

Significance
The WOPR-domain family of transcriptional regulators is deeply conserved in the fungal kingdom where the members function as master transcriptional regulators of cell morphology and pathogenesis. Despite the critical biological roles of WOPR-domain proteins, previous bioinformatic and structural prediction did not provide any significant matches between these proteins and any other type of protein. We describe a 2.6-Å–resolution structure of a WOPR domain in complex with its preferred DNA sequence. We also describe a set of biochemical experiments that confirms and rationalizes the importance of the protein–DNA contacts observed in the structure. Based on the structure, we conclude that the WOPR domain represents a new family of DNA-binding proteins, one with key roles for fungal morphogenesis and pathogenesis.
Keywords: fungal pathogenesis, transcription factor, transcriptional regulation, protein–DNA interaction, Candida albicans
Abstract
WOPR-domain proteins are found throughout the fungal kingdom where they function as master regulators of cell morphology and pathogenesis. Genetic and biochemical experiments previously demonstrated that these proteins bind to specific DNA sequences and thereby regulate transcription. However, their primary sequence showed no relationship to any known DNA-binding domain, and the basis for their ability to recognize DNA sequences remained unknown. Here, we describe the 2.6-Å crystal structure of a WOPR domain in complex with its preferred DNA sequence. The structure reveals that two highly conserved regions, separated by an unconserved linker, form an interdigitated β-sheet that is tilted into the major groove of DNA. Although the main interaction surface is in the major groove, the highest-affinity interactions occur in the minor groove, primarily through a deeply penetrating arginine residue. The structure reveals a new, unanticipated mechanism by which proteins can recognize specific sequences of DNA.
The WOPR family of transcriptional regulators [named for the members Wor1 (white-opaque regulator 1), Pac2 (pat1 compensator 2), and Ryp1 (required for yeast phase growth 1)] controls morphological changes and pathogenesis in a diverse group of fungal species (Fig. 1A), including the human pathogens Candida albicans (1–3), Histoplasma capsulatum (4), and Cryptococcus neoformans (5) and several plant pathogens (6–9). In C. albicans, Wor1 controls the process of white–opaque switching and mating (1–3), and it has a key role in promoting commensalism (10). In H. capsulatum, it controls the transition from the mycelia form (found in soil) and the yeast form (found in infected human hosts) (4). Recently, a series of in vitro and in vivo experiments demonstrated that the WOPR domain of Wor1 binds DNA in a sequence-specific fashion and defined the DNA sequence recognized by it (Fig. 1B) (11, 12). Muc1 expressed independent of TEC1 1 (Mit1) from Saccharomyces cerevisiae, a fourth WOPR-domain protein, and Ryp1 from H. capsulatum have since been shown to recognize this same DNA sequence, representing conservation of the WOPR domain–DNA sequence interactions over a period of 600 million to 1.2 billion years (13).
Fig. 1.

Overview of the WOPR family of conserved fungal transcriptional regulators. (A) Phylogenetic tree of species with experimentally characterized WOPR proteins. The tree is based on Wang et al. (35); branch lengths are not drawn to scale. Experimentally characterized functions in each species are indicated to the right (1–9, 13, 36–38). (B) Previously reported position-specific weight and affinity matrices for the DNA sequence recognized by Wor1 developed from ChIP-chip (Left) (12, 39) and microfluidic affinity analysis (Right) (12). The DNA sequence used for the structural determination was TTAGAGTTTA. (C) Alignment of the 12 experimentally characterized WOPR homologs from 10 fungal species. S. cerevisiae YHR177w is 453 aa long; the other proteins are drawn to scale. The green and light blue boxes represent the conserved regions R1 and R2, respectively.
Biochemical experiments indicated that the WOPR domain binds DNA in an unusual way. The WOPR domain consists of two regions conserved across many fungal species (referred to here as “R1” and “R2”), separated by a poorly conserved “linker” region of variable length (Fig. 1C). R1 is 80 aa in length, R2 is 50 aa, and the linker ranges from less than 25 to more than 100 aa, depending on the species. The WOPR domain alone can bind DNA tightly (∼5 nM) and specifically (11). Additional experiments demonstrated that neither R1 nor R2 alone could bind to DNA individually, but binding was observed—albeit at lower affinity—when the two regions were expressed as separate peptides and mixed in the presence of DNA. Two general models of binding to DNA were consistent with this result: either both regions form part of the interface with DNA, or only one region contacts the DNA and the second region induces or stabilizes a conformational change allowing the stable interaction with DNA (11).
