

Abstract
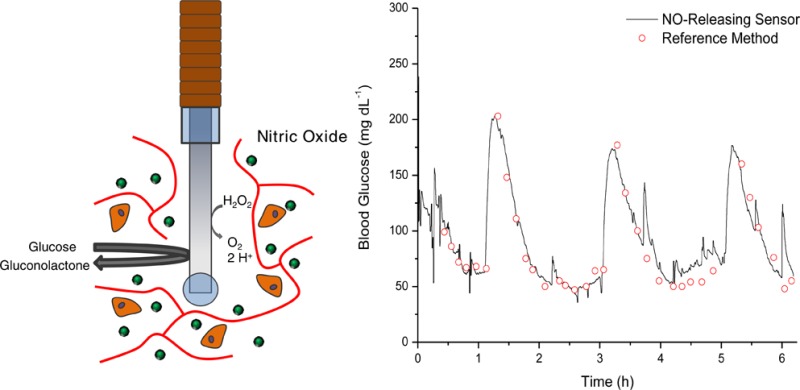
The in vivo analytical performance of percutaneously implanted nitric oxide (NO)-releasing amperometric glucose biosensors was evaluated in swine for 10 d. Needle-type glucose biosensors were functionalized with NO-releasing polyurethane coatings designed to release similar total amounts of NO (3.1 μmol cm–2) for rapid (16.0 ± 4.4 h) or slower (>74.6 ± 16.6 h) durations and remain functional as outer glucose sensor membranes. Relative to controls, NO-releasing sensors were characterized with improved numerical accuracy on days 1 and 3. Furthermore, the clinical accuracy and sensitivity of rapid NO-releasing sensors were superior to control and slower NO-releasing sensors at both 1 and 3 d implantation. In contrast, the slower, extended, NO-releasing sensors were characterized by shorter sensor lag times (<4.2 min) in response to intravenous glucose tolerance tests versus burst NO-releasing and control sensors (>5.8 min) at 3, 7, and 10 d. Collectively, these results highlight the potential for NO release to enhance the analytical utility of in vivo glucose biosensors. Initial results also suggest that this analytical performance benefit is dependent on the NO-release duration.
Despite the obvious benefits of continuous glucose monitoring (CGM) for the management of diabetes, the utility of in vivo amperometric glucose biosensors is limited to <1 week due to poor analytical performance, resulting primarily from the foreign body response (FBR).1,2 Insertion of the sensor damages vascularized tissue and results in a cascade of inflammatory events, many of which negatively impact glucose measurements.3 For example, the resulting passive adsorption of biomolecules (mainly <15 kDa protein fragments) to the sensor surface initiates an inflammatory response and is responsible for a dramatic decrease in sensor sensitivity (∼50%) following sensor implantation.3−6 Increased metabolic activity of inflammatory cells (i.e., macrophages and foreign body giant cells) at the sensor–tissue interface results in inordinate consumption of glucose and oxygen, decreasing their local concentrations and attenuating sensor performance.7 The hallmark of the FBR is the formation of a thick, avascular collagen capsule surrounding the sensor, isolating it from the tissue and obstructing mass transport of interstitial glucose to the sensor.3 Indeed, the FBR increases sensor response time, decreases sensitivity, and often results in device failure.
Efforts to improve the analytical performance of in vivo biosensors have largely focused on chemical or physical modifications to the outermost, tissue-contacting membrane to mitigate the FBR.8 Examples of such strategies include biomimicry (e.g., the attachment of phospholipids to coating surfaces),9 employing naturally derived materials as coatings,10 utilizing membranes that reduce cell adhesion,11 encouraging tissue ingrowth into porous coatings,12−14 and modulating cell behavior through coating topography.15 The active release of anti-inflammatory or pro-angiogenic bioactive agents such as dexamethasone (DX) and vascular endothelial growth factor (VEGF) has also been proposed as a viable option for improving glucose sensor function.16,17 However, in addition to the immune suppression associated with DX18 and pro-inflammatory roles of VEGF,19 the controlled release of these molecules from sensor coatings remains a major hurdle.
