

Abstract
Lemurs are among the world's most threatened mammals. The critically endangered black-and-white ruffed lemur (Varecia variegata), in particular, has recently experienced rapid population declines due to habitat loss, ecological sensitivities to habitat degradation, and extensive human hunting pressure. Despite this, a recent study indicates that ruffed lemurs retain among the highest levels of genetic diversity for primates. Identifying how this diversity is apportioned and whether gene flow is maintained among remnant populations will help to diagnose and target conservation priorities. We sampled 209 individuals from 19 sites throughout the remaining V. variegata range. We used 10 polymorphic microsatellite loci and ∼550 bp of mtDNA sequence data to evaluate genetic structure and population dynamics, including dispersal patterns and recent population declines. Bayesian cluster analyses identified two distinct genetic clusters, which optimally partitioned data into populations occurring on either side of the Mangoro River. Localities north of the Mangoro were characterized by greater genetic diversity, greater gene flow (lower genetic differentiation) and higher mtDNA haplotype and nucleotide diversity than those in the south. Despite this, genetic differentiation across all sites was high, as indicated by high average FST (0.247) and ΦST (0.544), and followed a pattern of isolation-by-distance. We use these results to suggest future conservation strategies that include an effort to maintain genetic diversity in the north and restore connectivity in the south. We also note the discordance between patterns of genetic differentiation and current subspecies taxonomy, and encourage a re-evaluation of conservation management units moving forward.
Keywords: Conservation genetics, dispersal, genetic diversity, lemur, Madagascar
Introduction
Lemurs are among the world's most endangered mammals (IUCN 2013). Currently, 93 of 103 lemur taxa (90%) are classified as at least vulnerable and the number of species listed as Critically Endangered (i.e., at extremely high risk of extinction in the wild) has tripled since 2008 (Schwitzer et al. 2013). Understanding the genetic structure of these threatened populations, particularly those that exist in degraded or fragmented habitats, is not only an urgent priority for conservation efforts (Schwartz et al. 2006; Frankham 2010), but also relevant to developing environmental and climate change models (e.g., Ikeda et al. 2012).
Because of its unique biota (Ganzhorn et al. 2001), Madagascar is routinely identified as a global conservation priority (Myers et al. 2000; Robinson 2006). Since the 1950s, more than half of Madagascar's remaining forest cover has been cleared and forest edges have quadrupled (Harper et al. 2007). In fact, some authorities estimate as much as 85–90% of primary vegetation has already been lost (e.g., Myers et al. 2000). Land use practices, including logging, mining, and slash and burn agriculture (tavy), continue to threaten Madagascar's unique flora and fauna (McConnell 2002; Kull 2004; Mittermeier et al. 2010). Such practices have led to habitat loss and fragmentation that may potentially restrict or eliminate gene flow between subpopulations and result in rapid population declines and genetic bottlenecks. As a consequence, populations may be susceptible to reduced genetic diversity via drift and inbreeding depression (Nei 1975). This in turn poses significant threats to small, isolated populations by limiting their genotypic and phenotypic flexibility and long-term resilience to environmental changes (Madsen et al. 1999; Reed and Frankham 2003). Determining the distribution of genetic diversity and whether subpopulations are in migratory contact can highlight important dispersal corridors, as well as identify isolated populations, thereby suggesting priority areas for conservation (Schwartz et al. 2006).
The critically endangered black-and-white ruffed lemur (Varecia variegata) provides an ideal case study with which to investigate these relationships. Ruffed lemurs are medium-sized (3–4 kg; Baden et al. 2008), arboreal obligate frugivores that live in large, spatially dispersed social groups, also known as communities (Morland 1991; Vasey 2003; Baden and Gerber in review). This species is among the most frugivorous of the Malagasy primates (74–90%, Balko 1998), making it particularly sensitive to habitat degradation; in fact, ruffed lemurs are among the first to disappear in the face of habitat loss (White et al. 1995). Furthermore, their boom-bust reproductive strategy (i.e., long, synchronous interbirth intervals followed by “booms” in reproduction, whereby all breeding females within a community bear litters of 2–3 offspring; Baden et al. 2013) and slow life histories result in a relatively low reproductive rate (Baden et al. 2013). Previous population estimates suggest that fewer than 10,000 V. variegata individuals remain (Mittermeier et al. 2010). However, the species’ patchwork distribution throughout Madagascar's remaining eastern rainforest corridor makes accurate population estimates difficult and suggests that the true population size of this taxon could be far less (Irwin et al. 2005). These remaining V. variegata individuals are fragmented into several geographically distinct localities with limited potential for reproductive contact and unknown population structure. Individuals within these localities are under continued threat from habitat loss and fragmentation, and more recently bushmeat hunting, particularly in the northern distribution of their range (Golden 2009).
Previous studies have found evidence of genetic isolation and population decline (Holmes et al. 2013), as well as low haplotype diversity (Wyner et al. 1999) at several ruffed lemur sites. Nevertheless, recent comparative genomics research indicates that ruffed lemurs have among the highest measures of genetic diversity for primates (Perry et al. 2013), implying that this critically endangered lemur species may still harbor considerable genetic variation throughout parts of its range. Given the risk of rapid decline and isolation of these populations, there is an immediate need to understand how this genetic variation is distributed and what geophysical and/or anthropogenic barriers influence gene flow among localities. Furthermore, it is important to evaluate the extent to which existing subspecies designations (V. v. subcincta, V. v. variegata, V. v. editorum; Table 1; Fig. 1) are concordant with population structure to help gauge conservation priorities and inform captive management programs.
Table 1.
Sampling localities, subspecies designations and sample sizes used in this study
| Site name | Site code | Subspecies2 | Latitude | Longitude | n | Nm | Nf |
|---|---|---|---|---|---|---|---|
| Nosy Mangabe S.R.1 | NOSY | V. v. subcincta | S15°30′11.7″ | E049°45′30.5″ | 9 | 4 | 5 |
| Marotandrano S.R. | TANDRA | V. v. subcincta | S16°16′8.25″ | E048°49′08.3″ | 9 | 4 | 5 |
| Mananara Nord N.P. | NARA | V. v. subcincta | S17°34′36.5″ | E049°57′20.8″ | 8 | 4 | 4 |
| Ambatovaky S.R. | VAK | V. v. variegata | S16°49′01.4″ | E049°16′24.5″ | 5 | 4 | 1 |
| Zahamena N.P., S.N.R. | ZAHA | V. v. variegata | S17°29′21.0″ | E048°44′50.0″ | 10 | 3 | 7 |
| Betampona S.N.R. | BET | V. v. variegata | S17°55′87.1″ | E049°12′20.0″ | 9 | 5 | 4 |
| Mangerivola S.R. | VOLA | V. v. variegata | S18°14′11.4″ | E048°54′27.5″ | 3 | 1 | 2 |
| Mantadia Andasibe N.P. | TAD | V. v. editorum | S18°48′49.0″ | E048°25′47.8″ | 14 | 9 | 5 |
| Torotorofotsy U.F. | TORO | V. v. editorum | S18°50′07.7″ | E048°21′03.9″ | 3 | 1 | 2 |
| Maromizaha U.F. | MIZA | V. v. editorum | S18°58′30.2″ | E048°27′43.5″ | 2 | 1 | 1 |
| Anosibe an'ala C.F. | ANOSIB | V. v. editorum | S19°13′76.8″ | E048°16′86.0″ | 8 | 3 | 5 |
| Fandriana U.F. | FAN | V. v. editorum | S20°23′40.2″ | E047°38′09.8″ | 11 | 5 | 6 |
| Vatoharanana (Ranomafana N.P.) | VATO | V. v. editorum | S21°14′90.0″ | E047°25′26.6″ | 10 | 5 | 5 |
| Mangevo (Ranomafana N.P.) | MGV | V. v. editorum | S21°22′22.8″ | E047°26′59.1″ | 30 | 14 | 16 |
| Kianjavato U.F. | KIAN | V. v. editorum | S21°21′43.4″ | E047°50′54.3″ | 32 | 18 | 14 |
| Vatovavy U.F. | VAVY | V. v. editorum | S21°24′20.0″ | E047°56′26.0″ | 21 | 10 | 11 |
| Lakia U.F. | LAKI | V. v. editorum | S21°28′52.5″ | E047°53′29.0″ | 10 | 4 | 6 |
| Tolongoina U.F. | TOL | V. v. editorum | S21°35′30.0″ | E047°29′06.0″ | 4 | 2 | 2 |
| Manombo S.R. | MAB | V. v. editorum | S23°01′69.5″ | E047°43′84.1″ | 11 | 6 | 5 |
| Total sample | 209 | 103 | 106 |
UF, unclassified forest; CF, classified forest; SR, Special Reserve; SNR, Strict Nature Reserve; NP, National Park, n: total sample; Nm: number of males sampled; Nf: number of females sampled.
Introduced population.
Figure 1.

Map illustrating the estimated species distribution and current subspecies designations (A) and results from population structure analysis (B) illustrating the proportional membership (Q) of each ruffed lemur in the two clusters identified. Animals are each represented by a single horizontal bar. Locality of origin is indicated to the left of each individual (see Table 1 for full site names). Structure results are consistent using both biparentally and maternally inherited markers.
As long-term gene flow and the apportionment of genetic diversity are linked to short-term natal dispersal, we also test hypotheses regarding sex-biased dispersal in this species. Previous studies found some behavioral evidence of male transfer between communities (Morland 1991; Balko 1998), and females are generally considered the philopatric sex (Kappeler 1997; but see Erhart and Overdorff 2008). Accordingly, we predicted that black-and-white ruffed lemur communities would consist of unrelated males and closely related females, although some molecular evidence from red ruffed lemurs (V. rubra) suggests that both sexes disperse (Razakamaharavo et al. 2010).
Here, we describe the population genetic structure and dispersal patterns of black-and-white ruffed lemurs (V. variegata) as inferred from microsatellite markers and mitochondrial DNA sequence variation. Our analysis is unusually in that our samples were collected across the extent of the species’ range and thereby provides a species-level view of genetic apportionment for a critically endangered primate.
