in the intestine needs to be further explored.
These simulations do not include all of the gastrointestinal variations that can occur and will affect the absorption rate along the intestine (CDER, 2000). However, it does provide support for a BCS sub-classification for classifying these complex oral absorption profiles. In summary, with in silico simulation methodology the expected in vivo dissolution profiles can be determined and can serve as a base for establishing in vitro dissolution profiles for Quality by Design and IVIVC methodologies.
While Bioperformance dissolution methodologies have received increasing scientific attention and use for specific pharmaceutical products, there is, as yet, no general scientific basis or paradigm for their development. Based on the BCS sub-classification and the results of these simulations some general suggestions can be made for the initial development of predictive dissolution methodologies.
4.2. In vivo predictive (IPD) dissolution methodology
The proposed BCS sub-classification (Table 3a) distinguishes drug substances based on their relative solubilities in fluids characteristic of the average fasted human stomach and the small intestine. Some generalizations about dissolution and permeation characteristics in vivo can be made based on these relative solubilities, as well as on permeability along the intestine, characteristics of drug substances, which can be used as a starting point for development of an in vivo biopredictive dissolution methodology (IPD) and IVIVC methodology. The in vivo predictive dissolution methodology must be developed and adapted to a particular drug product, and include the most relevant variables while not overly complicating the method. Careful selection of the best methodology can only be made after evaluating (including initial estimates and assumptions) the important drug substance and drug product characteristics, including particle size distribution, dose, and permeation rate across different segments of the GI tract, physical form, disintegration rate, etc. For use in product development and in vivo prediction, a considerable amount of additional research is necessary to define the gastrointestinal variables, in terms of both average values and statistical distributions. Prediction would then entail computational simulation, of Monte Carlo type, over the expected range of these variables. The main focus would be on ‘typical’ subjects in the fasted and fed states, with fed state requiring further consideration regarding meal type including compositions to represent real meals. Further, consideration would have to be given to disease states that can alter the gastrointestinal physiology e.g. diabetes, achlorhydria and drug–drug interactions that may be mediated by alterations of the gastrointestinal physiology (Budha et al., 2012, 2013).
In vivo predictive dissolution (IPD) methods will be considerably different from, and more complex than USP quality control (QC) methodologies. A USP type QC methodology, along with additional QC specifications is the standard for keeping a product within the Phase III product specifications, the most essential basis for the safety and efficacy label claims of the drug product (Amidon et al., 2011). The IPD methodology would be most useful for product development and eventually, when fully developed and validated, useful for product changes e.g. SUPAC type changes. The QC methodology would be a subset of the IPD method when the critical performance parameters have been defined. The validated IPD method would have great value in establishing Quality by Design (QbD) product performance specifications.
The sections below contain discussion regarding IPD initial (starting) recommendations, which are outlined in Table 3b, based on the proposed BCS sub-classification. It must be emphasized that these suggestions are to be used as a starting point for development of an in vivo predictive dissolution methodology for a particular drug and drug product.
Table 3b.
Proposed relevant bioperformance dissolution methodology (apparatus and media).
| BCS sub-classification | Gastric medium | Consider gastric compartment? | Intestinal luminal medium | Consider absorption compartment?a |
|---|---|---|---|---|
| I | 250 ml PGBe | Nob | 900 ml PIBc | No |
| IIa | Yesd | 100 ml PIB | Yes | |
| IIb | Yesd | 100 ml PIB | Yes | |
| IIc | Yesd | 100 ml PIB + bile acids/lipid | Yes | |
| III | Nob | 100 ml PIB | No | |
| IVa | Yesd | 100 ml PIB | Yes | |
| IVb | Yesd | 100 ml PIB | Yes | |
| IVc | Yesd | 100 ml PIB + bile acids/lipid | Yes |
An organic medium (such as 1-octanol) can be used for the absorption compartment when logDpH6.5 > than about 0.5–1 (Mudie et al., 2012). Otherwise, an alternative such as an artificial or caco2 membrane (Kataoka et al., 2003, 2006, 2012, 2013) may be used. Surface area of organic medium (or membrane) to volume ratio of luminal medium (A/V) must be chosen properly (Mudie et al., 2012).
Pretreat dosage form with gastric medium for 15 min and subsequently transfer dosage form + gastric medium to intestinal luminal medium. Determine contribution of gastric emptying rate to delay in onset of appearance in plasma computationally.
Physiological intestinal buffer (PIB), which should be pH 6.5 bicarbonate buffer with a buffer concentration between about 5–15 mM, or an aqueous buffer with an equivalent buffer capacity.
Empty gastric contents in a first order manner with a rate coefficient of 2.8 h−1 (half-emptying time of 15 min).
Physiological gastric buffer (PGB), which is 0.01 N HCl.
4.2.1. BCS Class I drugs
The proposed IPD (Table 3b) for BCS I drugs includes pretreatment of the dosage form in 250 ml of Physiological Gastric Buffer (PGB, comprising 0.01 N HCl) for 15 min, after which time the dosage form and PGB should be transferred to 900 ml of Physiological Intestinal Buffer (PIB). To represent fasted state conditions in the upper GI tract, PIB should be a pH 6.5 bicarbonate buffer with a concentration in the range of about 5–15 mM (Mudie et al., 2010), or a buffer with a buffer capacity ‘equivalent’ to that of a 5–15 mM bicarbonate buffer (Miller et al., 2011; Sheng et al., 2009). Although a BCS I drug with high solubility should dissolve equally well in PGB and PIB, exposure of the dosage form to both buffers is proposed as a conservative method to capture a possible influence of pH and an aqueous environment on potential physical changes of the drug substance or product , e.g. conversion from a salt to a free acid or base, crystal form changes, etc. (Miller et al., 2011). The high solubilities of these compounds should make the dissolution rate only marginally dependent upon the concentration of drug dissolved in the aqueous buffer. In addition, since the intestinal absorption of the drug is high, with a fast absorption rate across the intestinal membrane, conditions approaching that of a ‘sink-conditions’ would be maintained in the intestinal lumen. Therefore, dissolution testing may be carried out in a large volume of PIB without the need for adding an “absorption compartment” to the methodology.