The controlled release of nitric oxide (NO), an endogenous molecule with multiple roles in inflammation, wound healing, and angiogenesis, from polymeric coatings has been shown to minimize the FBR.1,20−22 Hetrick et al. examined the FBR to subcutaneously implanted NO-releasing xerogels coated on silicone elastomers in a murine model.23 Nitric oxide-releasing implants, which generated ∼1.35 μmol cm–2 NO over 72 h at fluxes >1 pmol cm–2 s–1, elicited only a mild FBR with reduced fibrous encapsulation (>25%) after 3 and 6 weeks compared to tissue near control implants. Concomitant with a reduced FBR, blood vessel density in the tissue surrounding the NO-releasing implants was greater (∼50%) than that observed surrounding control implants. Nichols et al. assessed glucose recovery as a function of NO release using percutaneously implanted microdialysis probes.24 A constant NO flux (162 pmol cm–2 s–1, 4.6 μmol cm–2 NO daily) was achieved from microdialysis probes by using a saturated NO solution as the perfusate. While the glucose recovery of control probes was severely diminished beyond 7 d, NO-releasing microdialysis probes exhibited near constant glucose recovery throughout the study. These results were correlated to tissue histology observations. Indeed, histological analysis of the tissue surrounding NO-releasing probes at 14 d revealed lower inflammatory cell counts and a thinner collagen capsule versus probes that did not release NO. The lessened FBR and increased glucose recovery suggest that NO release lowered tissue impedance to glucose transport. In a separate study, Nichols and co-workers investigated the effects of NO-release kinetics on the FBR to subcutaneous NO-releasing wire implants (i.e., mock glucose sensors) in a porcine model.25 Decreased collagen capsule thickness (>50%) was observed for substrates that released NO for extended durations (i.e., >14 d) versus wires that did not release NO. In contrast, substrates with shorter NO-release durations (12–24 h) were characterized by greater collagen density at the implant–tissue interface compared to the materials which released NO for extended durations. Collectively, this body of work highlights the dramatic effect of NO-release kinetics on the FBR and the potential to impact the analytical performance of in vivo glucose biosensors.
Despite extensive characterization of the host response to NO-releasing implants, the interplay between reduced FBR and actual sensor performance remains a critical void. To date, only one study has evaluated the in vivo performance of a NO-releasing glucose sensor. Gifford et al. reported improved clinical accuracy for NO-releasing needle-type glucose biosensors implanted in rats for 3 d.26 However, the NO release from the sensors was limited to 16 h and deterioration of sensor performance by day 3 was observed.27 Histological analysis of the surrounding tissues revealed suppressed inflammation at NO-releasing sensors on day 1 versus controls, but no benefits following depletion of the NO reservoir. Clearly, the role of NO release on sensor analytical performance should be studied in greater detail.
As the severity of the FBR to NO-releasing implants is dependent on release kinetics, we sought to investigate these effects on the performance of percutaneously implanted glucose biosensors. Given previous findings, it is hypothesized that by extending NO-release duration, sensor merits (i.e., accuracy, sensitivity, response time) may be maintained for longer implantation periods (>7 d). Herein, we report on the analytical performance of NO-releasing needle-type glucose biosensors in swine as a function of NO-release duration.