Methods
Sample collection and storage
We sampled a total of 209 adult individuals (103 males, 106 females) from 19 localities from across the existing V. variegata range (n = 2–32 individuals per locality; Table 1; Fig. 1). Distances between localities ranged from six to 860 km. To obtain these data, field assistants from Omaha's Henry Doorly Zoo and Aquarium (OHDZA) and the Madagascar Biodiversity Partnership (MBP) immobilized study individuals with 10 mg/kg estimated body weight of Telazol® (Fort Dodge Animal Health, IA), administered by Dan-Inject (Børkop, Denmark) Model JM CO2-powered projection rifle and 9 mm disposable Pneu-Darts™ (Williamsport, PA). Whole blood (1 mL/kg) samples were collected from the femoral vein and stored at room temperature in 5 mL of lysis buffer solution (0.1 mol/L Tris-HCl pH, 8.0, 0.1 mol/L EDTA, 0.01 mol/L NaCl, and 0.5% w/v SDS) (Seutin et al. 1991) until they were banked in a −80°C freezer at the OHDZA. Sample collection occurred under veterinary supervision and followed a strict protocol outlined by Glander (1993). All capture procedures occurred during nonreproductive seasons in the absence of infants and dependent offspring.
Additional noninvasive sampling occurred in two sites (Kianjavato: n = 20 individuals, and Vatovavy, n = 11 individuals). In these cases, SMH, MBP, and field assistants collected fecal samples from ruffed lemurs that had been previously collared for individual identification. Researchers and assistants collected 2–5 samples per lemur and removed seeds from fecal samples prior to preservation in RNAlater® (Life Technologies, Grand Island, NY) at a ratio of 1 mL feces to 5 mL RNAlater®. Samples were kept at room temperature for 15–105 days until transported to OHDZA, where they were stored at −20°C.
Importantly, samples were collected from only adult individuals to minimize the chance of sampling parent/offspring pairs, and samples were collected across different social groups. Furthermore, our analysis of relatedness and sex-biased dispersal did not indicate clusters of close relatives within sampling locales.
Immobilizations, handling, sample collections, and export/import protocols adhered to and were approved by the OHDZA's Institutional Animal Care and Use Committee (IACUC #97-001), Stony Brook University IACUC (#2005-20081449), University of Calgary Life and Environmental Sciences Animal Care Committee (BI11R-15), Malagasy wildlife authorities, Convention on International Trade in Endangered Species regulations, and US Fish & Wildlife Service.
DNA extraction
Total genomic DNA was extracted from blood and fecal samples using standard nucleic acid extraction kits (QIAamp® DNA Mini Kit & DNA Stool Mini Kit; QIAGEN, Valencia, CA) following the manufacturer's protocols. Ten microsatellite loci (Louis et al. 2005), which regularly and reliably amplified fecal DNA in initial tests, were used to genotype all individuals (see Table S1).
DNA from blood
Microsatellite loci were amplified in 25 μL reactions consisting of 2 μL DNA template (50–80 ng), 12.5 μL QiagenHotStarTaq Master Mix and 10 μmol/L of each primer. Amplification conditions were as follows: initial denaturation at 95°C for 15 min; 35 cycles of 30 sec at 94°C, 40 sec at 54 to 60°C (see Louis et al. 2005), 1 min at 72°C, and a final extension of 7 min at 72°C. The 5′ end of the forward primer was fluorescently labeled, and amplification products were separated using capillary electrophoresis (ABI 3730xl Genetic Analyzer).
DNA from feces
For the low-quality fecal DNA extracts, we carried out PCR amplifications in a 25 μL volume with 4 μL template (20–50 ng), 12.5 μmol/L of each primer, 1.5 mmol/L MgCl2, 200 μmol/L dNTP, 10 mmol/L Tris (pH 8.3), 50 mmol/L KCl, and 0.5 U Taq DNA polymerase (Promega; Madison, WI). Thermal cyclers profiles were as follows: 35 cycles of 30 sec at 95°C, 30 sec at 54 to 60°C, and 30 sec at 72°C with a 10 min final extension phase at 72°C (see also Table S1).
Microsatellite genotyping
Amplification products were separated using capillary electrophoresis (ABI 3730xl Genetic Analyzer), and alleles were sized relative to an internal size standard (ROX-500) using Gene Mapper software v. 4.0 (Applied Biosystems, Foster City, CA). To detect and avoid allelic dropout, multiple PCR replicates were performed according to the concentration of DNA in each sample (Morin et al. 2001). Final genotypes were scored based on multiple independent reactions (Taberlet et al. 1996); all heterozygotes were confirmed by a minimum of two separate reactions and homozygous genotypes were typically confirmed via five amplifications. DNA yields from fecal samples were substantially lower than from blood samples, thereby sometimes requiring greater numbers of replicates (range 3–11) to confirm homozygous genotypes (as in Morin et al. 2001). The observed probability of identity (PID; Paetkau and Strobeck 1994) for all markers was 9.51 × 10−15, demonstrating the very low probability that two individuals would share the same multilocus genotype.
mtDNA sequencing
For a geographically representative subset of individuals (n = 159), we amplified the D-loop or control region of the mitochondrial DNA (mtDNA) using primers dLp5 (Baker et al. 1993) and DLp1.5 (Wyner et al. 1999). We generated 555 bp fragments using 50 ng of DNA and the following conditions: 94°C for 4 min, 35 cycles of 94°C for 30 sec, 47°C for 45 sec, 72°C for 45 sec and 72°C for 10 min.
To exclude potential amplification of nuclear insertions, we subsequently generated the PCR products with a quick, efficient species independent technique derived from the degenerate oligonucleotide primed PCR method (DOP-PCR; Telenius et al. 1992). Adapting the long products from low-quantity DOP-PCR methodology (LL-DOP-PCR), we verified sequence data generated from overlapping segments for the D-loop, COII, 12S rRNA, and PAST PCR fragments. Amplifications were carried out on a MBS Satellite 0.2G Thermal Cycler (Thermo Electron Corporation; Waltham, MA) and verified by electrophoresing samples on a 1.2% agarose gel. We purified samples using the QIAquick PCR purification kit (Qiagen), cycle-sequenced them using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) and generated sequences with a 7% polyacrylamide gel by an ABI 377 automated sequencer (Applied Biosystems).
Individual sequences were analyzed, edited and aligned using Sequencher 4.9 (Gene Corp; Ann Arbor, MI). Final alignment of all sequences was performed using ClustalX software (Thompson et al. 1997) and checked by eye. Notably, each sample yielded a single clear, unambiguous sequence (i.e., no evidence of heteroplasmy), further indicating that amplicons were not a mixture of mitochondrial and nuclear targets.
Population genetic analyses
Genetic diversity
All loci were tested for the presence of null alleles using Micro-Checker (van Oosterhout et al. 2004) and linkage disequilibrium using Genepop v.3.4 (Raymond and Rousset 1995). Departures from equilibrium were evaluated with 10,000 permutations. We used GenoDive v.2.0b23 (Meirmans and Van Tienderen 2004) to calculate measures of genetic diversity, including the number of alleles per locus (nA), the mean number of alleles per sampling locality (MNA), allelic richness per locality (AR), and observed (HO) and expected (HE) heterozygosities (Nei 1978) for each sampling locality. To account for differences in sample size (Kalinowski 2004), allelic richness (AR) was standardized to the smallest sample size in the dataset using rarefaction implemented in HP-Rare 1.0 (Kalinowski 2005). Finally, we estimated Wright's FIS (a measure of deviation from Hardy–Weinberg equilibrium) according to Weir and Cockerham (1984) and tested populations with a sufficient sample set for significant deviations from equilibrium with 10,000 permutations.
Inferring population genetic structure
We used two methods to infer population structure from our sample of 209 ruffed lemur microsatellite genotypes. We first used the model-based Bayesian clustering method implemented in Structure v2.3.4 (Pritchard et al. 2000) to infer the optimal number of genetic populations (K) as suggested by the microsatellite data. This method uses the Markov Chain Monte Carlo (MCMC) approach to group individuals into K populations based on their multilocus genotypes without prior information regarding their sampling localities (i.e., USEPOPINFO was not specified). We also calculated the fractional membership of individuals within each population (Q). We evaluated the hypotheses K = 1–22, the number of sampling locations plus 3, following Evanno et al. (2005). From a pilot study, we determined that 50,000 iterations of burn-in followed by 100,000 iterations of Markov Chain Monte Carlo (MCMC) were sufficient to allow convergence of parameters prior to data collection; longer burn-in or MCMC did not result in significant changes in our results (data not shown). Because different runs produced different likelihood values, we carried out 20 runs for each value of K assuming correlated allele frequencies and admixture. Using the admixture model allowed us to estimate the number of natural genetic clusters and detect historical population admixture (Falush et al. 2003; Ostrowski et al. 2006). We identified the most likely number of populations (K) using the ΔK method (Evanno et al. 2005) implemented in Structure Harvester v0.6.93 (Earl and vonHoldt 2012). Using this method, optimum K is identified by the highest value of ΔK, or the second order rate of change in the likelihood of K, which corresponds to the most pronounced genetic subdivision present within the data. For the chosen value of K, we averaged Q across the 20 independent runs. As the ΔK method generally identifies the highest level of structure in the dataset, we took a two-step approach. First, we identified the most likely number of clusters within the overall sample (n = 209). We then ran subsequent analyses within each of the K clusters from the original run following Evanno et al. (2005) to evaluate whether further substructure existed.
To corroborate Structure results, we performed a principal coordinates analysis (PCoA) with a standard genetic distance matrix (Nei 1978) using GenAlEx v.6.5 (Peakall and Smouse 2012).
Finally, we performed a follow-up exclusion test (Cornuet et al. 1999) in Geneclass 2.0 (Piry et al. 2004). Using population simulations, we statistically tested whether one or more of the sampled localities could be ruled out as the area of origin for each individual. The probability of individual genotypes coming from each locality was calculated by comparing individual genotypes to 10,000 simulated individuals per locality. We used the default criteria for computation parameters and selected the Paetkau et al. (2004) simulation method.
Results from the Bayesian cluster assignments and PCoA guided subsequent analyses. First, we examined microsatellite population genetic structure with a locus-by-locus Analysis of Molecular Variance (AMOVA, Excoffier et al. 1992) implemented in GenoDive. We used permutation tests of 10,000 iterations to examine the distribution of genetic variation at four hierarchical levels: among populations (i.e., K clusters), among sampling localities within populations, among individuals within sampling localities, and within individuals. Distances were calculated using the Infinite Alleles Model (FST analog).
We also compared allelic diversity at each microsatellite marker between the inferred K populations using the log-likelihood G test of genotypic variation implemented in Genepop (Rousset 2008). Significance was calculated using 10,000 randomizations not assuming HWE (Goudet et al. 1996). Finally, we performed pairwise tests for population differentiation (FST) in GenoDive. Significance was calculated using 10,000 randomizations not assuming HWE and corrected for multiple comparisons (Bonferroni adjusted P = 0.0004).