Although the WOPR domain was shown to bind DNA and activate transcription (11), bioinformatic analyses have not produced matches between the WOPR family and any known family of transcriptional regulators. Likewise, structural prediction algorithms failed to map either conserved region to any published structure. Given the lack of a suitable model for understanding how the WOPR domain could recognize DNA, fundamental questions remained about this family of transcriptional regulators. By what mechanism do WOPR proteins recognize specific sequences of DNA? Why are both the conserved regions needed to bind DNA? Does the poorly conserved linker region serve only to tether the two conserved regions together (increasing their effective concentration), or does it play some other role in DNA binding? The lack of a structural model also prevented any broader evaluation of whether the WOPR family evolved de novo or is related to other, well-studied regulators.
Here, we use X-ray crystallography to solve the structure of a WOPR protein–DNA complex. We show that the two conserved regions are tightly bound to one another through an extensive interface of interdigitated β-strands, with the unconserved linker looped out away from the DNA. Using site-directed mutagenesis combined with biochemical experiments, we confirm and rationalize the importance of the individual protein–DNA contacts observed in the structure and assess the role of linker length in DNA-binding affinity. Although the individual parts comprising the WOPR protein–DNA interface are reminiscent of previously characterized DNA-binding domains, they are combined in a unique way in the WOPR domain and comprise a previously undescribed solution for recognizing specific DNA sequences.
Results
Crystal Structure of the WOPR Family Member YHR177w.
The crystal structure of the DNA-binding domain of S. cerevisiae WOPR protein YHR177w (a paralog of S. cerevisiae Mit1) in complex with an optimized 19-bp DNA site was solved at 2.6-Å resolution by multiwavelength anomalous diffraction (MAD) methods using a selenomethionine-substituted protein crystal (Table S1). The protein fragment (containing YHR177w residues 6–201) previously has been shown to bind DNA in a sequence-specific manner (13). The quality of the electron-density maps allowed the unambiguous placement of both water-mediated and direct contacts between the YHR177w WOPR domain and the DNA. The final structure contains the full oligonucleotide and 175 residues of the protein, lacking small portions of the linker region and the 11 C-terminal amino acids. We failed to produce crystals from several other WOPR domains that contained longer linkers between the two highly conserved regions, and we hypothesize that the diminished flexibility of the shorter linker in YHR177w (28 aa) aided the crystallization.
Overall Fold of the YHR177w-DNA Complex.
Consistent with previous biochemical studies, the WOPR domain binds B-form DNA as a monomer (11). The core of the protein is a mixed α-helix β-sheet structure with a central, six-stranded β-sheet composed of discontinuous, antiparallel strands. These strands are contributed by both R1 and R2 (the two highly conserved regions) and together form a β-sheet, the bottom edge of which is inserted in the major groove (Fig. 2 A–C and Fig. S1 A and B). Given the centrality of the β-sheet to the overall structure, R1 and R2 appear to form a single, interwoven globular domain rather than two distinct subdomains as originally suggested by the primary sequence (Fig. 2C).
Fig. 2.
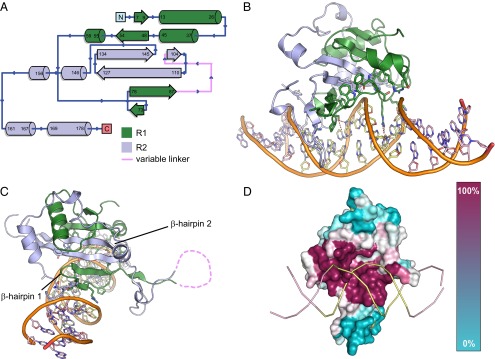
The structure of a WOPR domain bound to DNA. (A) Topology diagram showing the secondary structure composed of R1 (green) and R2 (blue). (B) Overview of the structure of the WOPR domain with secondary structure elements colored as in A. (C) Both R1 and R2 contribute to the six-stranded β-sheet at the core of the WOPR domain. β-hairpins from R1 and R2 are indicated. Secondary structure elements are colored as in A. The portion of the variable linker region disordered in the structure is indicated by a dashed pink line. (D) A color plot of the conservation of residues in the WOPR domain generated using the default settings in the program Consurf (31–33). Magenta represents the highest level of conservation; as shown, many of these residues contact DNA.