Experimental Section
Materials
Glucose oxidase (GOx; type VII from Aspergillus niger, >100,000 units g–1), d-(+)-glucose anhydrous, acetaminophen (AP), l-ascorbic acid (AA), urea (UA), phenol, diethylenetriaminepentaacetic acid (DTPA) and sodium methoxide (5.4 M in methanol) were purchased from Sigma (St. Louis, MO). Tetrahydrofuran (THF), ethanol (EtOH), aqueous ammonium hydroxide (30 wt %), and all salts were purchased from Fisher Scientific (St. Louis, MO), Tetraethyl orthosilicate (TEOS), (3-mercaptopropyl)trimethoxysilane (MPTMS), and (3-methylaminopropyl)trimethoxysilane (MAP3) were purchased from Gelest (Tullytown, PA). Methyltrimethoxysilane (MTMOS) was purchased from Fluka (Buchs, Switzerland). Cetyltrimethylammonium bromide (CTAB) was purchased from Acros Organics (Geel, Belgium). Hydrothane (AL25-80A) polyurethane (HPU) was a gift from AdvanSource Biomaterials (Wilmington, MA). Tecoflex (SG-85A) polyurethane (TPU) was a gift from Lubrizol (Cleveland, OH). Steel wire (356 μm diam) was purchased from McMaster-Carr (Atlanta, GA). Argon, nitrogen, oxygen, and nitric oxide calibration gas (25.87 ppm in nitrogen) were purchased from Airgas National Welders (Raleigh, NC). Nitric oxide gas was purchased from Praxair (Danbury, CT). Water was purified using a Millipore Milli-Q UV gradient A10 system (Bedford, MA) to a resistivity of 18.2 MΩ·cm and a total organic content of ≤6 ppb. All other chemicals were reagent grade and used as received.
Synthesis of NO-Releasing Silica Nanoparticles
Synthesis of NO-releasing silica nanoparticles was carried out as described previously.28 Briefly, MPTMS particles were synthesized via the co-condensation of MPTMS (70 mol %) and TEOS. The thiol-containing nanoparticles were nitrosated by reaction with acidified nitrite in the dark at 0 °C for 2 h. Mesoporous MAP3 silica nanoparticles were prepared via the surfactant-templated co-condensation of TEOS in the presence of CTAB, followed by removal of CTAB and surface-grafting of MAP3 to the particle surface. Subsequent N-diazeniumdiolation of the secondary amine-containing nanoparticles was carried out under high pressures of NO (10 atm) at room temperature for 3 d in the presence of sodium methoxide. Sizes and total NO-release payloads of the silica nanoparticles are included in the Supporting Information [SI] (Table S2).
Preparation of NO-Releasing Mock Sensors
Steel wire was cut in 7 cm pieces and cleaned by sonication in EtOH for 10 min. Polymer solutions containing the macromolecular NO-release scaffolds were prepared by dispersing MAP3 or MPTMS particles (72 and 48 mg mL–1, respectively) in an 80 mg mL–1 solution of 50:50 wt % HPU/TPU in 1:1 EtOH/THF. Wire substrates were modified by dip coating (5 mm s–1 with a 5-s hold time) four times into the particle-containing PU solution using a DipMaster 50 dip coater (Chemat Technology, Inc.; Northridge, CA) with 30 min drying periods under ambient conditions between dips. A final TPU topcoat was applied by dip coating into a 40 mg mL–1 TPU solution in THF.
Characterization of NO-Releasing Wire Substrates
Nitric oxide release from steel wire substrates was measured in real time using a Sievers 280i chemiluminescence NO analyzer (NOA; Boulder, CO). Generation of NO from PU films was detected indirectly by the formation of a chemiluminescent product (NO2*) upon reaction of NO with ozone. The NOA was calibrated using an atmospheric gas sample passed through a Sievers NO zero filter (0 ppb) and 25.9 ppm of NO in N2. Substrates were immersed in deoxygenated phosphate buffered saline (PBS; 0.01 M, pH 7.4) at 37 °C. The liberated NO from PU films was carried to the NOA by a stream of N2, bubbled into solution at a volumetric flow rate of 75 mL min–1. For films containing S-nitrosothiol NO donors (i.e., MPTMS particles), the sample flask was shielded from light, and 500 μM DTPA was added to the PBS buffer to chelate trace copper. Data output from the NOA was collected every 1 s, allowing for near real-time monitoring of NO generated from the films.