Dispersal or migration barriers can hinder gene flow, thereby increasing genetic distance, even in cases where populations are not geographically distant (Liu et al. 2009; Quéméré et al. 2009). Thus, to investigate the relationship between genetic distance among sampling localities and their geographic distances, we performed tests of spatial autocorrelation (isolation-by-distance) implemented in GenAlEx. We used the Geographic Distance Matrix Generator v.1.2.3 from the American Museum of Natural History (http://biodiversity-informatics.amnh.org/open_source/gdmg/) to calculate pairwise geographic distances (in km) between all sampling localities based on their decimal degree coordinates. This matrix was then compared with a matrix of Nei's genetic distances (described previously) using Mantel matrix correlations. Significance was evaluated based on 9,999 permutations.
To examine mtDNA population genetic structure, we used a standard AMOVA for haplotype data implemented in GenAlEx. We used permutation tests of 9,999 iterations, this time at three hierarchical levels (within individual comparisons are not possible in haplotype AMOVAs including only one locus): among populations, among sampling localities within populations, and among individuals within sampling localities. We calculated nucleotide and haplotype diversity using DnaSP v5 (Librado and Rozas 2009) and inferred haplotype networks of mtDNA sequences using a median-joining algorithm (Bandelt et al. 1999) implemented in Network v.2.2 (Fluxus, Clare, Suffolk, UK). Epsilon (ε) was set equal to zero and variable sites were weighted equally. Finally, we performed pairwise tests for population differentiation (ΦST) in GenoDive. Significance was calculated using 10,000 randomizations not assuming HWE and corrected for multiple comparisons (Bonferroni adjusted P = 0.0004).
Inferences of population dynamics
Sex-biased dispersal
We evaluated whether dispersal was sex-biased following methods described by Goudet et al. (2002) and implemented in Fstat 2.9.3 (Goudet 2001). For both males and females, we estimated and compared the following measures: levels of inbreeding (FIS); average relatedness (R); population differentiation (FST); mean Assignment Indices (mAIc); and variance in Assignment Indices (vAIc). FIS represents a measure of how well genotype frequencies within a population match expectations of Hardy–Weinberg Equilibrium (Hartl and Clark 1997) and can be used to detect a reduction in heterozygosity that is typically caused by population substructure. Because the dispersing sex in a population often includes a combination of both immigrants and residents, the admixture of these two populations should lead to a resultant heterozygote deficiency (and a positive FIS) within the dispersing sex. The dispersing sex should also have lower average relatedness (R) among postdispersal aged members of a population than members of the more philopatric sex because dispersal reduces the likelihood that relatives are living in close association (Greenwood 1980; Goudet et al. 2002; but see Lukas et al. 2004). Consequently, FST, or the measurable proportion of genetic variance attributable to among-population differentiation, should be lower in the dispersing sex because the less philopatric sex should be less differentiated in its allele frequencies among populations (i.e., increased gene flow yields fewer genetic differences between populations in the dispersing sex) (Hartl and Clark 1997). Finally, members of the dispersing sex should show significantly lower mean Assignment Indices, but higher variance than members of the more philopatric sex (Lawson Handley and Perrin 2007). Assignment Indices are statistics that summarize the likelihood that an individual's multilocus genotype originated in the population from which it was sampled and can be used to test for differences in the mean values (mAIc) and the variance (vAIc) of assignments between the sexes. These indices can then be standardized, subtracting the population mean AI from each individual's AI (Favre et al. 1997), such that animals with positive “corrected” assignment indices (AIc) are those which are more likely to have been born in the population, while immigrant genotypes are less likely to occur in the sample and should therefore have negative AIc values (Goudet et al. 2002). In sum, compared with the philopatric sex, the dispersing sex is predicted to have (1) positive FIS values, (2) lower average relatedness, (3) lower values of FST, (4) lower mean assignment scores (mAIc) and 5) greater variance in assignment (vAIc). We calculated two-tailed P-values using 10,000 randomizations, where sex was randomly assigned to genotypes while keeping the sex ratio and group identity constant thereby producing a null distribution (see Table 4). We also compared the average number of haplotypes shared by males and by females using a two-tailed Student's t-test.
Table 4.
Mean values and tests of sex-biased dispersal using microsatellite data from n = 92 adult males and n = 93 adult females
| Results | |||||
|---|---|---|---|---|---|
| Test | Predicted | Male | Female | Observed | P |
| FIS | + | 0.264 | 0.261 | = | 0.94 |
| R | − | 0.223 | 0.216 | = | 0.79 |
| FST | − | 0.153 | 0.148 | = | 0.77 |
| mAIc | − | 0.218 | −0.216 | + | 0.54 |
| vAIc | + | 24.853 | 20.539 | + | 0.48 |
| Haplotypes | + | 1.420 | 1.260 | = | 0.37 |
Predictions based on previous evidence of male-biased dispersal.
Results based on 10,000 randomizations in FSTAT.
Significance in haplotype number tested using a two-tailed Student's t-test.
Bottleneck analyses
Finally, we tested for genetic signatures of recent population decline using Bottleneck software (Luikart and Cornuet 1998; Piry et al. 1999). We used a Wilcoxon signed-ranks test to compare observed and expected heterozygosity at mutation-drift equilibrium (HEeq) because of its robusticity to small sample sizes (<30) and small numbers of loci (<20) (Piry et al. 1999). As the mutation model underlying the real data is never known and is likely to change from locus to locus, we used three models (IAM: infinite alleles model; SMM: stepwise mutation model; and TPM: two-phase mutation model) in parallel to assess whether departures from mutation-drift equilibrium were robust under all models or sensitive to model changes (e.g., Goossens et al. 2006). Default parameters were used and significance was evaluated with 10,000 replications.
Results
Genetic diversity
All ten loci were polymorphic with 4–13 alleles each (Table S1). We pooled individuals from across sampling localities and found no evidence of significant linkage disequilibrium across markers. Deviations from Hardy–Weinberg equilibrium (HWE) were present in 15 of 190 possible locus-site combinations (10 loci × 19 sites), likely reflecting the relatively small sample size for some localities (Table S1). Of these, five loci had positive values and two loci had negative values of FIS and no locus stood out as an outlier. For these reasons, all loci were kept in the analysis. Sampling localities averaged between 2.2–4.7 alleles (Table S2).
Population genetic structure
From our first Structure analysis of 209 individuals, we identified two genetic clusters, as indicated by the highest value of ΔK (Fig. 1; Fig. S1). These results were the same whether we used biparentally or maternally inherited markers. From the microsatellite analyses, Cluster 1 consisted exclusively of individuals from the eleven northern-most sampling localities (80 of 209 members; Table 1; Fig. 1), and Cluster 2 comprised only individuals sampled from the eight southernmost sampling localities (129 of 209 members; Table 1; Fig. 1). It is important to note that these genetic clusters did not correspond to the current taxonomy of the species that separates V. variegata into three separate subspecies (Table 1; Fig. 1).
We repeated the analysis with each of the K = 2 clusters separately following Evanno et al. (2005) and found that Cluster 1 (northern sampling localities) and Cluster 2 (southern sampling localities) could each be further subdivided into K = 2 clusters (Figs. S2 and S3). In both cases, sampling localities appear to cluster primarily by latitude (i.e., geographic location) and perhaps also according to habitat connectivity. Sampling localities from the original Cluster 1 (North) grouped into K = 2 distinct subpopulations (Subcluster 1.1: Nosy, Tand, Nara; Subcluster 1.2: Tad, Toro, Miza, Anosib). Sites located between these localities (Vak, Zaha, Bet, Vola) shared varying degrees of proportional membership with each of the K = 2 clusters. Furthermore, we observed a second, albeit small peak at K = 4 (Fig. S2). Subdivision still occurred mainly according to geographic proximity. The four southernmost populations (Tad, Toro, Miza, Anosib) still clustered into a single population. However, with the exception of Nosy, northern sampling localities (Tandra, Nara, Vak, Zaha, Bet, Vola) exhibited substantial admixture and shared varying levels of proportional membership among the remaining K = 3 clusters. It is interesting to note, however, that animals from Nosy, an island population whose founders were introduced in the 1930s (Petter and Peyreiras 1970), cluster almost exclusively with individuals from Nara, perhaps suggesting their provenance (Fig. S3).
Similar patterns of substructure were detected among populations in the south. Localities within the original Cluster 2 (South) grouped into K = 2 subpopulations. The two sampling localities from within Ranomafana National Park (Vato, Mgv) clustered together with the northernmost (Fan) and southernmost localities (Mab) in Subcluster 2.1, while the fragmented (and geographically proximate) habitats of Kian, Vavy, and Laki clustered together in a second subcluster (Subcluster 2.2). Tol exhibited substantial admixture of the two.
The principle coordinate analysis (PCoA; Fig. 2) corroborated Structure results, in that it showed a clear separation along axis 1 (PCo1) that grouped sampling localities into two clusters. Clusters showed minimal overlap and appeared to separate according to geographic location. Sampling localities north of the Mangoro River clustered together (bottom right), as did sampling localities to the south of the river (top left). This component accounted for 35.2% of the total molecular variance. Another 17.9% of the variance was explained by Axis 2 (PCo2), which seemed to primarily separate sampling localities within the south. It is interesting to note that, again, the genetic clustering did not correspond to subspecies status (Fig. 2).
Figure 2.
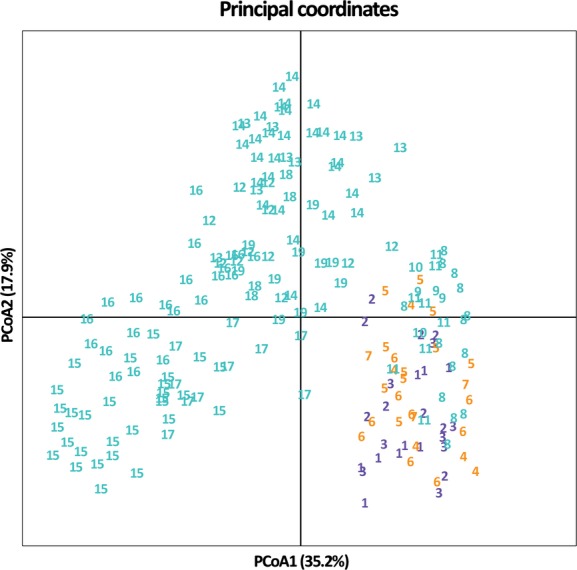
Principle coordinate analysis (PCoA). Data points are represented by numbers that correspond to sampling locality (1–19) and are color coded according to current subspecies assignments (purple: Varecia variegata subcincta; orange: V. v. variegata; blue: V. v. editorum).