Generally the solubility classification, highest dose strength in 250 ml of water throughout the physiological pH range, is conservative. While a sub-classification of BCS Class I drugs is not proposed, sub-classification may be necessary for drugs on the borderline of the solubility and permeability criteria for Class I. It has been observed that about 5–10% of the oral drug products change classification between the lowest and highest FDA approved doses (Takagi et al., 2006). These drugs products would be the most likely candidates for a BCS Class I sub-classification. Depending upon the relevant ‘near-by’ sub-classification (IIa, b, or c, or IVa, b, c) the IPD recommended for BCS Class II or IV sub-classification could be used as a guideline for developing the proper an in vivo predictive dissolution methodology.
Further considerations would be given for transport mechanism for BCS I drugs, particularly the involvement of carrier mediated transport, either for absorption or secretion along the gastrointestinal tract. BCS Class I drugs are generally non polar and eliminated by metabolism (Wu and Benet, 2005). However, if they are polar and absorbed via carrier mediated transport mechanisms, e.g. PEPT1 transport of valacyclovir (Kim et al., 2003; Lai et al., 2008) and β-lactam antibiotics(Sinko and Amidon, 1989) or ACE inhibitors (Friedman and Amidon, 1989), then they would be expected to have position dependent absorption (Dahan and Amidon, 2009) limited or no metabolism other than prodrug activation and potential phase I or II metabolism of the prodrug. Variation in plasma levels for typical BCS Class I drugs is expected to be mainly due to metabolism variation along the gastrointestinal tract and potentially variation in absorption rate along the GI tract, i.e. permeability along the GIT, or when metabolism or an exporter e.g. P-gp is nonlinear and position dependent along the GI tract.
4.2.2. BCS Class II
The sub-classification, a: for acids (pKa < ~5), b: for bases (pKa > ~5, and c: for neutral drugs (pKa < 0 or ~ >8) is the clearest starting point for distinguishing and making recommendations for evaluation of low solubility drug substances. While other properties such a particle size, solid state form e.g. crystalline, amorphous etc. clearly affect drug product dissolution both in vitro and in vivo, the BCS sub-classification is based on fundamental properties determining equilibrium solubility of the drug over the physiological pH range. Non equilibrium solubility and amorphous or ‘high energy’ forms, while clearly having a potentially significant impact on dissolution and drug oral delivery in vivo, do not change the actual BCS classification (or sub-classification) of a drug and product.
The proposed IPD for subclasses IIa, IIb, and IIc are included in Table 3b. For IIa drugs, which would be virtually non-ionized and thus poorly soluble near pH 2, low gastric pH coupled with relatively short gastric residence time would lead to a limited extent of dissolution in the stomach in vivo. However, particularly when the Dose Number is high,1 it may be important to include a separate gastric compartment in the dissolution test and transfer gastric contents (including both dissolved & non-dissolved drug as well as PGB) to the intestinal compartment in a physiologically relevant manner, such as at a first-order rate with a half emptying time of about 15 min (Oberle et al., 1990). Using this transfer method, as opposed to simply adding drug as a bolus into the intestinal compartment, should assist in maintaining aqueous saturation conditions in the intestinal compartment more similar to those expected in the intestinal lumen in vivo, leading to a more physiologically relevant dissolution rate. Introduction of a gastric compartment, such as the “artificial stomach duodenum” system, into the dissolution test as has been discussed (Carino et al., 2006) would be a dissolution methodology more reflective of in vivo conditions.
When the Class IIa drug reaches the small intestine, its high extent of ionization and corresponding high solubility should likely lead to relatively fast absorption. The high permeability classification (and likely moderate to high absorption rate of drug across the intestinal membrane) should act to keep the percent drug saturation in the luminal fluid relatively low for cases when Do is low and low to moderate for cases when Do is high. Since dissolution rate should be relatively independent of a low degree of drug saturation when Do is low, simply using 900 ml of PIB in the intestinal compartment should be adequate. However, for cases when Do is high, and the surface area high, it is possible that the intestinal luminal fluid is moderately saturated with drug in vivo, and for some BCS IIa compounds this saturation could cause the dissolution rate to be a function of saturation solubility and also be impacted by a confinement effect (8Wang et al., 2012). Especially when Do is high it is recommended to perform dissolution in a physiologically relevant volume of medium (100 ml) and add an “absorption compartment” to draw dissolved drug out of the PIB, which, when scaled properly (Mudie et al., 2010, 2012) , should create more physiologically relevant saturation conditions than would be achieved using a large volume (e.g. 900 ml) of medium. Several options exist for creation of an absorption compartment in the dissolution methodology. A two-phase (or “biphasic”) apparatus, which comprises an aqueous medium into which the drug dissolves and a second, immiscible organic medium (such as 1-octanol) into which the drug partitions has been cited quite a few times in the literature (Hoa and Kinget, 1996; Mudie et al., 2012). Other methods of introducing an absorption compartment include use a Caco2 membrane separating two distinct buffers (Kataoka et al., 2003). For cases when it is recommended to add both a stomach compartment as well as an absorption compartment, such as with BCS IIa drugs with Do > 4, it would be necessary to devise such an apparatus. For example, a system for which the ASD and two-phase systems are coupled together would be appropriate. Gu et al. developed a multi-compartment dissolution system containing a gastric compartment, intestinal compartment, absorption compartment and reservoir compartment to study a poorly soluble weak base. While this system had some drawbacks (including real-time pumping of non-dissolved drug particles between the gastric and intestinal compartments) it could be used as a starting point for development of a more in vivo predictive system (Gu et al., 2005).
For Class IIb drug substances, it is proposed to carry out the IPD using a separate aqueous compartment containing 250 ml of PGB with controlled, first order emptying of contents into 100 ml of PIB with an absorption compartment (especially when Do is high). Use of both PGB and PIB including transfer of drug in a kinetically-appropriate manner is especially important for Class IIb bases due to the high solubility and likely high extent of dissolution in the stomach, followed by low solubility and likely precipitation in the small intestine in vivo. Use of a gastric compartment for BCS IIb drug products has been utilized several numerous times (Carl-ert et al., 2010, 2012; Kostewicz et al., 2002, 2004). Carlert et al. investigated the accuracy of both in vitro and in vitro/in silico methods for prediction of the in vivo intestinal precipitation of basic BCS Class II drugs in humans. One of the conclusions was that “reduction of luminal drug concentration by time due to absorption in vivo must be included in predictive methods,” and that without it intestinal precipitation in humans in vivo may be less of a limitation than would be predicted in a simple in vitro method for which an absorption compartment is not included. Similarly, Bhattachar et al. stated that, when used without an absorption compartment, in vitro results in the ASD system over predicted the extent of exposure differences for a BCS II weak base that was dosed using different methods of stomach pretreatment (including low and high pH) in dogs (Bhattachar et al., 2011a,b).