Both the R1 and R2 conserved regions make specific contacts with DNA. The amino acids most conserved across diverse species are in contact with DNA and also form the β-sheet at the core of the protein; the less-conserved residues are positioned on surfaces not involved in DNA interactions (Fig. 2D). This observation explains the preservation of WOPR–DNA binding specificity over long periods of evolutionary time (13). The poorly conserved linker between R1 and R2 extends away from the protein core and DNA interfaces, representing an extended loop between two of the β-strands forming the β-sheet. Despite the lack of conservation in this region, the linker region possesses a degree of structural organization, forming several short, antiparallel β-strands (Fig. 2C). The loop is pointed away from the DNA, and longer loops would not be expected to contact the DNA.
The WOPR domain forms interactions with both the major and minor grooves of DNA as well as the phosphate backbones that flank the grooves. Fifteen WOPR residues form hydrogen bonds with DNA; four of these bonds are base-specific interactions, and the rest are to the phosphate backbone. Up to 33 residues interact with DNA, with a total buried surface area of ∼1,146 Å2 across a highly positively charged surface (Fig. S1C and Table S2).
Minor-Groove Interactions.
The conserved region of YHR177w interacts with the minor groove through an extended loop running parallel to the groove (Fig. 3A). Arg62, which extends from the N terminus of this loop, is inserted directly into the minor groove, where it forms multiple hydrogen bonds with the base of bT13 (DNA chain B, residue T13) and the phosphate backbone of bA15. This loop continues from the minor groove, crosses the phosphate backbone (which it contacts through a water-mediated aG9 position), and joins the β-sheet in the major groove. Additional interactions (Arg35–bA15, Lys41–bC16, and Asp38–bG17) stabilize this loop and orient the main Arg62 interaction. Additional details of the minor-groove interactions are described in SI Results, along with biochemical experiments that verify the importance of individual minor-groove contacts by measuring the affinities of a series of mutant proteins (Fig. 4, Fig. S2, and Table S3).
Fig. 3.

Specific DNA-binding interactions. (A) R62 makes a bifurcated hydrogen bond with base bT13. Additional hydrogen bonds are formed between this residue and the DNA backbone (Table S2). R62 is positioned at the end of a long loop that wraps around the DNA backbone into the major groove. This loop ends with S72, which makes base-specific hydrogen bonds to aG11 in the major groove. (B) Close-up of major-groove interactions, highlighting the base-specific interactions with S72, N183, and R185.
Fig. 4.

Summary of biochemical experiments that assess contributions of individual amino acid side chains to binding affinity. The effects of WOPR domain single-point mutants (all to alanine) on DNA-binding affinity were measured by gel mobility shift experiments in which the concentration of protein was varied and are expressed as kds relative to that of wild type. Effects of the mutations are grouped by colors: brown type indicates no observable change (kd ∼4 nM), cyan type indicates a moderate effect (kd ∼10–100 nM), and magenta type indicates a strong effect (kd >100 nM). The line between the amino acid and the DNA indicates the type of interaction: solid lines indicate hydrophobic interactions, and dashed lines indicate hydrogen bonding.
Major-Groove Interactions.
Moving C-terminally from the minor-groove interactions, the protein makes a series of base-specific, water-mediated, and backbone-mediated interactions in the adjacent major groove. Both R1 and R2 contribute to the major-groove DNA interactions (Figs. 3B and 4). A total of 17 residues (e.g., Ser72, Leu79, and Tyr81) bury surface area at the protein–DNA interface in or adjacent to the major groove. These residues, particularly Tyr81 through a water-mediated hydrogen bond, appear to stabilize the position of the Arg62 loop as it extends from the minor groove into the major groove.