From the microsatellite data, an exclusion test with resampling accurately assigned 91.4% of individuals to both their correct sampling locality and cluster when considering the locality of highest probability (Fig. 3). In the 8.6% of cases where individuals were incorrectly assigned, most were assigned to localities belonging to the same cluster (7.7%). Less than 1% of individuals were incorrectly assigned to both sampling locality and cluster/population. However, in 97.9% of the correct assignments, additional localities other than that with highest probability could not be ruled out as the source population. This was true for individuals in the northern cluster (73 of 73 individuals; 100%) and the southern cluster (114 of 118; 96.6%).
Figure 3.
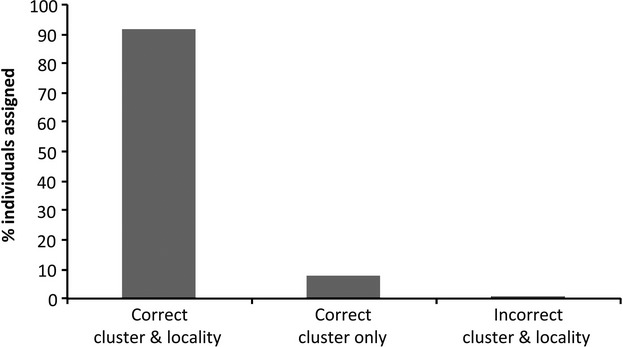
Distribution of highest probability assignments as determined using the resampling procedure in GENECLASS.
Allelic diversity within the two clusters ranged from four to 13 alleles (mean = 8.9); four to 11 alleles (mean = 8) in the north and three to 13 (mean = 6.6) in the south (Table 2). Genotypic differentiation between the two clusters was highly statistically significant overall (P < 0.0001), as well as for each of the 10 markers individually (Table 2). Results from the locus-by-locus AMOVA revealed pronounced levels of structure in the microsatellite data (Table 3). Although the highest percentage of variation was within individuals, due to high levels of heterozygosity, there was significant variation apportioned to all hierarchical levels of V. variegata.
Table 2.
Comparison of number of alleles between northern (n = 80) and southern (n = 129) populations of V. variegata. P-values correspond to 10,000 randomizations of log-likelihood G tests of population differentiation for each marker
| No. alleles | ||||
|---|---|---|---|---|
| Marker | North | South | Total | G test P-value |
| 51HDZ20 | 11 | 8 | 13 | <0.001 |
| 51HDZ25 | 5 | 3 | 5 | <0.001 |
| 51HDZ204 | 4 | 3 | 4 | <0.001 |
| 51HDZ247 | 10 | 7 | 10 | <0.001 |
| 51HDZ560 | 9 | 7 | 10 | <0.001 |
| 51HDZ598 | 8 | 7 | 9 | <0.001 |
| 51HDZ691 | 11 | 11 | 13 | <0.001 |
| 51HDZ790 | 4 | 4 | 5 | <0.001 |
| 51HDZ816 | 10 | 8 | 10 | <0.001 |
| 51HDZ988 | 8 | 8 | 10 | <0.001 |
| Mean | 8.0 | 6.6 | 8.9 | |
Table 3.
Locus-by-locus AMOVA of 10 microsatellite markers for 209 V. variegata individuals and standard AMOVA for haplotype data of mtDNA d-loop sequences (n = 159). P is based on 10,000 permutations. df = degrees of freedom, SS = sum of squared deviations, MS = mean of squared deviations
| Variance component | df | SS | MS | Variation | Proportion of total variation | Statistic | P |
|---|---|---|---|---|---|---|---|
| Locus-by-locus microsatellite AMOVA | |||||||
| Among populations (northern and southern) | 1 | 126.97 | – | 0.543 | 0.136 | Fct | <0.001 |
| Among sites within populations | 17 | 266.55 | – | 0.601 | 0.150 | Fsc | <0.001 |
| Among individuals within sites | 190 | 585.79 | – | 0.233 | 0.058 | FIS | <0.001 |
| Within individuals | 209 | 547.00 | – | 2.617 | 0.655 | FIT | <0.001 |
| Standard mtDNA haplotype AMOVA | |||||||
| Among populations (northern and southern) | 1 | 253.45 | 253.454 | 2.703 | 0.328 | PhiRT | <0.001 |
| Among sites within populations | 17 | 574.06 | 33.768 | 3.905 | 0.474 | PhiPR | <0.001 |
| Among individuals within sites | 140 | 227.60 | 1.626 | 1.626 | 0.197 | PhiPT | <0.001 |
Pairwise FST comparisons suggest the same pattern of relationships as the Bayesian methods and are consistent with results from the locus-by-locus AMOVA suggesting significant levels of genetic differentiation both between northern and southern clusters and among sampling localities (Table S3). Overall, genetic differentiation among sampling localities as measured by FST was significant in 101 of 171 cases (Table S3). Pairwise values of FST among sampling localities ranged from 0.002 to 0.442, with a mean of 0.241, though these are viewed with caution given the range of sample sizes (N = 2–32, Table 1). Within-cluster FST values were almost always lower than between-cluster comparisons (data not shown). Somewhat contrary to the individual-based analyses, most sampling localities show significant divergence from one another; however, this is more prevalent among southern sampling localities (24 of 28 pairs differed significantly; 85.7%) than among localities in the north (17 of 55 pairs; 30.9%).
In addition, we found strong evidence of isolation-by-distance (i.e., a significant positive correlation between genetic and geographic distance matrices among individuals; r2 = 0.501 P < 0.0001). This same pattern held true when looking among individuals within either of the two clusters (North: r2 = 0.424, P < 0.0001; South: r2 = 0.428, P < 0.0001).
Aligning 159 V. variegata mtDNA sequences (North n = 83; South n = 76), we found 19 haplotypes and 44 polymorphic sites (Table S4). Of these 44 single nucleotide polymorphisms (SNPs), there were no fixed differences and no haplotype sharing between northern and southern clusters. The mitochondrial DNA of the northern cluster was much more diverse (43 SNPs; 16 haplotypes) than the southern cluster (2 SNPs; 3 haplotypes). These results were reflected in median-joining network analysis, which again shows clear clustering into northern and southern groups (Fig. 4).
Figure 4.
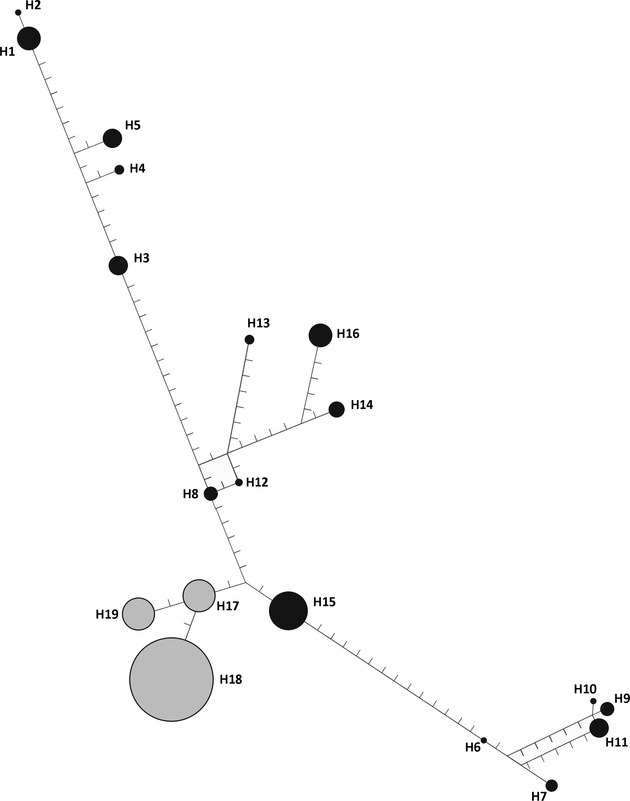
Haplotype networks of V. variegata mtDNA d-loop sequences created using a median-joining algorithm implemented in NETWORK. Shading indicates populations identified from STRUCTURE analyses (black = northern; gray = southern). Size of the node corresponds to the frequency of that haplotype among sampled individuals. Internal nodes represent reconstructed median haplotypes. Notches represent nucleotide differences between haplotypes.
The standard AMOVA for haplotype data shows strong differentiation among all hierarchical levels of the analysis (Table 3). Population differentiation is stronger for mtDNA than microsatellite markers, with variation among populations explaining 33% of the total variation in mtDNA, while only 14% of total variation in microsatellite markers is explained by variation among populations. Similarly, variation among sites within populations explains nearly half (48%) of the total variation in mtDNA, whereas among-site variation explains only 17% in microsatellites (Table 3).
As with measures of pairwise FST, comparisons of ΦST suggest the same pattern of relationships as the Bayesian methods and correspond with results from the standard AMOVA suggesting significant levels of genetic differentiation both within clusters and among sampling localities (Table S5).
Population dynamics, sex-biased dispersal and gene flow
We did not find evidence of sex-biased dispersal (Table 4). Estimates of R, FST, and FIS did not differ significantly between males and females, suggesting that both sexes are equally likely to disperse. Although mAIc was positive for males (mean = 0.218) and negative for females (mean = −0.216), this difference was not significant. Moreover, variance in AIc was very high for both sexes, and differences were not significant. Furthermore, males and females did not differ significantly in their number of haplotypes across sampling localities (two-tailed Student t-test, P = 0.369).
Finally, using Bottleneck, we found evidence of deviations from mutation-drift equilibrium at 5 of 9 sampling localities when either the IAM or TPM models were assumed (Table 5). However, 10 of 19 sampling localities were excluded from this analysis due to small sample sizes.
Table 5.
Analysis of past population bottleneck events under each of three mutation models
| Mutation model | |||||
|---|---|---|---|---|---|
| Site code | n | IAM | TPM | SMM | Mode shift |
| NOSY | 9 | – | – | – | – |
| TANDRA | 9 | – | – | – | – |
| NARA | 8 | – | – | – | – |
| VAK | 5 | – | – | – | – |
| ZAHA | 10 | 0.053 | 0.246 | 0.500 | Shifted |
| BET | 9 | – | – | – | – |
| VOLA | 3 | – | – | – | – |
| TAD | 14 | 0.001 | 0.001 | 0.019 | Shifted |
| TORO | 3 | – | – | – | – |
| MIZA | 2 | – | – | – | – |
| ANOSIB | 8 | – | – | – | – |
| FAN | 11 | 0.065 | 0.348 | 0.920 | Normal |
| VATO | 10 | 0.116 | 0.246 | 0.652 | Normal |
| MGV | 30 | 0.000 | 0.001 | 0.053 | Shifted |
| KIAN | 32 | 0.216 | 0.500 | 0.839 | Normal |
| VAVY | 21 | 0.001 | 0.019 | 0.213 | Normal |
| LAKI | 10 | 0.001 | 0.001 | 0.001 | Shifted |
| TOL | 4 | – | – | – | – |
| MAB | 11 | 0.012 | 0.053 | 0.116 | Shifted |
| Total | 209 | ||||
IAM: infinite allele model; TPM: two-phase model; SMM: stepwise mutation model Significant P-values (bold) indicate an excess of heterozygosity under each of three mutation models. Mode shift provide qualitative description of shifts from low to medium frequency alleles in a population. Significance calculated using one-tailed Wilcoxon signed-ranks test. P < 0.05. Samples with fewer than 10 samples were not included in this analysis.