The IPD for BCS IIc drugs is the same for Class IIa in terms of apparatus type, but differs in the recommended intestinal medium. The dosage form should be exposed to 250 ml of PGB and transferred at a first order rate into 100 ml of PIB containing a physiological relevant concentration of surfactant e.g. bile acids and lipids with an added absorption compartment, particularly when Do is high. As frequently noted, in vivo bile acids and lipids can enhance solubility for some IIc drugs, making use of a medium such as a fasted simulated intestinal fluid (Vertzoni et al., 2004, 2005) particularly relevant for in vivo prediction. For cases when bile acids and lipids are used (or a solubility pharmaceutical surfactant matching in vivo solubility) with addition of an organic medium as the absorption compartment, the solubility of and dissolution rate of drug in the surfactant solution saturated with the organic medium, as well as the distribution coefficient of the drug in organic medium saturated with the aqueous medium and surfactant should be evaluated, since long-chain alcohols such as 1-octanol can form mixed micelles with ionic surfactants (Moya et al., 1999). For BCS IIa, IIb, and IIc drugs that may be on the borderline between low and high permeability, it is recommended to evaluate the corresponding dosage forms using IPD guidelines suggested for BCS IVa, IVb, and IVc drugs.
Finally, food effects for BCS Class II drugs will further depend on BCS sub-class and the actual solubility and pKa’s of the API, leading to variable food effects on absorption for BCS Class II drugs. For example, the in vivo dissolution of a Class IIa API will depend on the actual (intrinsic, uncharged) solubility and pKa in the gastrointestinal physiological range. The food effect on a Class IIb API is even more complex and dynamic, depending on the above, the nucleation and crystal growth in vivo and the local intestinal permeability (i.e. the absorption rate relative to the precipitation rate in vivo). While for a Class IIc drug (not ionized) dissolution will depend on the variable in vivo solubilization along the gastrointestinal tract. While the solubilization itself may be matched with pharmaceutical surfactant media, the micelle and emulsion solubilization in vivo is complex and evidencing this matching to gastrointestinal physiological conditions require further scientific investigation. Never-the-less, in many (most) cases we can come close to matching gastrointestinal physiological processes in vitro, and ensure in vivo bioperformance.
4.2.3. BCS Class III
The proposed IPD for Class III drugs includes pretreatment of the dosage form in 250 ml of PGB for 15 min with subsequent transfer of the dosage form and PGB into 100 ml of PIB. The rationale behind the chosen dissolution medium is similar to that for BCS I compounds due to the similar solubility characteristics. However, for BCS III drugs the low permeability (and expected low permeation rate through the intestinal membrane) in vivo could lead to moderate to high luminal concentrations of drug depending on Do. If the solubility of drug in the intestinal lumen in vivo is high, then the expected higher luminal concentrations of drug due to slow absorption may not affect the dissolution rate. A smaller volume of PIB (100 ml) was chosen for the intestinal compartment (which when combined with the 250 ml of PGB would be equal to a total volume of 350 ml) for Class III drugs to more accurately capture aqueous drug concentration, at least compared to 900 ml. If solubility of the drug in the intestinal lumen is such that the dissolution rate is decreases due to non-sink conditions, then a two-phase dissolution system maybe more appropriate IPD. If a separate absorption compartment is needed, use of a layer of Caco2 cells (Kataoka et al., 2013; Yamashita et al., 1997) or other membrane may be more appropriate than a two-phase system utilizing an organic solvent such as 1-octanol, since some Class III drugs have low logP and logDpH6.5 values. Use of a separate absorption compartment would allow for better evaluation of the confinement effect on dissolution rate. Further, since low permeability drugs must have, based on their incomplete oral absorption, an intestinal position (jejunum, ileum, colon) dependent permeability, an IPD-method needs to carefully consider drug pH dependent properties, and potential carrier mediated, position dependent absorption from the GI tract, in developing an IPD dissolution method.
4.2.4. BCS Class IV
As with BCS Class III, the low intestinal permeability may lead to high luminal drug concentrations relative to the drug’s solubility and thus non-sink dissolution conditions. Coupled with the low expected solubilities for most BCS IV compounds in the intestinal lumen, these probable non-sink conditions would possibly lead to an impact of saturation solubility and a confinement effect on dissolution rate. Therefore, it is recommended to evaluate all BCS IV drugs (a, b, and c) in 250 ml of PGB contained in a separate compartment, with controlled transfer to 100 ml of PIB (IVa and IVb) or 100 ml of PIB + bile acids and lipids (IVc) with a separate absorption compartment. As with BCS II drugs, possible food effects will depend on the actual solubility and pKa’s of the API, leading to variable food effects on absorption.
5. Summary
In this manuscript we use in silico results to support the proposed BCS Subclassification and present some general recommendations for dissolution methodologies. These subclasses are based on fundamental drug properties and can serve as a basis for developing in vivo predictive dissolution and absorption methodology. An in vivo predictive dissolution methodology when combined with a permeability profile of the gastrointestinal tract would be predictive of drug absorption as a function of time (and position along the GI tract). The drug product quality, i.e. safety and efficacy, would be assured by a dissolution methodology that predicts this in vivo dissolution. The central hypothesis underlying this conclusion is that stated in the original BCS paper (Amidon et al., 1995).
“If two drug products have the same in vivo dissolution profile under all luminal conditions, they will have the same rate and extent of drug absorption”.
Since the rate and extent of metabolism depends on the rate of absorption over time, the same can be stated for metabolism. Thus the pivotal dissolution standard is that of the clinically tested Phase III product and matching this in vivo dissolution over the life time of a drug product is the most important and essential pharmaceutical quality standard for oral products (Amidon et al., 2011). The BCS sub-classification and IPD methodology provides a starting point for the development of this pharmaceutical quality standard.
We believe the BCS sub-classification approach to setting oral dissolution methodology will be preferable, and occupy a middle ground between the…‘one size fits all’ approach and an unnecessarily large number of almost independent methods…one or more for each oral ‘drug product’…depending on the specific product.
Table 3a.
Solubility and permeability characteristics of drug substances based on BCS sub-classification.
| BCS sub- classification |
Solubility at pH 2 |
Solubility at pH 6.5 |
Permeability |
|---|---|---|---|
| I | High | High | High |
| IIaa | Low | High | High |
| IIbb | High | Low | High |
| IIcc | Low | Low | High |
| III | High | High | Low |
| IVaa | Low | High | Low |
| IVbb | High | Low | Low |
| IVcc | Low | Low | Low |
With a pKa < 5.