The stability of silica particles in PU films was assessed using inductively coupled plasma optical emission spectrometry (ICP-OES). Modified wire substrates were immersed in PBS buffer and incubated at 37 °C for 10 d. The degree of particle leaching into soak solutions was determined by monitoring the silicon emission intensity at 251.611 nm using a Prodigy high dispersion ICP (Teledyne Leeman Laboratories; Hudson, NH).
Fabrication and Benchtop Performance of NO-Releasing Needle-Type Glucose Sensors
Bare needle-type glucose sensors (Pinnacle Technology, Inc., Lawrence, KS), composed of an integrated silver/silver chloride (Ag/AgCl) pseudoreference electrode wound around a 90:10 platinum/iridium (Pt/Ir) working electrode (127 μm diam, ∼1 mm length), were functionalized by the successive deposition of a polyphenol selectivity layer, a GOx enzyme layer, a NO-releasing flux-limiting membrane, and a polyurethane topcoat, as described previously.29−31 Experimental details regarding biosensor sterilization, functionalization, and benchtop testing are provided in the SI. Following deposition of the selectivity and enzyme layers, sensors were coated with a PU diffusion-limiting/NO-releasing layer by dip-coating into a particle-containing PU solution. A TPU topcoat was then applied as an additional layer. Control sensors were coated using PU solutions containing MAP3 or MPTMS nanoparticles (72 and 48 mg mL–1, respectively) that were not functionalized with N-diazeniumdiolate or S-nitrosothiol NO donors.
In Vivo Protocol for Assessing Biosensor Analytical Performance
The animal protocol used in this study was approved by the IACUC at Synchrony, LLC (Durham, NC). The in vivo performance of glucose biosensors was evaluated in Yorkshire-type piglets (n = 10; Palmetto Research Swine; Reevesville, SC) weighing approximately 7–15 kg. Details regarding sensor implantation and operation are provided in the SI. Biosensor performance was evaluated on 0, 1, 3, 7, and 10 d after sensor implantation. A peripherally inserted central catheter was placed in an external jugular vein for blood draws. Reference blood glucose (BG) concentrations were measured every 10 min for 6–8 h using a One Touch Ultra glucometer (LifeScan, Inc.; Milpitas, CA) for comparison to sensor data. During glucose sensor evaluation, pigs were fasted and sedated with propofol (2 mg kg–1 h–1) administered through a catheter in a peripheral ear vein. Once on the day of implantation and three times daily thereafter, the swine were challenged with an intravenous glucose tolerance test (IVGTT; 0.7 g kg–1, 50 wt % dextrose, 1–1.5 h duration), administered over 30 s through the peripheral catheter, to assess the ability of glucose sensors to track changing blood glucose concentrations. On day 10, pigs were euthanized and the sensors were explanted by removal of the surrounding tissue en bloc. Postexplantation, sensors were imaged using environmental scanning electron microscopy (ESEM; FEI Quanta 200 Field Emission Gun; Hillsboro, OR).
Data Analysis
Sensor current traces were filtered and analyzed using custom MATLAB scripts (Mathworks, Inc.; Natick, MA). A finite impulse response (FIR) filter was used to attenuate large noise spikes caused by pig motion and potentiostat RF transmitter dropout.32 A 1 min median filter was used to further smooth the data before pairing sensor current traces with reference measurements. Glucose sensors were calibrated with respect to reference BG measurements once per day using a two-point retrospective calibration.33,34 One point for calibration was taken at a stable glucose baseline (i.e., prior to the first IVGTT), while the second point was taken at a stable point after the first dextrose administration with at least a 15 mg dL–1 difference between BG concentrations. The slope of a linear trend line connecting these two points was taken as the apparent in vivo biosensor sensitivity on each day, expressed as mean values ± standard deviation. The method of Poincaré was used to approximate the time delay at which the correlation between the reference and calibrated sensor signals was greatest, using R2 as the agreement criterion.35,36 This delay was determined at ∼5 min and used to correct sensor data on each day for the physiological time lag that characterizes mass transfer of glucose from blood to tissue.37 After sensor implantation, the “run-in” time (i.e., the time required for sensors to achieve a stable background current) was estimated by determining the period over which two consecutive sensor measurements agreed with their respective reference measurements within 20%.