Discussion
Genetic diversity and population structure
Together, our analyses show significant genetic differentiation among sampling localities, with a primary division north and south of the Mangoro River (Figs. 1B, 2 and 4). This division does not, however, correspond to the current taxonomy (Fig. 1A; see below). We also found some degree of substructure within each cluster, though this was more pronounced in southern versus northern sampling localities; in the south, substructure was inconsistent with geographic clustering of sites (Fig. S3). While Structure cannot detect fewer than K = 2 genetic clusters, given that PCoA and exclusion analyses both support this same pattern of clustering, we regard K = 2 populations as the best-supported hypothesis at this time (see also Evanno et al. 2005).
Localities characterized as forest fragments (i.e., unclassified & classified forests, Table 1) show clear patterns of isolation-by-distance (IBD) both within and between northern and southern clusters. Over time, genetic drift is expected to eliminate patterns of IBD. Therefore, the spatial autocorrelation observed in this study could be indicative of relatively recent interconnectivity among localities via forest corridors (e.g., Ranomafana–Andringitra Corridor; Mittermeier et al. 2005).
Despite high levels of of FST and IBD, sampling localities clustered together, regardless of whether samples derived from forest fragments or national parks within larger forest blocks, and these patterns were true whether we used biparentally or maternally inherited genetic markers. This is in contrast to previous studies that have found comparatively more substructure among fragmented habitats versus continuous forest sites, despite being separated by comparable geographic distances (Olivieri et al. 2008; Oaklander et al. 2010; Schneider et al. 2010; Holmes et al. 2013). Together, our results suggest that while forest fragmentation and habitat loss have increased genetic differentiation among sampling localities, it may have occurred recently enough that genetic differentiation has not yet increased beyond the drift effects of pure isolation-by-distance.
Looking within each of the two genetic clusters, we found that V. variegata individuals located in northern sampling localities are characterized by significantly higher allelic diversity, greater genetic and haplotypic diversity, and higher levels of gene flow than V. variegata individuals located within the southern cluster of sampling localities. It is possible that different environmental and/or landscape conditions are operating in the two geographic regions to produce these divergent results. Further investigation (e.g., landscape genetic analysis) will help to better understand the patterns observed herein.
Nevertheless, our results clearly indicate that gene flow among localities is limited. Average FST for this species is the highest observed in any lemur study to date (Table 6). Moreover, although ruffed lemurs are distributed over a relatively narrow geographic range (i.e., the eastern rainforest corridor of Madagascar), their level of genetic differentiation is an order of magnitude greater than chimpanzees distributed across all of (western to eastern) equatorial Africa (average FST = 0.014, Langergraber et al. 2011). These results highlight the importance of taking a species-specific approach when identifying potential dispersal barriers (i.e., barriers to some species may not hinder dispersal in others; Baguette and Van Dyck 2007).
Table 6.
Population differentiation according to FST
| Sampling locality | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | Species | Sample | Marker | Sample N | Entire range? | N | Min FST | Max FST | Mean FST ± SD | Inferred K clusters | Primary structuring factors |
| Daubentoniidae | Daubentonia madagascariensis1 | Tissue | Genomic DNA (666,256 SNPs) | 12 | No | 8 | (0.129) | (0.194) | (0.164 ± 0.033) | 3 | – |
| Cheirogaleidae | Microcebus bongolavensis2 | Tissue | 8 microsats | 45 | No | 3 | 0.057 | 0.102 | 0.076 ± 0.023 | 1 | Rivers & geographic distance |
| Microcebus danfossi2 | Tissue | 8 microsats | 78 | No | 7 | 0.025 | 0.195 | 0.096 ± 0.049 | 2 | Rivers & geographic distance | |
| Microcebus murinus3 | Tissue | 10 microsats | 167 | No | 3 | 0.004 | 0.016 | – | 1 | – | |
| Microcebus ravelobensis4 | Tissue | 8 microsats | 187 | No | 12 | −0.002 | 0. 122 | 0.052 ± 0.027 | 3 | Road or other (unknown) & geographic distance | |
| Microcebus ravelobensis2 | Tissue | 8 microsats | 205 | No | 8 | 0.006 | 0.156 | 0.072 ± 0.035 | 2 | Rivers & geographic distance | |
| Lepilemuridae | Lepilemur mustelinus5 | Blood | 3 enzyme loci | 72 | No | 4 | −0.026 | 0.133 | 0.055 ± 0.080 | – | – |
| Indriidae | Propithecus tattersali6 | Feces | 13 microsats | 82 | No | 3 | 0.136 | 0.160 | (0.147) | 2 | Rivers & geographic distance; Road not a barrier |
| Propithecus tattersali7 | Feces | 13 microsats | 230 | Yes | 9 | 0.010 | 0.300 | 0. 119 ± 0.067 | 3 | Rivers & geographic distance; Road not a barrier | |
| Propithecus verreauxi8* | Tissue | 7 microsats | 77–131 | No | 10–28 | 0.024 | 0.075 | 0.052 | – | – | |
| Lemuridae | Eulemur cinereiceps9 | Tissue | 26 microsats | 53 | No | 4 | 0.020 | 0.076 | 0.054 ± 0.019 | – | Geographic distance |
| Varecia rubra10 | Tissue | 15 microsats | 32 | No | 2 | – | – | 0.077 | – | – | |
| Varecia variegata11 | Blood, feces | 16 microsats | 55 | No | 5 | 0.039 (0.193) | 0.291 (0.229) | 0.197 ± 0.084 (0.212) | 3 | – | |
| Varecia variegata12 | Blood, feces | 10 microsats | 209 | Yes | 19 | 0.002 | 0.441 | 0.247 ± 0.094 (0.163) | 2 | Rivers & geographic distance | |
Perry et al. (2013).
Olivieri et al. (2008).
Fredsted et al. (2005).
Radespiel et al. (2008).
Tomiuk et al. (1997).
Quéméré et al. (2009).
Quéméré et al. (2010).
Lawler et al. (2003).
Brenneman et al. (2011).
Razakamaharavo et al. (2010).
Holmes et al. (2013).
this study.
FST comparisons are among sampling localities unless otherwise noted. Values in parentheses denote comparisons among K inferred clusters.
Denotes groupwise comparisons within a single population.
Barriers to gene flow
The northern and southern clustering occurred on either side of the Mangoro River (Fig. 1B), the largest river in eastern Madagascar. These results support the principles of Martin (1972) and are in accordance with long-standing hypotheses regarding Malagasy microendemism and patterns of population structure (e.g., Martin 1972; Wilmé et al. 2006; Craul et al. 2007). Similar patterns have been found among many of the Malagasy strepsirrhines, including other large-bodied, diurnal species of Propithecus and Eulemur (Ganzhorn et al. 2006). Interestingly, in both of these examples, populations that were once considered subspecies occurring on either side of the Mangoro River have since been elevated to full species status (North of the Mangoro: P. diadema and E. fulvus; South of the Mangoro: P. edwardsi and E. rufifrons: Mittermeier et al. 2010; see also Markolf and Kappeler 2013).
Population dynamics and patterns of gene flow
Ruffed lemurs, like most mammals (Greenwood 1980), are generally assumed to exhibit female philopatry. That is, males within the species are considered the predominantly dispersing sex (Kappeler 1997; Morland 1991; but see Balko 1998; Erhart and Overdorff 2008; S. M. Holmes, S. E. Johnson, E. E. Louis, pers. obs.). By contrast, our analyses did not detect significant differences between males and females, instead indicating a lack of sex-biased dispersal in V. variegata (Table 4). However, simulation studies have shown that tests based on mAIc and FST can only reliably detect sex biases in dispersal when the bias is quite large and only with exhaustive sampling (Goudet et al. 2002). Although our sampling strategy was geographically extensive, sample sizes across some localities were limited.
Bottleneck analyses detected significant deviations from mutation-drift equilibrium in 50% of sites tested (5 of 10) under two (IAM, TPM) of the three models, but only two sites under the SMM. Earlier work has suggested that the TPM may be the most appropriate model for microsatellites given its intermediate status between the more conservative SMM and the rather unconstrained IAM (Di Rienzo et al. 1994; Piry et al. 1999). The general approach implemented in Bottleneck software is known to lack power simply because summary statistics do not use the genetic information very efficiently (Felsenstein 1992). Thus, if we had detected significant signals of population bottlenecks across all mutation models, this would have suggested that the signal was strong enough to be detected using a summary approach, as was found in orangutans by Goossens et al. (2006). Unfortunately, tests of excess heterozygosity have limited power with small sample sizes (Peery et al. 2012). Thus, our results should be viewed with caution, particularly among northern sampling localities. Furthermore, several studies have now shown that population structure can generate spurious bottleneck signals (e.g., Wakeley 1999; Chikhi et al. 2010). Thus, future work that seeks to identify population declines within this species will benefit from more sophisticated methods, such as those used by Olivieri et al. (2008) and Craul et al. (2009).
Conservation applications
Results from this study have important implications for lemur conservation. Conservation genetics provides a powerful tool with which to identify important conservation priorities and also monitor the fate of populations (Schwartz et al. 2006, Frankham 2010). Effective conservation management often depends on the identification of management units (MUs), which are usually defined as demographically independent populations whose population dynamics (e.g., population growth rate) depend largely on local birth and death rates rather than on immigration. The identification of MUs is central to the short-term management and conservation of natural populations and is typically used to delineate entities for monitoring (Schwartz et al. 2006) and regulating the effects of human activity upon the abundance of populations and species. In the absence of population genetic and/or long-term demographic information, however, MUs are often identified on the basis of taxonomic (i.e., subspecies) designations to target conservation priorities and assess potential translocations and/or reintroductions (Templeton 1986; Lynch 1996).