With a pKa > 6.
With no pKa(s) or pKa(s) < 0 or >8.
Acknowledgement
This work was supported by NIH Grant GM-2R01GM037188.
Footnotes
Further research is needed to define low and high Do to study its impact on dissolution rate for a range of drug substance properties, including particle size and permeation rate and make recommendations for a suitable IPD.
References
- Abernethy DR, Greenblatt DJ. Ibuprofen disposition in obese individuals. Arthritis Rheum. 1985;28:1117–1121. doi: 10.1002/art.1780281006. [DOI] [PubMed] [Google Scholar]
- Agrawal S, Panchagnula R. Implication of biopharmaceutics and pharmacokinetics of rifampicin in variable bioavailability from solid oral dosage forms. Biopharm. Drug Dispos. 2005;26:321–334. doi: 10.1002/bdd.464. [DOI] [PubMed] [Google Scholar]
- Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- Amidon KS, Langguth P, Lennernas H, Yu L, Amidon GL. Bioequivalence of oral products and the Biopharmaceutics Classification System: science, regulation, and public policy. Clin. Pharmacol. Ther. 2011;90:467–470. doi: 10.1038/clpt.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anby MU, Williams HD, McIntosh M, Benameur H, Edwards GA, Pouton CW, Porter CJ. Lipid digestion as a trigger for supersaturation: evaluation of the impact of supersaturation stabilization on the in vitro and in vivo performance of self-emulsifying drug delivery systems. Mol. Pharm. 2012 doi: 10.1021/mp300164u. [DOI] [PubMed] [Google Scholar]
- Avdeef A, Box KJ, Comer JE, Hibbert C, Tam KY. PH-metric logP 10. Determination of liposomal membrane-water partition coefficients of ionizable drugs. Pharm. Res. 1998;15:209–215. doi: 10.1023/a:1011954332221. [DOI] [PubMed] [Google Scholar]
- Baxter JG, Brass C, Schentag JJ, Slaughter RL. Pharmacokinetics of ketoconazole administered intravenously to dogs and orally as tablet and solution to humans and dogs. J. Pharm. Sci. 1986;75:443–447. doi: 10.1002/jps.2600750504. [DOI] [PubMed] [Google Scholar]
- Becker C, Dressman JB, Junginger HE, Kopp S, Midha KK, Shah VP, Stavchansky S, Barends DM. Biowaiver monographs for immediate release solid oral dosage forms: rifampicin. J. Pharm. Sci. 2009;98:2252–2267. doi: 10.1002/jps.21624. [DOI] [PubMed] [Google Scholar]
- Beetge E, du Plessis J, Muller DG, Goosen C, van Rensburg FJ. The influence of the physicochemical characteristics and pharmacokinetic properties of selected NSAID’s on their transdermal absorption. Int. J. Pharm. 2000;193:261–264. doi: 10.1016/s0378-5173(99)00340-3. [DOI] [PubMed] [Google Scholar]
- Bhattachar SN, Perkins EJ, Tan JS, Burns LJ. Effect of gastric pH on the pharmacokinetics of a BCS Class II compound in dogs: utilization of an artificial stomach and duodenum dissolution model and GastroPlus, simulations to predict absorption. J. Pharm. Sci. 2011a;100:4756–4765. doi: 10.1002/jps.22669. [DOI] [PubMed] [Google Scholar]
- Bhattachar SN, Risley DS, Werawatganone P, Aburub A. Weak bases and formation of a less soluble lauryl sulfate salt/complex in sodium lauryl sulfate (SLS) containing media. Int. J. Pharm. 2011b;412:95–98. doi: 10.1016/j.ijpharm.2011.04.018. [DOI] [PubMed] [Google Scholar]
- Budha NR, Frymoyer A, Smelick GS, Jin JY, Yago MR, Dresser MJ, Holden SN, Benet LZ, Ware JA. Drug absorption interactions between oral targeted anticancer agents and PPIs: is pH-dependent solubility the Achilles heel of targeted therapy? Clin. Pharmacol. Ther. 2012;92:203–213. doi: 10.1038/clpt.2012.73. [DOI] [PubMed] [Google Scholar]
- Budha NR, Benet LZ, Ware JA. Response to “Drug interactions produced by proton pump inhibitors: not simply a pH effect”. Clin. Pharmacol. Ther. 2013;93:151. doi: 10.1038/clpt.2012.206. [DOI] [PubMed] [Google Scholar]
- Butler JM, Dressman JB. The developability classification system: application of biopharmaceutics concepts to formulation development. J. Pharm. Sci. 2010;99:4940–4954. doi: 10.1002/jps.22217. [DOI] [PubMed] [Google Scholar]
- Carino SR, Sperry DC, Hawley M. Relative bioavailability estimation of carbamazepine crystal forms using an artificial stomach-duodenum model. J. Pharm. Sci. 2006;95:116–125. doi: 10.1002/jps.20495. [DOI] [PubMed] [Google Scholar]
- Carino SR, Sperry DC, Hawley M. Relative bioavailability of three different solid forms of PNU-141659 as determined with the artificial stomach-duodenum model. J. Pharm. Sci. 2010;99:3923–3930. doi: 10.1002/jps.22236. [DOI] [PubMed] [Google Scholar]
- Carlert S, Palsson A, Hanisch G, von Corswant C, Nilsson C, Lindfors L, Lennernas H, Abrahamsson B. Predicting intestinal precipitation – a case example for a basic BCS Class II drug. Pharm. Res. 2010;27:2119–2130. doi: 10.1007/s11095-010-0213-8. [DOI] [PubMed] [Google Scholar]
- Carlert S, Akesson P, Jerndal G, Lindfors L, Lennernas H, Abrahamsson B. In vivo dog intestinal precipitation of mebendazole: a basic BCS Class II drug. Mol. Pharm. 2012;9:2903–2911. doi: 10.1021/mp300224h. [DOI] [PubMed] [Google Scholar]
- CDER, U.S.D.o.H.a.H.S.F.a.D.A.C.f.E.a.R. CDER/FDA; 2000. Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics Classification System, FDA Guidance for Industry. 2000. U.S. Department of Health and Human Services Food and Drug Administration Center for Evaluation and Research (CDER). Guidances for industry: Waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a Biopharmaceutics Classification System. [Google Scholar]
- Cordero JA, Alarcon L, Escribano E, Obach R, Domenech J. A comparative study of the transdermal penetration of a series of nonsteroidal antiinflammatory drugs. J. Pharm. Sci. 1997;86:503–508. doi: 10.1021/js950346l. [DOI] [PubMed] [Google Scholar]