The extreme C terminus of R2, chiefly Asn183 and Arg185, also contributes to the binding, making additional, base-specific hydrogen bonds with the bases of bT6 and bG5. Ser186, Asn187, and Pro188 also form hydrogen bonds with the DNA backbone. These sets of interactions further anchor the protein into the major groove. To assess the relative importance of these protein–DNA interactions, we mutated the key amino acids and measured the effects on protein–DNA affinity (Fig. 4, Fig. S2, and Table S3). Taken as a whole, the results of the mutagenesis studies indicate that, although both the major- and minor-groove interfaces contribute to the WOPR domain’s affinity for DNA, mutation of the residues interacting with the minor groove leads to the largest decreases in binding affinity (Fig. 4 and Fig. S2B).
Role of the WOPR Linker.
The poorly conserved linker between R1 and R2 ranges in size across species from less than 25 to more than 100 aa, and we tested whether these differences in length affected DNA-binding affinity in vitro. First we note that the WOPR domains of YHR177w (linker length, 28 aa) and Wor1 (linker length, 113 aa) bind to the same DNA sequence with approximately the same affinity (kd ∼2 nM and ∼3 nM, respectively). To test the role of the linker length on protein–DNA affinity more directly, we constructed a chimeric WOPR domain in which the short linker of YHR177w was replaced with the longer linker from Wor1 (Fig. 5A). As shown in Fig. 5B and Table S3, the affinity for DNA did not change significantly. Likewise, we shortened the linker in Wor1 by deleting 79 aa (reducing the number to 34) and 91 aa (reducing the number to 22) and did not observe a significant change in DNA-binding affinity (Fig. 5B and Table S3). These results demonstrate that neither linker region length nor its precise amino acid sequence have a major impact on the WOPR domain monomer binding to DNA in vitro. This idea makes sense, because the structural organization would allow the insertion of variable loops into a compact, conserved core structure that binds DNA.
Fig. 5.

Changes in linker length do not affect WOPR affinity for DNA. (A) Diagrams of the C. albicans WOR1 and S. cerevisiae YHR177w constructs used in B. Wor1 sequences are shown in black, and YHR177w sequences shown in gray. Conserved regions R1 and R2 are represented by the boxes. (B) EMSAs using the same DNA fragment for all of the experiments. Concentrations of protein (in nanomoles) are indicated above each lane.
We next tested whether the linker is important for the function of the protein in vivo. In the first set of experiments, we induced Wor1 expression ectopically in C. albicans and monitored conversion of the white cell type to the opaque cell type. White and opaque cells differ in many features (including mating ability, metabolic specialization, recognition by the innate immune system, and preference for different niches in the host), and Wor1 is the master regulator of this transition. In accordance with prior work (1–3), ectopic expression of full-length Wor1 converts white cells to opaque cells en mass (100%) (Fig. 6A, lines 3 and 4, and Fig. 6B). When full-length Wor1 was replaced by a version with a shortened linker (Δ104–196), the conversion of white cells to opaque cells was reduced significantly (Fig. 6A, line 6, and Fig. 6B). In a second set of experiments, a test promoter controlled by a Wor1 cis-regulatory sequence was monitored in S. cerevisiae. Full-length Wor1 activated transcription from this construct efficiently, whereas two versions with shortened linkers showed significant decreases (Fig. 6C). Taken together with the DNA-binding experiments, the results show that changing the linker length does not affect the intrinsic DNA binding of the WOPR domain but does affect the function of the protein in vivo.
Fig. 6.

Shortening the WOPR linker affects white-to-opaque switching and transcriptional activation. (A) Frequency of white-to-opaque switching for strains with different Wor1 ectopic overexpression constructs. Full-length Wor1 (under control of an inducible pMET3 promoter) converts 100% of white cells to opaque cells under conditions that activate the pMET3 promoter. Replacing full-length Wor1 with a version missing a portion of the linker results in only partial conversion, as shown by both the small number of purely opaque colonies observed and the intermediate appearance of many colonies, which are mixtures of white and opaque cells. (B) Light microscope images of cells in the experiment in A. The Wor1 construct with the shortened linker produces a range of intermediate colony types, with some made up primarily of white cells (row 3, Center) and some with mixtures of white and opaque cells (row 3, Right). (C) Activation of test promoters controlled by full-length Wor1 and two versions of Wor1 missing part of the linker.