The pattern of genetic differentiation found in this study contradicts expectations based on current taxonomy and thus calls into question the appropriateness of treating the three Varecia subspecies as discrete units for existing in situ and captive population management plans. Although they do not differ morphometrically (Baden et al. 2008), subspecies exhibit a wide variety of pelage variation (i.e., the patterning of saddles; “lightness” or “redness” of coat color) (Vasey and Tattersall 2002). There is, however, little indication that coat color pattern corresponds to either geographic location or genetic type (Wyner et al. 1999; Vasey and Tattersall 2002). Although the goal of this study was not to re-evaluate the taxonomic status of V. variegata subspecies, we found that the current subspecific taxonomy provides a misleading view of population differentiation (Fig. 1A and B). Both microsatellite and mtDNA sequence data grouped V. variegata into northern and southern genetic clusters, much like patterns identified previously by Wyner et al. (1999). We therefore propose that future conservation efforts should consider treating genetic clusters (such as those identified herein), not current subspecies, as distinct MUs, as genetic variation is arguably a more biologically accurate metric. We also hope the results presented herein will prompt a re-evaluation of the existing subspecies designations (Fig. 1).
Beyond identifying units for conservation management, this analysis has also allowed us to understand patterns of genetic diversity and thus suggest targeted conservation strategies. Until recently, northern V. variegata sites have likely experienced the greatest connectivity (i.e., gene flow) and genetic diversity among sampling localities; however, this is also where a majority of the illegal hunting and timber extraction has occurred due to recent political unrest (Barrett et al. 2010; Jenkins et al. 2011; Allnutt et al. 2013). Varecia variegata populations, among other lemur species, are currently being hunted at unsustainable levels (Golden 2009). On the other hand, animals from sampling localities within the southern cluster have significantly lower allelic, genetic, and haplotypic diversity than sites in the north. Most southern localities exhibit significant genetic differentiation (FST), and there is some evidence that several have undergone recent population declines (Holmes et al. 2013; this study). We therefore propose that future conservation efforts should focus on maintaining genetic diversity in northern sampling localities by focusing on reducing hunting pressures and forest loss, while also increasing connectivity among southern localities to encourage gene flow among isolated populations. Ongoing efforts initiated by EEL are already underway to link fragments in Kianjavato/Vatovavy area via grassroots reforestation projects such as the Education Promoting Reforestation Project (Manjaribe et al. 2013).
Finally, our results raise the possibility of returning confiscated animals to their likely region of origin, and estimating the likely provenance of some captive animal populations. Nearly, all individuals (91%) within our study were assigned to their source localities with high statistical certainty across genetically differentiated sampling localities. Even in cases where individuals could not be assigned to their particular locality of origin, they could be successfully assigned to their appropriate genetic cluster (9%). Interestingly, Structure analyses clustered individuals from Nosy Mangabe (Nosy), an isolated V. variegata population originally introduced to the island in the 1930s (Petter and Peyreiras 1970), exclusively with individuals from Mananara Nord (Nara). Although, to the best of our knowledge, no known records exist regarding their true origins (I. Porton, pers. comm.), our results suggest that the founder population for the Nosy Mangabe individuals might have come from Mananara Nord, a coastal mainland site located ∼312 km south of the island. Thus, sampling animals and localities from across the full species’ range – as we have done in this study – provides not only a comprehensive picture of genetic diversity, but also useful tools for wildlife forensics.
Acknowledgments
We would like to thank the Madagascar Biodiversity Partnership, MICET, ANGAP/MNP, and the Centre ValBio for logistical support and sample collection. Laboratory support was generously provided by the Omaha's Henry Doorly Zoo and Aquarium and Gary Aronsen at Yale's Molecular Anthropology Lab. Ute Radespiel graciously provided raw data included in our comparisons of lemur population differentiation. We would like to thank Rachel Jacobs and two anonymous reviewers for helpful suggestions on earlier versions of this manuscript. Funding for this research was provided by: National Science Foundation (ALB, DDIG BSC-0725975), J. William Fulbright Foundation (ALB), the L.S.B. Leakey Foundation (ALB), the Natural Sciences and Engineering Research Council of Canada (SMH), the Government of Alberta (SMH), the International Primatological Society (SMH), the Calgary Zoological Society (SMH), Conservation International and Margot Marsh Biodiversity Fund (ALB, EEL, SEJ), MMBF Primate Action Fund (ALB, SMH, SEJ), Primate Conservation, Inc. (ALB, SEJ, SMH), Stony Brook University (ALB), Yale University (ALB, BJB), the University of Calgary (SEJ, SMH), and Omaha's Henry Doorly Zoo (EEL).
Data accessibility
DNA sequences have been deposited in GenBank (accessions KJ700486-KJ700626; AF495865-AF495895, AY588494, and AF493669-AF493671; see also Louis et al. 2005, 2006; Andriantompohavana et al. 2006).
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. ΔK values, a measure of the rate of change in the Structure likelihood function, as a function of K, the number of putative iterations. Results from both microsatellite (A) and mtDNA (B) analyses both indicate K = 2 populations.
Figure S2. ΔK values as a function of K. Results from subsequent Structure analysis within each of the K = 2 previously defined (A) northern and (B) southern clusters indicate further population substructure.
Figure S3. Results from subsequent Structure analysis within each of the K = 2 previously defined (A) northern and (B) southern clusters indicate further population substructure. Each animal is represented by a single horizontal bar. Locality of origin is indicated to the far left of Q plots. Left: Original Structure analysis (n = 129 adults) identifying K = 2 populations. Center: Subsequent Structure analysis within each of the K = 2 previously defined clusters (Cluster 1: North; Cluster 2: South) identifying further population substructure in both northern (K = 2) and southern (K = 2) populations. Right: Structure analysis identifying K = 4 clusters among the northern 8 sampling localities. The posterior probability of K = 4 was less than that of K = 2 among northern sampling localities (see Fig. S2) and was thus not selected using the Evanno et al. (2005) method.
Table S1. Characteristics of 10 microsatellite markers amplified in 209 V. variegata samples, including number of alleles per locus (nA), observed (Ho) and expected (He) heterozygosity, and deviations from Hardy–Weinberg Equilibrium (HWE). Significant values at P < 0.05 are shown in bold.
Table S2. Allelic diversity within each of 19 sampling localities, including mean number of alleles per locus (MNA), allelic richness (AR), observed (Ho) and expected (He) heterozygosity, inbreeding coefficient (FIS) and significant deviations from Hardy–Weinberg Equilibrium (HWE) calculated using 10,000 iterations. Significant values at P < 0.05 are indicated in bold. ‘-’ indicates populations with <10 samples that were not tested from deviations from HWE.
Table S3. Pairwise FST values (above diagonal) and significance values (below diagonal) among sampling localities and populations of V. variegata. * indicates significant values at P < 0.0003 after Bonferroni corrections.
Table S4. Measures of haplotype diversity across sampling localities, including the number of samples analyzed (n), the number of polymorphic sites (S), haplotype diversity (h), and nucleotide diversity (π).
Table S5. Pairwise ΦPT values (FST analog; above diagonal) and significance values (below diagonal) among sampling localities and populations of V. variegata. * indicates significant values at P < 0.0003 after Bonferroni corrections.
References
- Allnutt TF, Asner GP, Golden CD, Powell GVN. Mapping recent deforestation and forest disturbance in northeastern Madagascar. Trop. Conserv. Sci. 2013;6:1–15. [Google Scholar]
- Andrainarivo C, Andriaholinirina V, Feistner AT, Felix T, Ganzhorn JU, Garbut N, et al. 2009. Varecia variegata. IUCN. Available at http://www.iucnredlist.org. (accessed 7 January 2014)
- Andriantompohavana R, Zaonarivelo JR, Engberg SE, Randriamampionona R, McGuire SM, Shore GD, et al. The mouse lemurs of northwestern Madagascar with a description of a new species at Lokobe Special Reserve. Occas. Pap. Tex. Tech. Univ. Mus. 2006;259:1–23. [Google Scholar]
- Baden AL, Gerber BD. Spatial ecology of black-and-white ruffed lemurs (Varecia variegata. Am. J. Primatol. (In review) [Google Scholar]
- Baden AL, Brenneman RA, Louis EE., Jr Morphometrics of wild blackandwhite ruffed lemurs [Varecia variegata; Kerr, 1792] Am. J. Primatol. 2008;70:913–926. doi: 10.1002/ajp.20583. [DOI] [PubMed] [Google Scholar]
- Baden AL, Wright PC, Louis EEJ, Bradley BJ. Communal nesting, kinship, and maternal success in a social primate. Behav. Ecol. Sociobiol. 2013;67:1939–1950. [Google Scholar]
- Baguette M, Van Dyck H. Landscape connectivity and animal behavior: functional grain as a key determinant for dispersal. Landscape Ecol. 2007;22:1117–1129. [Google Scholar]
- Baker CS, Perry A, Bannister JL, Weinrich MT, Abernethy RB, Calambokidis J, et al. Abundant mitochondrial DNA variation and world-wide population structure in humpback whales. Proc. Natl Acad. Sci. USA. 1993;90:8239–8243. doi: 10.1073/pnas.90.17.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balko EA. Syracuse, NY: State University of New York; 1998. A behaviorally plastic response to forest composition and logging disturbance by Varecia variegata variegata in Ranomafana National Park, Madagascar. PhD Dissertation. [Google Scholar]
- Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Barrett MA, Brown JL, Morikawa MK, Labat J, Yoder AD. CITES designation for endangered rosewood in Madagascar. Science. 2010;328:1109–1110. doi: 10.1126/science.1187740. [DOI] [PubMed] [Google Scholar]