- Dahan A, Amidon GL. Small intestinal efflux mediated by MRP2 and BCRP shifts sulfasalazine intestinal permeability from high to low, enabling its colonic targeting. Am. J. Physiol. Gastroint. Liver Physiol. 2009;297:G371–G377. doi: 10.1152/ajpgi.00102.2009. [DOI] [PubMed] [Google Scholar]
- Davies NM. Clinical pharmacokinetics of flurbiprofen and its enantiomers. Clin. Pharmacokinet. 1995;28:100–114. doi: 10.2165/00003088-199528020-00002. [DOI] [PubMed] [Google Scholar]
- Davies NM. Clinical pharmacokinetics of ibuprofen. The first 30 years. Clin. Pharmacokinet. 1998;34:101–154. doi: 10.2165/00003088-199834020-00002. [DOI] [PubMed] [Google Scholar]
- Davies NM, Anderson KE. Clinical pharmacokinetics of naproxen. Clin. Pharmacokinet. 1997;32:268–293. doi: 10.2165/00003088-199732040-00002. [DOI] [PubMed] [Google Scholar]
- Davis SS, Hardy JG, Fara JW. Transit of pharmaceutical dosage forms through the small intestine. Gut. 1986;27:886–892. doi: 10.1136/gut.27.8.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson PA, Lee WW, Stott PW, Townsend AI, Smart JP, Ghahramani P, Hammett T, Billett L, Behn S, Gibb RC, Abrahamsson B. Clinical relevance of dissolution testing in quality by design. AAPS J. 2008;10:380–390. doi: 10.1208/s12248-008-9034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanska U, Pobudkowska A, Pelczarska A, Gierycz P. pKa and solubility of drugs in water, ethanol, and 1-octanol. J. Phys. Chem. B. 2009;113:8941–8947. doi: 10.1021/jp900468w. [DOI] [PubMed] [Google Scholar]
- Dressman JB, Lennernäs H. Marcel Dekker. New York: 2000. Oral Drug Absorption: Prediction and Assessment. [Google Scholar]
- Dressman JB, Reppas C. In vitro–in vivo correlations for lipophilic, poorly water-soluble drugs. Eur. J. Pharm. Sci.: Official J. Eur. Fed. Pharm. Sci. 2000;11(Suppl. 2):S73–S80. doi: 10.1016/s0928-0987(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Dressman JB, Hempenstall J, Reppas J, Reppas C. The BCS: where do we go from here? Pharm. Technol. 2001;25:68–76. [Google Scholar]
- Faassen F, Vromans H. Biowaivers for oral immediate-release products: implications of linear pharmacokinetics. Clin. Pharmacokinet. 2004;43:1117–1126. doi: 10.2165/00003088-200443150-00004. [DOI] [PubMed] [Google Scholar]
- Fadda HM, Sousa T, Carlsson AS, Abrahamsson B, Williams JG, Kumar D, Basit AW. Drug solubility in luminal fluids from different regions of the small and large intestine of humans. Mol. Pharm. 2010;7:1527–1532. doi: 10.1021/mp100198q. [DOI] [PubMed] [Google Scholar]
- Fei Y, Kostewicz ES, Sheu MT, Dressman JB. Analysis of the enhanced oral bioavailability of fenofibrate lipid formulations in fasted humans using an in vitro–in silico–in vivo approach. Eur. J. Pharm. Biopharm.: Official J. Arbeitsgemein. Pharm. Verfahrenstech. e.V. 2013 doi: 10.1016/j.ejpb.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Friedman DI, Amidon GL. Passive and carrier-mediated intestinal absorption components of two angiotensin converting enzyme (ACE) inhibitor prodrugs in rats: enalapril and fosinopril. Pharm. Res. 1989;6:1043–1047. doi: 10.1023/a:1015978420797. [DOI] [PubMed] [Google Scholar]
- Geisslinger G, Menzel S, Wissel K, Brune K. Pharmacokinetics of ketoprofen enantiomers after different doses of the racemate. Br. J. Clin. Pharmacol. 1995;40:73–75. doi: 10.1111/j.1365-2125.1995.tb04537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GlaxoSmithKline COREG tablets prescribing information. 2009.
- Gu CH, Rao D, Gandhi RB, Hilden J, Raghavan K. Using a novel multicompartment dissolution system to predict the effect of gastric pH on the oral absorption of weak bases with poor intrinsic solubility. J. Pharm. Sci. 2005;94:199–208. doi: 10.1002/jps.20242. [DOI] [PubMed] [Google Scholar]
- Hoa NT, Kinget R. Design and evaluation of two-phase partition–dissolution method and its use in evaluating artemisinin tablets. J. Pharm. Sci. 1996;85:1060–1063. doi: 10.1021/js960115u. [DOI] [PubMed] [Google Scholar]
- Holtmann G, Kelly DG, Sternby B, DiMagno EP. Survival of human pancreatic enzymes during small bowel transit: effect of nutrients, bile acids, and enzymes. Am. J. Physiol. 1997;273:G553–G558. doi: 10.1152/ajpgi.1997.273.2.G553. [DOI] [PubMed] [Google Scholar]
- Hooper WD, Eadie MJ, Dickinson RG. Single oral dose pharmacokinetics and comparative bioavailability of danazol in humans. Biopharm. Drug Dispos. 1991;12:577–582. doi: 10.1002/bdd.2510120804. [DOI] [PubMed] [Google Scholar]
- Huang YC, Colaizzi JL, Bierman RH, Woestenborghs R, Heykants J. Pharmacokinetics and dose proportionality of ketoconazole in normal volunteers. Antimicrob. Agents Chemother. 1986;30:206–210. doi: 10.1128/aac.30.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibekwe VC, Fadda HM, McConnell EL, Khela MK, Evans DF, Basit AW. Interplay between intestinal pH, transit time and feed status on the in vivo performance of pH responsive ileo-colonic release systems. Pharm. Res. 2008;25:1828–1835. doi: 10.1007/s11095-008-9580-9. [DOI] [PubMed] [Google Scholar]
- Ige PP, Baria RK, Gattani SG. Fabrication of fenofibrate nanocrystals by probe sonication method for enhancement of dissolution rate and oral bioavailability. Colloids Surf., B: Biointerf. 2013;108:366–373. doi: 10.1016/j.colsurfb.2013.02.043. [DOI] [PubMed] [Google Scholar]