Sensor performance was determined using numerical and clinical accuracy metrics. The mean absolute relative deviation (MARD) for a data set collected by a single sensor (∼25–35 measurements) was used to characterize sensor numerical accuracy at each time point.38 Sensor MARD was calculated using eq 1, where CGM and BG are the blood glucose values determined by the sensor and reference glucometer, respectively.
| 1 |
Additionally, the International Standards Organization (ISO) criteria for glucose monitor performance was used to assess sensor numerical accuracy by separately calculating the percentage of glucose measurements determined by sensors that were within (1) ±15 mg dL–1 of the paired reference determination when BG was ≤70 mg dL–1 and (2) ±20% of the paired reference determination when BG was >70 mg dL–1.38 Sensor clinical accuracy was determined using Clarke error grid analysis (EGA) by quantifying the percentage of blood glucose determinations falling in zones A and B of the error grid.39 Cross-correlation of the reference signals and raw sensor current traces was used to estimate sensor lag time, with possible lag times restricted to >100 s.40,41 Values for MARD and lag time are expressed as mean values ± standard error of the mean. Differences in median values for sensor MARD, lag time, and sensitivity between NO-releasing and control sensors were analyzed using a two-tailed nonparametric Mann–Whitney U test.42
Results and Discussion
Nitric oxide-releasing polyurethanes were selected as sensor coatings for evaluating the effect of NO-release duration on in vivo glucose biosensor performance. Total NO payloads sufficient for minimizing inflammation (i.e., >1 μmol cm–2)23 with varied NO-release durations (<1 h to >14 d) were achieved by tuning the PU properties (i.e., water uptake) and NO donor type.30 We have shown previously that sensor response is not negatively affected by NO release from PU coatings at a working electrode potential of +600 mV vs Ag/AgCl.30 The versatile NO-release kinetics and compatibility with amperometric glucose sensing make NO-releasing polyurethanes an ideal platform for assessing the effects of NO release on in vivo glucose biosensor performance.
In Vitro Characterization of NO-Releasing Glucose Sensors
Wire substrates, selected to mimic the geometry and size of a needle-type glucose sensor, were modified with NO-releasing PU coatings via a dip-coating procedure. A hydrophobic TPU topcoat was employed to both minimize any leaching of the macromolecular NO donors and eliminate the surface roughness introduced by nanoparticle dopants. Undoubtedly, the physical properties (i.e., roughness) of an implant surface will affect the FBR.15,43 The stability of the nanoparticle-doped PU coatings in PBS was investigated over 10 d by analyzing the silicon content of soak solutions using ICP-OES. While silica is intrinsically biocompatible and considered nontoxic,23,44 the resulting changes in coating structure or potential tissue inflammation may affect the performance of glucose sensors in vivo. For coatings doped with NO-releasing MPTMS-RSNO particles as well as controls, leaching of silica particles from the PU matrix was undetectable (<2%). Slight leaching (10.8 ± 2.9% of the total incorporated silica) was observed from coatings containing NO-releasing MAP3/NO particles. Interestingly, the majority of the observed leaching occurred during the first 4 h, suggesting some instability associated with encapsulating the charged N-diazeniumdiolate NO donor moieties within the polyurethane coating (data not shown).