- Brenneman RB, Johnson SE, Bailey CA, Ingraldi C, Delmore KE, Wyman TM, et al. Population genetics and abundance of the endangered grey-headed lemur Eulemur cinereiceps in south-east Madagascar: assessing risks for fragmented and continuous populations. Oryx. 2011;46:298–307. [Google Scholar]
- Chikhi L, Sousa VC, Luisi P, Goossens B, Beaumont MA. The confounding effects of population structure, genetic diversity, and the sampling scheme on the detection and quantification of population size changes. Genetics. 2010;186:983–995. doi: 10.1534/genetics.110.118661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornuet JM, Piry S, Luikart G, Estoup A, Solignac M. New methods employing multilocus genotypes to select or exclude populations as origins of individuals. Genetics. 1999;153:1989–2000. doi: 10.1093/genetics/153.4.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craul M, Zimmermann E, Rasoloharijaona S, Randrianambinina B, Radespiel U. Unexpected species diversity of Malagasy primates (Lepilemur spp.) in the same biogeographical zone: a morphological and molecular approach with the description of two new species. BMC Evol. Biol. 2007;7:83. doi: 10.1186/1471-2148-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craul M, Chikhi L, Sousa V, Olivieri GL, Rabesandratana A, Zimmermann E, et al. Influence of forest fragmentation on an endangered large-bodied lemur in northwestern Madagascar. Biol. Conserv. 2009;142:2862–2871. [Google Scholar]
- Di Rienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl Acad. Sci. USA. 1994;91:3166–3170. doi: 10.1073/pnas.91.8.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012;4:359–361. [Google Scholar]
- Erhart EM, Overdorff DJ. Rates of agonism by diurnal lemuroids: implications for female social relationships. Int. J. Primatol. 2008;29:1227–1247. [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard JK. Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre L, Balloux F, Goudet J, Perrin N. Female-biased dispersal in the monogamous mammal Crocidura russula: evidence from field data and microsatellite patterns. Proc. Biol. Sci. 1997;264:127–132. doi: 10.1098/rspb.1997.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Estimating effective population size from samples of sequences: inefficiency of pairwise and segregating sites as compared to phylogenetic estimates. Genet. Res. 1992;59:139–147. doi: 10.1017/s0016672300030354. [DOI] [PubMed] [Google Scholar]
- Frankham R. Challenges and opportunities of genetic approaches to biological conservation. Biol. Conserv. 2010;143:1919–1927. [Google Scholar]
- Fredsted T, Pertoldi C, Schierup MH, Kappeler PM. Microsatellite analyses reveal fine-scale genetic structure in grey mouse lemurs (Microcebus murinus. Mol. Ecol. 2005;14:2363–2372. doi: 10.1111/j.1365-294X.2005.02596.x. [DOI] [PubMed] [Google Scholar]
- Ganzhorn JU, Lowry PP, Schatz GE, Sommer G. The biodiversity of Madagascar: one of the world's hottest hotspots on its way out. Oryx. 2001;35:346–348. [Google Scholar]
- Ganzhorn JU, Goodman SM, Nash S, Thalmann U. Lemur biogeography. In: Lehman SM, Fleagle JG, editors. Primate biogeography: progress and prospects. Springer: New York; 2006. pp. 229–254. [Google Scholar]
- Glander KE. Capture and marking techniques for arboreal primates. In: Estrada A, Rodriguez-Luna E, Lopez-Wilchis R, Coates-Estrada R, editors. Estudios primatologicos en Mexico. Vol. 1. Mexico: Universidad Veracruzana; 1993. pp. 299–304. [Google Scholar]
- Golden CD. Bushmeat hunting and use in the Makira Forest, northeastern Madagascar: a conservation and livelihoods issue. Oryx. 2009;43:386–392. [Google Scholar]
- Goossens B, Setchell JM, James SS, Funk SM, Chikhi L, Abulani A, et al. Philopatry and reproductive success in Bornean orang-utans (Pongo pygmaeus. Mol. Ecol. 2006;15:2577–2588. doi: 10.1111/j.1365-294X.2006.02952.x. [DOI] [PubMed] [Google Scholar]
- Goudet J. Lausanne: University of Lausanne; 2001. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3) Available from http://www.unil.ch/dee/page6767_en.html. [Google Scholar]
- Goudet J, Raymond M, de Meeus T, Roussett F. Testing differentiation in diploid populations. Genetics. 1996;144:1933–1940. doi: 10.1093/genetics/144.4.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet J, Perrin N, Waser P. Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol. Ecol. 2002;11:1103–1114. doi: 10.1046/j.1365-294x.2002.01496.x. [DOI] [PubMed] [Google Scholar]
- Greenwood PJ. Mating systems, philopatry and dispersal in birds and mammals. Anim. Behav. 1980;28:1140–1162. [Google Scholar]
- Harper GJ, Steininger MK, Tucker CJ, Juhn D, Hawkins F. Fifty years of deforestation and forest fragmentation in Madagascar. Environ. Conserv. 2007;34:1–9. [Google Scholar]
- Hartl DL, Clark AG. Principles of population genetics. Sunderland, MA: Sinauer Associates, Inc., Publishers; 1997. [Google Scholar]
- Holmes SM, Baden AL, Brenneman RA, Engberg SE, Louis EEJ, Johnson SE. Patch size and isolation influence genetic patterns in black-and-white ruffed lemur (Varecia variegata) populations. Conserv. Genet. 2013;14:615–624. [Google Scholar]
- Ikeda DH, Bothwell HM, Lau MK, O'Neill GA, Grady KC, Whitham TG. Climate change and species range shifts: a genetics-based Universal Community Transfer Function for predicting the impacts of climate change on future communities. Funct. Ecol. 2012;28:65–74. [Google Scholar]
- Irwin MT, Johnson SE, Wright PC. The state of lemur conservation in south-eastern Madagascar: population and habitat assessments for diurnal and cathemeral lemurs using surveys, satellite imagery and GIS. Oryx. 2005;39:204–218. [Google Scholar]
- Jenkins RKB, Keane A, Rakotoarivelo AR, Rakotomboavonjy V, Randrianandrianina FH, Razafimanahaka HJ, et al. Analysis of patterns of bushmeat consumption reveals extensive exploitation of protected species in eastern Madagascar. PLoS ONE. 2011;6:e27570. doi: 10.1371/journal.pone.0027570. doi: 10.1371/journal.pone.0027570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski ST. Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv. Genet. 2004;5:539–543. [Google Scholar]
- Kalinowski ST. HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes. 2005;5:187–189. [Google Scholar]
- Kappeler PM. Determinants of primate social organization: comparative evidence and new insights from Malagasy lemurs. Biol. Rev. 1997;72:111–151. doi: 10.1017/s0006323196004999. [DOI] [PubMed] [Google Scholar]
- Kull CA. Isle of fire: the political ecology of landscape burning in Madagascar. London: University of Chicago Press; 2004. [Google Scholar]
- Langergraber K, Schubert G, Rowney C, Wrangham R, Zommers Z, Vigilant L. Genetic differentiation and the evolution of cooperation in chimpanzees and humans. Proc. Biol. Sci. 2011;278:2546–2552. doi: 10.1098/rspb.2010.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler RR, Richard AF, Riley MA. Genetic population structure of the white sifaka (Propithecus verreauxi verreauxi) at Beza Mahafaly Special Reserve, southwest Madagascar (1992-2001) Mol. Ecol. 2003;12:2307–2317. doi: 10.1046/j.1365-294x.2003.01909.x. [DOI] [PubMed] [Google Scholar]
- Lawson Handley LJ, Perrin N. Advances in our understanding of mammalian sex-biased dispersal. Mol. Ecol. 2007;16:1559–1578. doi: 10.1111/j.1365-294X.2006.03152.x. [DOI] [PubMed] [Google Scholar]
- Librado R, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Liu Z, Ren B, Wu R, Zhao L, Hao Y, Wang B, et al. The effect of landscape features on population genetic structure in Yunnan snub-nosed monkeys (Rhinopithecus bieti) implies an anthropogenic genetic discontinuity. Molec. Ecol. 2009;18:3831–3846. doi: 10.1111/j.1365-294X.2009.04330.x. [DOI] [PubMed] [Google Scholar]
- Louis EE, Jr, Ratsimbazafy JH, Razakamaharavo VR, Pierson DJ, Barber RC, Brenneman RA. Conservation genetics of black-and-white ruffed lemurs, Varecia variegata, from Southeastern Madagascar. Anim. Conserv. 2005;8:105–111. [Google Scholar]
- Louis EE, Jr, Coles MS, Andriantompohavana R, Sommer JA, Engberg SE, Zaonarivelo JR, et al. Revision of the mouse lemurs (Microcebus) of eastern Madagascar. Int. J. Primatol. 2006;27:347–389. [Google Scholar]
- Luikart G, Cornuet JM. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv. Biol. 1998;12:228–237. [Google Scholar]
- Lukas D, Bradley BJ, Nsubuga AM, Doran-Sheehy D, Robbins MM, Vigilant L. Major histocompatibility complex and microsatellite variation in two populations of wild gorillas. Mol. Ecol. 2004;13:3389–3404. doi: 10.1111/j.1365-294X.2004.02353.x. [DOI] [PubMed] [Google Scholar]
- Lynch M. A quantitative-genetic perspective on conservation issues. In: Avise JC, Hamrick JL, editors. Conservation genetics: casse histories from nature. New York: Chapman & Hall; 1996. pp. 471–501. [Google Scholar]
- Madsen T, Shine R, Olssen M, Wittzell H. Restoration of an inbred adder population. Nature. 1999;402:34–35. [Google Scholar]
- Manjaribe C, Frasier CL, Rakouth B, Louis EEJ. Ecological restoration and reforestation of fragmented forests in Kianjavato, Madagascar. J. Ecol. 2013;2013:726275. doi: 10.1155/2013/726275. [Google Scholar]
- Markolf M, Kappeler PM. Phylogeographic analysis of the true lemurs (genus Eulemur) underlines the role of river catchments for the evolution of micro-endemism in Madagascar. Front. Zool. 2013;10:70. doi: 10.1186/1742-9994-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RD. Review lecture: adaptive radiation and behaviour of the Malagasy lemurs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1972;264:295–352. doi: 10.1098/rstb.1972.0013. [DOI] [PubMed] [Google Scholar]
- McConnell WJ. Madagascar: emerald isle or paradise lost? Environment. 2002;44:10–22. [Google Scholar]
- Meirmans PG, Van Tienderen PH. GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes. 2004;4:792–794. [Google Scholar]
- Mittermeier RA, Hawkins F, Rajaobelina S, Langrand O. Wilderness conservation in a biodiversity hotspot. Int. J. Wilderness. 2005;11:42–45. [Google Scholar]
- Mittermeier RA, Louis EEJ, Richardson M, Schwitzer C, Langrand O, Rylands AB, et al. Lemurs of Madagascar. 3rd edn. Washington, DC: Conservation International; 2010. p. 767. [Google Scholar]
- Morin PA, Chambers KE, Boesch C, Vigilant L. Quantitative polymerase chain reaction analysis of DNA from noninvasive samples for accurate microsatellite genotyping of wild chimpanzees (Pan troglodytes verus. Molec. Ecol. 2001;10:1835–1844. doi: 10.1046/j.0962-1083.2001.01308.x. [DOI] [PubMed] [Google Scholar]
- Morland HS. New Haven, CT: Yale University; 1991. Social organization and ecology of black-and-white ruffed lemurs (Varecia variegata variegata) in lowland rain forest, Nosy Mangabe, Madagascar. PhD Dissertation. [Google Scholar]