- Jinno J, Kamada N, Miyake M, Yamada K, Mukai T, Odomi M, Toguchi H, Liversidge GG, Higaki K, Kimura T. In vitro–in vivo correlation for wet-milled tablet of poorly water-soluble cilostazol. J. Controlled Release. 2008;130:29–37. doi: 10.1016/j.jconrel.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Kambayashi A, Dressman JB. An in vitro–in silico–in vivo approach to predicting the oral pharmacokinetic profile of salts of weak acids: case example dantrolene. Eur. J. Pharm. Biopharm.: Official J. Arbeitsgemein. Pharm. Verfahrenstech. e.V. 2013;84:200–207. doi: 10.1016/j.ejpb.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernas H, Hussain AS, Junginger HE, Stavchansky SA, Midha KK, Shah VP, Amidon GL. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Masaoka Y, Yamazaki Y, Sakane T, Sezaki H, Yamashita S. In vitro system to evaluate oral absorption of poorly water-soluble drugs: simultaneous analysis on dissolution and permeation of drugs. Pharm. Res. 2003;20:1674–1680. doi: 10.1023/a:1026107906191. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Masaoka Y, Sakuma S, Yamashita S. Effect of food intake on the oral absorption of poorly water-soluble drugs: in vitro assessment of drug dissolution and permeation assay system. J. Pharm. Sci. 2006;95:2051–2061. doi: 10.1002/jps.20691. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Sugano K, da Costa Mathews C, Wong JW, Jones KL, Masaoka Y, Sakuma S, Yamashita S. Application of dissolution/permeation system for evaluation of formulation effect on oral absorption of poorly water-soluble drugs in drug development. Pharm. Res. 2012;29:1485–1494. doi: 10.1007/s11095-011-0623-2. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Yano K, Hamatsu Y, Masaoka Y, Sakuma S, Yamashita S. Assessment of absorption potential of poorly water-soluble drugs by using the dissolution/permeation system. Eur. J. Pharm. Biopharm.: Official J. Arbeitsgemein. Pharm. Verfahrenstech. e.V. 2013 doi: 10.1016/j.ejpb.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Kaus LC, Fell JT. Effect of stress on the gastric emptying of capsules. J. Clin. Hosp. Pharm. 1984;9:249–251. doi: 10.1111/j.1365-2710.1984.tb01083.x. [DOI] [PubMed] [Google Scholar]
- Keating GM, Croom KF. Fenofibrate: a review of its use in primary dyslipidaemia, the metabolic syndrome and type 2 diabetes mellitus. Drugs. 2007;67:121–153. doi: 10.2165/00003495-200767010-00013. [DOI] [PubMed] [Google Scholar]
- Kim I, Chu XY, Kim S, Provoda CJ, Lee KD, Amidon GL. Identification of a human valacyclovirase: biphenyl hydrolase-like protein as valacyclovir hydrolase. J. Biol. Chem. 2003;278:25348–25356. doi: 10.1074/jbc.M302055200. [DOI] [PubMed] [Google Scholar]
- Kortejarvi H, Urtti A, Yliperttula M. Pharmacokinetic simulation of biowaiver criteria: the effects of gastric emptying, dissolution, absorption and elimination rates. Eur. J. Pharm. Sci.: Official J. Eur. Fed. Pharm. Sci. 2007;30:155–166. doi: 10.1016/j.ejps.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Kostewicz ES, Brauns U, Becker R, Dressman JB. Forecasting the oral absorption behavior of poorly soluble weak bases using solubility and dissolution studies in biorelevant media. Pharm. Res. 2002;19:345–349. doi: 10.1023/a:1014407421366. [DOI] [PubMed] [Google Scholar]
- Kostewicz ES, Wunderlich M, Brauns U, Becker R, Bock T, Dressman JB. Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J. Pharm. Pharmacol. 2004;56:43–51. doi: 10.1211/0022357022511. [DOI] [PubMed] [Google Scholar]
- Kovacevic I, Parojcic J, Homsek I, Tubic-Grozdanis M, Langguth P. Justification of biowaiver for carbamazepine, a low soluble high permeable compound, in solid dosage forms based on IVIVC and gastrointestinal simulation. Mol. Pharm. 2009;6:40–47. doi: 10.1021/mp800128y. [DOI] [PubMed] [Google Scholar]
- Lai L, Xu Z, Zhou J, Lee KD, Amidon GL. Molecular basis of prodrug activation by human valacyclovirase, an alpha-amino acid ester hydrolase. J. Biol. Chem. 2008;283:9318–9327. doi: 10.1074/jbc.M709530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird BD, Peak D, Siciliano SD. The effect of residence time and fluid volume to soil mass (LS) ratio on in vitro arsenic bioaccessibility from poorly crystalline scorodite. J. Environ. Sci. Health A: Tox. Hazard Subst. Environ. Eng. 2010;45:732–739. doi: 10.1080/10934521003648958. [DOI] [PubMed] [Google Scholar]
- Le VN, Leterme P, Gayot A, Flament MP. Influence of granulation and compaction on the particle size of ibuprofen–development of a size analysis method. Int. J. Pharm. 2006;321:72–77. doi: 10.1016/j.ijpharm.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Lin C, Lim J, DiGiore C, Gural R, Symchowicz S. Comparative bioavailability of a microsize and ultramicrosize griseofulvin formulation in man. J. Int. Med. Res. 1982;10:274–277. doi: 10.1177/030006058201000415. [DOI] [PubMed] [Google Scholar]
- Lindfors KK, Boone JM, Nelson TR, Yang K, Kwan AL, Miller DF. Dedicated breast CT: initial clinical experience. Radiology. 2008;246:725–733. doi: 10.1148/radiol.2463070410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftsson T, Vogensen SB, Desbos C, Jansook P. Carvedilol: solubilization and cyclodextrin complexation: a technical note. AAPS PharmSciTech. 2008;9:425–430. doi: 10.1208/s12249-008-9055-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen JL, Krogsgaard OW. Gastrointestinal scintiscanning: dosimetry. Eur. J. Nucl. Med. 1989;15:260–261. doi: 10.1007/BF00257544. [DOI] [PubMed] [Google Scholar]