Comparison with Other Transcriptional Regulators and Possible Evolutionary Origins of the WOPR Family.
Although no significant sequence similarity exists between the WOPR domain and any known protein, a search of databases using the WOPR structure revealed some structural similarities to other transcription regulators (14). The highest-scoring match was to the glia cell missing (GCM) family of transcriptional regulators from metazoans (structure from Drosophila melanogaster, z-score 4.7). We also noted weaker matches to the NAC and WRKY families of transcriptional regulators from plants (maximum z scores of 3.9 and 3.5, respectively; both structures are from Arabidopsis thaliana). All three of these families are characterized by major-groove–interacting β-sheets with a secondary topology reminiscent of that observed for YHR177w. This feature appears to drive the matches identified by DALI (14), because the remainder of the structures differ greatly (rmsd of 3.1 and only 8% sequence identity for GCM) (Fig. S3) (15–18). Given the magnitude of these differences, it is clear that the WOPR proteins represent a new family of sequence-specific DNA-binding domains (19, 20).
Discussion
Understanding the WOPR Family.
Here, we present what, to our knowledge, is the first structure of the WOPR domain, a 175-aa domain found in many fungal regulators of morphology and pathogenesis, in complex with its preferred DNA sequence. Perhaps the most unusual feature of the WOPR domain comes from inspection of its amino acid sequence across all fungal species. It contains two highly conserved regions of 80 and 50 aa, respectively, connected by a linker that varies greatly in length and sequence from one species to the next. The structure reveals that the two conserved regions are tightly bound to each other through a β-sheet, with β-strands from one conserved region interdigitated with those from the other. This composite β-sheet inserts into the major groove at an angle, where it makes base-specific contacts. However, the most important contacts with DNA are made by an adjacent loop (contributed by R1), which is inserted into an especially narrow minor groove where it makes base-specific and backbone contacts.
The variable-length linker extends in a loop outward from this structure, and we show that neither its length nor its sequence impacts DNA-binding affinity in vitro. However, the linker is important for tethering the two conserved regions, because cutting the linker reduced DNA-binding affinity significantly (11). Although linker length and sequence are not important for DNA-binding affinity in vitro, we show that the loop sequence does affect the ability of WOPR proteins to drive transcriptional programs properly in vivo. One hypothesis is that the linker, through posttranslational modifications or interactions with additional proteins, may modulate DNA binding. However, the exact function of the linker in vivo remains to be determined.
Combining Common Features in an Uncommon Way.
The individual contacts between the WOPR domain and DNA have precedents in other structural classes of DNA-binding proteins. For example, a β-sheet in the major groove occurs in the GCM family (15), and an arginine side chain in a narrow minor groove is seen in the Hox family (21). However, no previously described DNA-binding domain combines the various elements in the specific manner seen in the WOPR domain. Moreover, the organization of the WOPR domain—two interdigitated conserved regions connected by an unconserved linker—further distinguishes the WOPR domain from other DNA-binding structures. If the WOPR family is ancestrally related to these other families, any evidence for such a relationship at the primary sequence level has vanished.
The WOPR domain is found in virtually all fungal species and thus has been conserved over approximately 1 billion years of evolution. Even its DNA-binding specificity has not changed significantly across species ranging from H. capsulatum to S. cerevisiae and C. albicans, which span a large portion of the fungal kingdom (estimated at 600 million to 1.2 billion years since divergence from the last common ancestor). In nearly every species in which they have been studied, WOPR-domain proteins regulate morphological changes, and in fungal species that infect plants or animals these proteins are required for pathogenesis. We can only speculate why this might be the case. Perhaps, like the role of MADS-domain proteins in plant development or the homeodomain proteins in animal development, WOPR-domain proteins became associated with fungal morphology and aspects of pathogenesis early in evolution. Although the detailed morphologies and host preferences differ enormously among modern fungal species, we propose that these transcription regulators remained master regulators of these two fundamental properties, even though the genes they control have changed as modern species radiated from their ancestors.