- Myers N, Mittermeier RA, Mittermeier CG, de Fonseca GAB, Kent J. Biodiversity hotspots for conservation priorities. Nature. 2000;403:853–858. doi: 10.1038/35002501. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular population genetics and evolution. Amsterdam: North-Holland Publishing Company; 1975. [PubMed] [Google Scholar]
- Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89:583–590. doi: 10.1093/genetics/89.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oaklander LI, Koalewski MM, Corach D. Genetic consequences of habitat fragmentation in black-and-gold howler (Alouatta caraya) populations from northern Argentina. Int. J. Primatol. 2010;3:813–832. [Google Scholar]
- Olivieri GL, Sousa V, Chikhi L, Radespiel U. From genetic diversity and structure to conservation: genetic signature of recent population declines in three mouse lemur species (Microcebus spp.) Biol. Conserv. 2008;141:1257–1271. [Google Scholar]
- van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes. 2004;4:535–538. [Google Scholar]
- Ostrowski MF, David J, Santoni S, Mckhann H, Rebound X, Le Corre V, et al. Evidence for a large-scale population structure among accessions of Arabidopsis thaliana: possible causes and consequences for the distribution of linkage disequilibrium. Mol. Ecol. 2006;15:1507–1517. doi: 10.1111/j.1365-294X.2006.02865.x. [DOI] [PubMed] [Google Scholar]
- Paetkau D, Strobeck C. Microsatellite analysis of genetic variation in black bear populations. Mol. Ecol. 1994;3:489–495. doi: 10.1111/j.1365-294x.1994.tb00127.x. [DOI] [PubMed] [Google Scholar]
- Paetkau D, Slade R, Burden M, Estoup A. Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol. Ecol. 2004;13:55–65. doi: 10.1046/j.1365-294x.2004.02008.x. [DOI] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics. 2012;28:2537–2539. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Be¨er E, Robinson S, et al. Reliability of genetic bottleneck tests for detecting recent population declines. Mol. Ecol. 2012;21:3403–3418. doi: 10.1111/j.1365-294X.2012.05635.x. [DOI] [PubMed] [Google Scholar]
- Perry GH, Melsted P, Marioni JC, Wang Y, Bainer R, Pickrell JK, et al. Comparative RNA sequencing reveals substantial genetic variation in endangered primates. Genome Res. 2013;22:602–610. doi: 10.1101/gr.130468.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petter JJ, Peyreiras A. Nouvelle contribution a l'edtude d'un lemurien Malagache, le aye-aye (Daubentonia madagascariensis, E. Geoffroy) Mammalia. 1970;34:167–193. [Google Scholar]
- Piry S, Luikart G, Cornuet JM. BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999;90:502–503. [Google Scholar]
- Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudouin L, Estoup A. GeneClass2: a software for genetic assignment and first-generation migrant detection. J. Hered. 2004;95:536–539. doi: 10.1093/jhered/esh074. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quéméré E, Louis EE, Jr, Ribéron A, Chikhi L, Crouau-Roy B. Non-invasive conservation genetics of the critically endangered golden-crowned sifaka (Propithecus tattersalli): high diversity and significant genetic differentiation over a small range. Conserv. Genet. 2009;11:675–684. [Google Scholar]
- Quéméré E, Crouau-Roy B, Rabarivola C, Louis EE, Jr, Chikhi L. Landscape genetics of an endangered lemur (Propithecus tattersalli) within its entire fragmented range. Molec. Ecol. 2010;19:1606–1621. doi: 10.1111/j.1365-294X.2010.04581.x. [DOI] [PubMed] [Google Scholar]
- Radespiel U, Rakotondravony R, Chikhi L. Natural and anthropogenic determinants of genetic structure in the largest remaining population of the endangered golden-brown mouse lemur, Microcebus ravelobensis. Am. J. Primatol. 2008;70:860–870. doi: 10.1002/ajp.20574. [DOI] [PubMed] [Google Scholar]
- Raymond M, Rousset F. Genepop (version 1.2): population genetics software for exact tests and ecumenicism. J. Hered. 1995;86:248–249. [Google Scholar]
- Razakamaharavo VR, McGuire SM, Vasey N, Louis EE, Brenneman RA. Genetic architecture of two red ruffed lemur (Varecia rubra) populations of Masoala National Park. Primates. 2010;51:53–61. doi: 10.1007/s10329-009-0171-0. [DOI] [PubMed] [Google Scholar]
- Reed DH, Frankham R. Correlation between fitness and genetic diversity. Conserv. Biol. 2003;17:230–237. [Google Scholar]
- Robinson JG. Conservation biology and real world conservation. Conserv. Biol. 2006;20:658–669. doi: 10.1111/j.1523-1739.2006.00469.x. [DOI] [PubMed] [Google Scholar]
- Rousset F. Genepop'007: a complete reimplementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- Schneider N, Chikhi L, Currat M, Radespiel U. Signals of recent spatial expansions in the grey mouse lemur (Microcebus murinus. BMC Evol. Biol. 2010;10:105. doi: 10.1186/1471-2148-10-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MK, Luikart G, Waples R. 2006. Genetic monitoring as a promising tool for conservation and management. Publications, Agencies and Staff of the U.S. Department of Commerce. Paper 482.
- Schwitzer C, Mittermerier RA, Davies N, Johnson S, Ratsimbazafy J, Razafindramanana J, et al. Bristol, UK: IUCN SSC Primate Specialist Group Report, Bristol Conservation and Science Foundation, and Conservation International; 2013. Lemurs of Madagascar: A strategy for their conservation 2013-2016; p. 185. [Google Scholar]
- Seutin G, White BN, Boag PT. Preservation of avian blood and tissue samples for DNA analyses. Can. J. Zool. 1991;69:82–90. [Google Scholar]
- Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escarvage N, et al. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 1996;24:3189–3194. doi: 10.1093/nar/24.16.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenius H, Carter NP, Bebb CE, Nordenskjob M, Ponder AJ, Tunnacliffe A. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718–725. doi: 10.1016/0888-7543(92)90147-k. [DOI] [PubMed] [Google Scholar]
- Templeton AR. Coadaptation and outbreeding depression. In: Soule ME, editor. Conservation biology: the science of scarcity and diversity. Sunderland, MA: Sinauer Associates Inc; 1986. pp. 105–116. [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;24:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiuk J, Bachmann L, Peipoldt M, Ganzhorn JU, Ries R, Weis M, et al. Genetic diversity of Lepilemur mustelinus ruficaudatus, a nocturnal lemur of Madagascar. Conserv. Biol. 1997;11:491–497. [Google Scholar]
- Vasey N. Varecia, Ruffed lemurs. In: Goodman SM, Benstead J, editors. The natural history of Madagascar. Chicago: The University of Chicago Press; 2003. pp. 1332–1336. [Google Scholar]
- Vasey N, Tattersall I. Do ruffed lemurs for a hybrid zone? Distribution and discovery of Varecia, with systematic and conservation implications. Am. Mus. Novit. 2002;3376:1–26. [Google Scholar]
- Wakeley J. Nonequilibrium migration in human history. Genetics. 1999;153:1863–1871. doi: 10.1093/genetics/153.4.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- White FJ, Overdorff DJ, Balko EA, Wright PC. Distribution of ruffed lemurs (Varecia variegata) in Ranomafana National Park, Madagascar. Folia Primatol. 1995;64:124–131. [Google Scholar]
- Wilmé L, Goodman SM, Ganzhorn JU. Biogeographic evolution of Madagascar's microendemic biota. Science. 2006;312:1063–1065. doi: 10.1126/science.1122806. [DOI] [PubMed] [Google Scholar]
- Wyner YM, Amato G, DeSalle R. Captive breeding, reintroduction, and the conservation genetics of black-and-white ruffed lemurs, Varecia variegata variegata. Mol. Ecol. 1999;8:S107–S115. doi: 10.1046/j.1365-294x.1999.00815.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. ΔK values, a measure of the rate of change in the Structure likelihood function, as a function of K, the number of putative iterations. Results from both microsatellite (A) and mtDNA (B) analyses both indicate K = 2 populations.
Figure S2. ΔK values as a function of K. Results from subsequent Structure analysis within each of the K = 2 previously defined (A) northern and (B) southern clusters indicate further population substructure.
Figure S3. Results from subsequent Structure analysis within each of the K = 2 previously defined (A) northern and (B) southern clusters indicate further population substructure. Each animal is represented by a single horizontal bar. Locality of origin is indicated to the far left of Q plots. Left: Original Structure analysis (n = 129 adults) identifying K = 2 populations. Center: Subsequent Structure analysis within each of the K = 2 previously defined clusters (Cluster 1: North; Cluster 2: South) identifying further population substructure in both northern (K = 2) and southern (K = 2) populations. Right: Structure analysis identifying K = 4 clusters among the northern 8 sampling localities. The posterior probability of K = 4 was less than that of K = 2 among northern sampling localities (see Fig. S2) and was thus not selected using the Evanno et al. (2005) method.
Table S1. Characteristics of 10 microsatellite markers amplified in 209 V. variegata samples, including number of alleles per locus (nA), observed (Ho) and expected (He) heterozygosity, and deviations from Hardy–Weinberg Equilibrium (HWE). Significant values at P < 0.05 are shown in bold.
Table S2. Allelic diversity within each of 19 sampling localities, including mean number of alleles per locus (MNA), allelic richness (AR), observed (Ho) and expected (He) heterozygosity, inbreeding coefficient (FIS) and significant deviations from Hardy–Weinberg Equilibrium (HWE) calculated using 10,000 iterations. Significant values at P < 0.05 are indicated in bold. ‘-’ indicates populations with <10 samples that were not tested from deviations from HWE.
Table S3. Pairwise FST values (above diagonal) and significance values (below diagonal) among sampling localities and populations of V. variegata. * indicates significant values at P < 0.0003 after Bonferroni corrections.
Table S4. Measures of haplotype diversity across sampling localities, including the number of samples analyzed (n), the number of polymorphic sites (S), haplotype diversity (h), and nucleotide diversity (π).
Table S5. Pairwise ΦPT values (FST analog; above diagonal) and significance values (below diagonal) among sampling localities and populations of V. variegata. * indicates significant values at P < 0.0003 after Bonferroni corrections.
Data Availability Statement
DNA sequences have been deposited in GenBank (accessions KJ700486-KJ700626; AF495865-AF495895, AY588494, and AF493669-AF493671; see also Louis et al. 2005, 2006; Andriantompohavana et al. 2006).