- Mannisto PT, Mantyla R, Nykanen S, Lamminsivu U, Ottoila P. Impairing effect of food on ketoconazole absorption. Antimicrob. Agents Chemother. 1982;21:730–733. doi: 10.1128/aac.21.5.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciani L, Cox EF, Hoad CL, Pritchard S, Totman JJ, Foley S, Mistry A, Evans S, Gowland PA, Spiller RC. Postprandial changes in small bowel water content in healthy subjects and patients with irritable bowel syndrome. Gastroenterology. 2010;138:469–477. 477, e461. doi: 10.1053/j.gastro.2009.10.055. [DOI] [PubMed] [Google Scholar]
- Miller JM, Beig A, Krieg BJ, Carr RA, Borchardt TB, Amidon GE, Amidon GL, Dahan A. The solubility–permeability interplay: mechanistic modeling and predictive application of the impact of micellar solubilization on intestinal permeation. Mol. Pharm. 2011;8:1848–1856. doi: 10.1021/mp200181v. [DOI] [PubMed] [Google Scholar]
- Moya S, Sukhorukov GB, Auch M, Donath E, Mohwald H. Microencapsulation of organic solvents in polyelectrolyte multilayer micrometer-sized shells. J. Colloid Interface Sci. 1999;216:297–302. doi: 10.1006/jcis.1999.6286. [DOI] [PubMed] [Google Scholar]
- Mudie DM, Amidon GL, Amidon GE. Physiological parameters for oral delivery and in vitro testing. Mol. Pharm. 2010;7:1388–1405. doi: 10.1021/mp100149j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudie DM, Shi Y, Ping H, Gao P, Amidon GL, Amidon GE. Mechanistic analysis of solute transport in an in vitro physiological two-phase dissolution apparatus. Biopharm. Drug Dispos. 2012;33:378–402. doi: 10.1002/bdd.1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller DG, Stella V, Lotter AP. Factors influencing the precipitation time of phenytoin in the presence of DDMS, one of its prodrugs. Int. J. Pharm. 1991;75:201–209. [Google Scholar]
- Mutalik S, Anju P, Manoj K, Usha AN. Enhancement of dissolution rate and bioavailability of aceclofenac: a chitosan-based solvent change approach. Int. J. Pharm. 2008;350:279–290. doi: 10.1016/j.ijpharm.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Oberle RL, Amidon GL. The influence of variable gastric emptying and intestinal transit rates on the plasma level curve of cimetidine; an explanation for the double peak phenomenon. J. Pharmacokinet. Biopharm. 1987;15:529–544. doi: 10.1007/BF01061761. [DOI] [PubMed] [Google Scholar]
- Oberle RL, Chen TS, Lloyd C, Barnett JL, Owyang C, Meyer J, Amidon GL. The influence of the interdigestive migrating myoelectric complex on the gastric emptying of liquids. Gastroenterology. 1990;99:1275–1282. doi: 10.1016/0016-5085(90)91150-5. [DOI] [PubMed] [Google Scholar]
- Oliary J, Tod M, Nicolas P, Petitjean O, Caille G. Pharmacokinetics of ibuprofen enantiomers after single and repeated doses in man. Biopharm. Drug Dispos. 1992;13:337–344. doi: 10.1002/bdd.2510130505. [DOI] [PubMed] [Google Scholar]
- Panchagnula R, Agrawal S. Biopharmaceutic and pharmacokinetic aspects of variable bioavailability of rifampicin. Int. J. Pharm. 2004;271:1–4. doi: 10.1016/j.ijpharm.2003.11.031. [DOI] [PubMed] [Google Scholar]
- Peeters K, De Maesschalck R, Bohets H, Vanhoutte K, Nagels L. In situ dissolution testing using potentiometric sensors. Eur. J. Pharm. Sci.: Official J. Eur. Fed. Pharm. Sci. 2008;34:243–249. doi: 10.1016/j.ejps.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Perez de la Cruz Moreno M, Moreno M, Oth M, Deferme S, Lammert F, Tack J, Dressman J, Augustijns P. Characterization of fasted-state human intestinal fluids collected from duodenum and jejunum. J. Pharm. Pharmacol. 2006;58:1079–1089. doi: 10.1211/jpp.58.8.0009. [DOI] [PubMed] [Google Scholar]
- Planinsek O, Kovacic B, Vrecer F. Carvedilol dissolution improvement by preparation of solid dispersions with porous silica. Int. J. Pharm. 2011;406:41–48. doi: 10.1016/j.ijpharm.2010.12.035. [DOI] [PubMed] [Google Scholar]
- Prajapati HN, Dalrymple DM, Serajuddin AT. A comparative evaluation of mono-, di- and triglyceride of medium chain fatty acids by lipid/surfactant/water phase diagram, solubility determination and dispersion testing for application in pharmaceutical dosage form development. Pharm. Res. 2012;29:285–305. doi: 10.1007/s11095-011-0541-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy SM, Sinha VR, Reddy DS. Novel oral colon-specific drug delivery systems for pharmacotherapy of peptide and nonpeptide drugs. Drugs Today (Barc.) 1999;35:537–580. doi: 10.1358/dot.1999.35.7.548266. [DOI] [PubMed] [Google Scholar]
- Rinaki E, Dokoumetzidis A, Valsami G, Macheras P. Identification of biowaivers among Class II drugs: theoretical justification and practical examples. Pharm. Res. 2004;21:1567–1572. doi: 10.1023/b:pham.0000041450.25106.c8. [DOI] [PubMed] [Google Scholar]
- Schiller C, Frohlich CP, Giessmann T, Siegmund W, Monnikes H, Hosten N, Weitschies W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2005;22:971–979. doi: 10.1111/j.1365-2036.2005.02683.x. [DOI] [PubMed] [Google Scholar]
- Sheng JJ, Sirois PJ, Dressman JB, Amidon GL. Particle diffusional layer thickness in a USP dissolution apparatus II: a combined function of particle size and paddle speed. J. Pharm. Sci. 2008;97:4815–4829. doi: 10.1002/jps.21345. [DOI] [PubMed] [Google Scholar]
- Sheng JJ, McNamara DP, Amidon GL. Toward an in vivo dissolution methodology: a comparison of phosphate and bicarbonate buffers. Mol. Pharm. 2009;6:29–39. doi: 10.1021/mp800148u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinko PJ, Amidon GL. Characterization of the oral absorption of beta-lactam antibiotics. II. Competitive absorption and peptide carrier specificity. J. Pharm. Sci. 1989;78:723–727. doi: 10.1002/jps.2600780904. [DOI] [PubMed] [Google Scholar]