Materials and Methods
Plasmid and Strain Construction.
Lists of plasmids, strains, and oligonucleotides used in this study can be found in Table S4. Details of strain and plasmid construction can be found in SI Materials and Methods.
EMSA.
Full details of EMSAs can be found in SI Materials and Methods. In brief, protein was purified for use in EMSAs as previously described (13). EMSAs were performed as previously described (11, 13), except that NaCl was reduced to 50 mM, and Poly(dIdC) was omitted. In all assays, 0.4 nM of labeled DNA was used.
Protein Purification and Crystallization.
Full details of the protein-purification process for crystallization can be found in SI Materials and Methods. In brief, YHR177 for crystallography was expressed for 20 h at 25 °C with 0.4 mM isopropyl β-d-1-thiogalactopyranoside in the BL21 background. Protein was purified on a Ni-NTA agarose column, followed by cleavage of the 8xHis tag with PreCission Protease (GE Healthcare). Cleaved protein was purified on a HiTrap SP FF column (GE Healthcare) followed by further purification over a Superdex 200 10/300 GL column (GE Healthcare).
Crystallization was conducted in 24-well plates (Hampton Research), with a drop size of 2 µL. Drops were a 1:1 mixture of protein/DNA stock and well solution; two different well solutions were used. Crystallization of the protein/DNA1 mixture, used for the Seleno-Met YHR177 mutant (crystal form 1, I 21 21 21) used 200 mM KCl, 22.5% (vol/vol) PEG MME 550, 50 mM Tris (pH7.5), 5 mM spermine, 7.5% (vol/vol) ethylene glycol. Cryoprotection involved a 3:7 mixture of 80% (vol/vol) ethylene glycol and well solution. Crystallization of the protein/DNA2 mixture, used for the Native P 21 21 21 structure (crystal form 2), used 9 mM calcium chloride, 10 mM spermine, 50 mM sodium cacodylate (pH 7), 4.5% (vol/vol) isopropanol, 3.5% (vol/vol) ethylene glycol. Cryoprotection involved a 4:6 mixture of 25% (vol/vol) glucose/ethylene glycol and well solution.
Data Collection and Processing.
Datasets were collected at the Advanced Light Source beamline 8.3.1 as detailed in Table S1. The data were processed using HKL2000 (22) and the automated procedures in Elves (23), which uses the programs mosflm (24) and scala (25).
Phasing and Model Building.
The model was built using data from a combination of two crystal forms, crystal form 1 (DNA1) in a body-centered, orthorhombic lattice and crystal form 2 (DNA2) in space group P212121. We initially collected a three-wavelength MAD dataset on a selenomethionine-containing crystal (DNA1, crystal form 1) and calculated the phases using the program autoSHARP. With these data, we were able to produce a series of maps at 2.8-Å resolution in which the DNA could be placed unambiguously, but the protein density was very poor for the expected resolution and appeared to have a large amount of internal disorder. In the initial structure, we could see that Arg62 was bound in a TTT region at aT13; however in DNA1 two stretches of TTT resulted from using the consensus cis-regulatory sequence as determined in ChIP-Chip experiments (11, 12). We reasoned that Arg62 could bind to the minor groove in two different orientations, thus leading to statistical disorder in the crystal that could not be modeled. From independent experiments with binding assays in vitro (11, 12), we knew one of the instances of TTT could be mutated to TCT without compromising binding. We thus created DNA2, which binds strongly to YHR177w and crystallized in a different lattice. We used molecular replacement of our partial model to solve the structure of this second crystal form. The resulting maps allowed the unambiguous placement of all the residues in the protein except amino acids 1–4, 91–97 (in the linker), 130–131 (in the loop in the R2 β-hairpin), and 192–202. Model bias was reduced by using the protein and DNA alone to calculate independent difference maps as a guide to model building. The model was built with the program Coot (26) and refined with the Phenix suite (27) to a current Rfree of 23.5%. DNA analysis was performed using the program suite 3DNA (28, 29). Model visualization and vacuum electrostatic charge predictions were performed using the PyMOL Molecular Graphics System, version 1.5.0.4 (Schrödinger, LLC). Fig. S1 shows examples of the experimental and calculated density maps.