- Sugano K. Aqueous boundary layers related to oral absorption of a drug: from dissolution of a drug to carrier mediated transport and intestinal wall metabolism. Mol. Pharm. 2010;7:1362–1373. doi: 10.1021/mp1001119. [DOI] [PubMed] [Google Scholar]
- Takagi T, Ramachandran C, Bermejo M, Yamashita S, Yu LX, Amidon GL. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006;3:631–643. doi: 10.1021/mp0600182. [DOI] [PubMed] [Google Scholar]
- Takano R, Furumoto K, Shiraki K, Takata N, Hayashi Y, Aso Y, Yamashita S. Rate-limiting steps of oral absorption for poorly water-soluble drugs in dogs; prediction from a miniscale dissolution test and a physiologically-based computer simulation. Pharm. Res. 2008;25:2334–2344. doi: 10.1007/s11095-008-9637-9. [DOI] [PubMed] [Google Scholar]
- Takano R, Kataoka M, Yamashita S. Integrating drug permeability with dissolution profile to develop IVIVC. Biopharm. Drug Dispos. 2012;33:354–365. doi: 10.1002/bdd.1792. [DOI] [PubMed] [Google Scholar]
- Tsume Y, Amidon GL. The biowaiver extension for BCS Class III drugs: the effect of dissolution rate on the bioequivalence of BCS Class III immediate-release drugs predicted by computer simulation. Mol. Pharm. 2010;7:1235–1243. doi: 10.1021/mp100053q. [DOI] [PubMed] [Google Scholar]
- Tubic-Grozdanis M, Bolger MB, Langguth P. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. AAPS J. 2008;10:213–226. doi: 10.1208/s12248-008-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertzoni M, Fotaki N, Kostewicz E, Stippler E, Leuner C, Nicolaides E, Dressman J, Reppas C. Dissolution media simulating the intralumenal composition of the small intestine: physiological issues and practical aspects. J. Pharm. Pharmacol. 2004;56:453–462. doi: 10.1211/0022357022935. [DOI] [PubMed] [Google Scholar]
- Vertzoni M, Dressman J, Butler J, Hempenstall J, Reppas C. Simulation of fasting gastric conditions and its importance for the in vivo dissolution of lipophilic compounds. Eur. J. Pharm. Biopharm.: Official J. Arbeitsgemein. Pharm. Verfahrenstech. e.V. 2005;60:413–417. doi: 10.1016/j.ejpb.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Vertzoni M, Diakidou A, Chatzilias M, Soderlind E, Abrahamsson B, Dressman JB, Reppas C. Biorelevant media to simulate fluids in the ascending colon of humans and their usefulness in predicting intracolonic drug solubility. Pharm. Res. 2010;27:2187–2196. doi: 10.1007/s11095-010-0223-6. [DOI] [PubMed] [Google Scholar]
- Wang Y, Abrahamsson B, Lindfors L, Brasseur JG. Comparison and analysis of theoretical models for diffusion-controlled dissolution. Mol. Pharm. 2012;9:1052–1066. doi: 10.1021/mp2002818. [DOI] [PubMed] [Google Scholar]
- Williams HD, Anby MU, Sassene P, Kleberg K, Bakala-N’Goma JC, Calderone M, Jannin V, Igonin A, Partheil A, Marchaud D, Jule E, Vertommen J, Maio M, Blundell R, Benameur H, Carriere F, Mullertz A, Pouton CW, Porter CJ. Toward the establishment of standardized in vitro tests for lipid-based formulations. 2. The effect of bile salt concentration and drug loading on the performance of type I, II, IIIA, IIIB, and IV formulations during in vitro digestion. Mol. Pharm. 2012a;9:3286–3300. doi: 10.1021/mp300331z. [DOI] [PubMed] [Google Scholar]
- Williams HD, Sassene P, Kleberg K, Bakala-N’Goma JC, Calderone M, Jannin V, Igonin A, Partheil A, Marchaud D, Jule E, Vertommen J, Maio M, Blundell R, Benameur H, Carriere F, Mullertz A, Porter CJ, Pouton CW. Toward the establishment of standardized in vitro tests for lipid-based formulations, Part 1: method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J. Pharm. Sci. 2012b;101:3360–3380. doi: 10.1002/jps.23205. [DOI] [PubMed] [Google Scholar]
- Williams HD, Trevaskis NL, Charman SA, Shanker RM, Charman WN, Pouton CW, Porter CJ. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013;65:315–499. doi: 10.1124/pr.112.005660. [DOI] [PubMed] [Google Scholar]
- Willmann S, Thelen K, Becker C, Dressman JB, Lippert J. Mechanism-based prediction of particle size-dependent dissolution and absorption: cilostazol pharmacokinetics in dogs. Eur. J. Pharm. Biopharm.: Official J. Arbeitsgemein. Pharm. Verfahrenstech. e.V. 2010;76:83–94. doi: 10.1016/j.ejpb.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Wilson WI, Peng Y, Augsburger LL. Comparison of statistical analysis and Bayesian Networks in the evaluation of dissolution performance of BCS Class II model drugs. J. Pharm. Sci. 2005;94:2764–2776. doi: 10.1002/jps.20358. [DOI] [PubMed] [Google Scholar]
- Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm. Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Tanaka Y, Endoh Y, Taki Y, Sakane T, Nadai T, Sezaki H. Analysis of drug permeation across Caco-2 monolayer: implication for predicting in vivo drug absorption. Pharm. Res. 1997;14:486–491. doi: 10.1023/a:1012103700981. [DOI] [PubMed] [Google Scholar]
- Yazdanian M, Briggs K, Jankovsky C, Hawi A. The “high solubility” definition of the current FDA Guidance on Biopharmaceutical Classification System may be too strict for acidic drugs. Pharm. Res. 2004;21:293–299. doi: 10.1023/b:pham.0000016242.48642.71. [DOI] [PubMed] [Google Scholar]
- Yu LX. Pharmaceutical quality by design: product and process development, understanding, and control. Pharm. Res. 2008;25:781–791. doi: 10.1007/s11095-007-9511-1. [DOI] [PubMed] [Google Scholar]
- Yun HY, Joo Lee E, Youn Chung S, Choi SO, Kee Kim H, Kwon JT, Kang W, Kwon KI. The effects of food on the bioavailability of fenofibrate administered orally in healthy volunteers via sustained-release capsule. Clin. Pharmacokinet. 2006;45:425–432. doi: 10.2165/00003088-200645040-00007. [DOI] [PubMed] [Google Scholar]