Structural Analysis and Bioinformatics.
Protein–DNA interactions in the WOPR structural model were identified using the PISA server (30), and comparisons to published structures were conducted using the DALI server (14). Conservation maps were generated using the ConSurf server (31–33). Minor groove widths in Table S5 were calculated using the 3DNA suite of programs (28, 29, 34) and established criteria for a narrow minor groove (34).
Supplementary Material
Acknowledgments
We thank Louis Metzger, Akram Alian, and Sarah Griner for technical assistance; Victor Hanson-Smith for help with bioinformatics; Liron Noiman and Sarah Foss for comments on the manuscript; James Holton and the staff at the Advanced Light Source beamline 8.3.1 for invaluable support and advice; and Chris Waddling and the resources of the University of California, San Francisco Molecular Structure Group, which were indispensable to this work. We also used the resources of the Stanford Synchrotron Radiation Lightsource beamline BL12.2 and the ID-24 (Northeastern Collaborative Access Team) at the Advanced Photon Source. This study was supported primarily by National Institutes of Health (NIH) Grant R01AI049187 (to A.D.J.). O.S.R. was supported by NIH Award K08 AI091656. J.S.F.-M. and R.M.S. were supported by NIH Grant GM51232 (to R.M.S.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The crystallography, atomic coordinates, and structure factors reported in this paper have been deposited at the Research Collaboratory for Structural Bioinformatics Protein Data Bank, http://pdb.rcsb.org/pdb/home/home.do (ID code 4M8B).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1410110111/-/DCSupplemental.
References
- 1.Zordan RE, Galgoczy DJ, Johnson AD. Epigenetic properties of white-opaque switching in Candida albicans are based on a self-sustaining transcriptional feedback loop. Proc Natl Acad Sci USA. 2006;103(34):12807–12812. doi: 10.1073/pnas.0605138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang G, et al. Bistable expression of WOR1, a master regulator of white-opaque switching in Candida albicans. Proc Natl Acad Sci USA. 2006;103(34):12813–12818. doi: 10.1073/pnas.0605270103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Srikantha T, et al. TOS9 regulates white-opaque switching in Candida albicans. Eukaryot Cell. 2006;5(10):1674–1687. doi: 10.1128/EC.00252-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen VQ, Sil A. Temperature-induced switch to the pathogenic yeast form of Histoplasma capsulatum requires Ryp1, a conserved transcriptional regulator. Proc Natl Acad Sci USA. 2008;105(12):4880–4885. doi: 10.1073/pnas.0710448105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu OW, et al. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell. 2008;135(1):174–188. doi: 10.1016/j.cell.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michielse CB, et al. The nuclear protein Sge1 of Fusarium oxysporum is required for parasitic growth. PLoS Pathog. 2009;5(10):e1000637. doi: 10.1371/journal.ppat.1000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jonkers W, Dong Y, Broz K, Kistler HC. The Wor1-like protein Fgp1 regulates pathogenicity, toxin synthesis and reproduction in the phytopathogenic fungus Fusarium graminearum. PLoS Pathog. 2012;8(5):e1002724. doi: 10.1371/journal.ppat.1002724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santhanam P, Thomma BP. Verticillium dahliae Sge1 differentially regulates expression of candidate effector genes. Mol Plant Microbe Interact. 2013;26(2):249–256. doi: 10.1094/MPMI-08-12-0198-R. [DOI] [PubMed] [Google Scholar]
- 9.Michielse CB, et al. The Botrytis cinerea Reg1 protein, a putative transcriptional regulator, is required for pathogenicity, conidiogenesis, and the production of secondary metabolites. Mol Plant Microbe Interact. 2011;24(9):1074–1085. doi: 10.1094/MPMI-01-11-0007. [DOI] [PubMed] [Google Scholar]
- 10.Pande K, Chen C, Noble SM. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat Genet. 2013;45(9):1088–1091. doi: 10.1038/ng.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lohse MB, Zordan RE, Cain CW, Johnson AD. Distinct class of DNA-binding domains is exemplified by a master regulator of phenotypic switching in Candida albicans. Proc Natl Acad Sci USA. 2010;107(32):14105–14110. doi: 10.1073/pnas.1005911107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernday AD, et al. Structure of the transcriptional network controlling white-opaque switching in Candida albicans. Mol Microbiol. 2013;90(1):22–35. doi: 10.1111/mmi.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cain CW, Lohse MB, Homann OR, Sil A, Johnson AD. A conserved transcriptional regulator governs fungal morphology in widely diverged species. Genetics. 2012;190(2):511–521. doi: 10.1534/genetics.111.134080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holm L, Rosenström P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server Issue):W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen SX, et al. Structure of the GCM domain-DNA complex: A DNA-binding domain with a novel fold and mode of target site recognition. EMBO J. 2003;22(8):1835–1845. doi: 10.1093/emboj/cdg182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamasaki K, et al. Solution structure of an Arabidopsis WRKY DNA binding domain. Plant Cell. 2005;17(3):944–956. doi: 10.1105/tpc.104.026435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan MR, et al. DNA binding mechanism revealed by high resolution crystal structure of Arabidopsis thaliana WRKY1 protein. Nucleic Acids Res. 2007;35(4):1145–1154. doi: 10.1093/nar/gkm001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welner DH, et al. DNA binding by the plant-specific NAC transcription factors in crystal and solution: A firm link to WRKY and GCM transcription factors. Biochem J. 2012;444(3):395–404. doi: 10.1042/BJ20111742. [DOI] [PubMed] [Google Scholar]
- 19.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247(4):536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 20.Weirauch MT, Hughes TR. In: A Catalogue of Eukaryotic Transcription Factor Types, Their Evolutionary Origin, and Species Distribution. A Handbook of Transcription Factors, Subcellular Biochemistry. Hughes TR, editor. Vol 52. Dordrecht, The Netherlands: Springer Netherlands; 2011. pp. 25–73. [DOI] [PubMed] [Google Scholar]
- 21.Joshi R, et al. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell. 2007;131(3):530–543. doi: 10.1016/j.cell.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 23.Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci USA. 2004;101(6):1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leslie AGW, Powell HR. In: Processing Diffraction Data with Mosflm Evolving Methods for Macromolecular Crystallography, NATO Science Series. Read RJ, Sussman JL, editors. Vol 245. Dordrecht: Springer Netherlands; 2007. pp. 41–51. [Google Scholar]
- 25.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu XJ, Olson WK. 3DNA: A versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc. 2008;3(7):1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng G, Lu XJ, Olson WK. Web 3DNA–a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res. 2009;37(Web Server Issue):W240–246. doi: 10.1093/nar/gkp358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 31.Glaser F, et al. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19(1):163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- 32.Landau M, et al. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33(Web Server Issue):W299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(Web Server Issue):W529–533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rohs R, et al. The role of DNA shape in protein-DNA recognition. Nature. 2009;461(7268):1248–1253. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang H, Xu Z, Gao L, Hao B. A fungal phylogeny based on 82 complete genomes using the composition vector method. BMC Evol Biol. 2009;9:195. doi: 10.1186/1471-2148-9-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan O, et al. Global gene deletion analysis exploring yeast filamentous growth. Science. 2012;337(6100):1353–1356. doi: 10.1126/science.1224339. [DOI] [PubMed] [Google Scholar]
- 37.Caspari T. Onset of gluconate-H+ symport in Schizosaccharomyces pombe is regulated by the kinases Wis1 and Pka1, and requires the gti1+ gene product. J Cell Sci. 1997;110(Pt 20):2599–2608. doi: 10.1242/jcs.110.20.2599. [DOI] [PubMed] [Google Scholar]
- 38.Kunitomo H, Sugimoto A, Wilkinson CR, Yamamoto M. Schizosaccharomyces pombe pac2+ controls the onset of sexual development via a pathway independent of the cAMP cascade. Curr Genet. 1995;28(1):32–38. doi: 10.1007/BF00311879. [DOI] [PubMed] [Google Scholar]
- 39.Zordan RE, Miller MG, Galgoczy DJ, Tuch BB, Johnson AD. Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans. PLoS Biol. 2007;5(10):e256. doi: 10.1371/journal.pbio.0050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.