The effect of NO-release duration on in vivo sensor performance was studied using two different macromolecular NO release systems: N-diazeniumdiolate NO donors and S-nitrosothiol-modified silica nanoparticles (MAP3/NO and MPTMS-RSNO, respectively). Briefly, N-diazeniumdiolate NO donors undergo proton-initiated decomposition in aqueous milieu to generate NO.45 Conversely, NO release from S-nitrosothiols may be triggered using light or Cu(I), but S-nitrosothiols also decompose sluggishly through thermal mechanisms in vivo.28,46 To simulate in vivo conditions, NO release from PU films was measured in PBS at 37 °C. For MPTMS-RSNO coatings, thermal decomposition of the S-nitrosothiol moieties was achieved using a light-shielded sample flask and the addition of DTPA to chelate trace copper. By appropriate selection of the nanoparticle dopant concentration (72 and 48 mg mL–1 for MAP3/NO and MPTMS-RSNO particles, respectively) we attained similar total NO payloads (∼3.1 μmol cm–2) for both coating formulations (Table 1). Of note, NO payloads from these coatings were more than 2 times greater than that from the xerogel coatings utilized by Hetrick et al. (∼1.35 μmol cm–2) and similar in magnitude to those employed by Nichols et al. (2.7–9.3 μmol cm–2)—both of which proved effective at reducing the FBR to subcutaneous implants.23,25
Table 1. Nitric Oxide Release from Polyurethane Coatings Doped with NO-Releasing MPTMS-RSNO and MAP3/NO Nanoparticles.
| NO-release merits | MPTMS-RSNO | MAP3/NO |
|---|---|---|
| [NO]max (pmol cm–2 s–1) | 551.4 ± 130.0 | 685.8 ± 11.4 |
| tmax (min)a | 1.68 ± 0.20 | 23.80 ± 7.17 |
| t1/2 (h)b | 6.29 ± 2.07 | 0.93 ± 0.17 |
| [NO]8h (pmol cm–2 s–1) | 14.0 ± 3.9 | 13.0 ± 3.2 |
| [NO]12h (pmol cm–2 s–1) | 9.8 ± 3.8 | 3.7 ± 1.5 |
| [NO]24h (pmol cm–2 s–1) | 3.3 ± 0.2 | 0c |
| [NO]48h (pmol cm–2 s–1) | 1.0 ± 0.1 | 0c |
| [NO]72h (pmol cm–2 s–1) | 0.5 ± 0.0 | 0c |
| [NO]168h (pmol cm–2 s–1) | 0.5 ± 0.0 | 0c |
| [NO]T (μmol cm–2)d | 3.14 ± 0.26e | 3.11 ± 0.27 |
| td (h)f | 74.6 ± 16.6 | 16.0 ± 4.4 |
Time required to reach maximum NO flux.
Half-life for NO-release from PU films.
Nitric oxide release was below the limit of detection of the NOA.
Total amount of NO released.
Measured by irradiation of the sample flask with 200 W light.
Determined at the time at which 99% of the total NO was released.
Upon immersion in PBS, MAP3/NO films exhibited a large initial NO flux ([NO]max = 685.8 ± 11.4 pmol cm–2 s–1) and released 99% of their total NO payload within ∼16 h, with no additional NO release measurable beyond 24 h. Such duration (16 h) was similar to that reported to improve glucose sensor accuracy by Gifford and co-workers (12–18 h).26 Similarly, MPTMS-RSNO films showed a large initial NO flux ([NO]max = 551.4 ± 130.0 pmol cm–2 s–1), with a rapid decrease to ∼14.0 pmol cm–2 s–1 at 8 h. In contrast to the MAP3/NO films, MPTMS-RSNO coatings required ∼3.1 d to release 99% of their total NO payload, with NO release (0.5 pmol cm–2 s–1) still measurable at ∼7 d. Even such low levels of NO are physiologically relevant, as vascular endothelial cells release NO at 1–7 pmol cm–2 s–1 to prevent platelet activation.27,47 Additionally, similar NO fluxes (1.5–30 pmol cm–2 s–1) inhibit in vitro bacterial adhesion to surfaces.48,49
As expected, NO release from the outer glucose sensor membrane did not impact biosensor response. After an initial hydration period of 3–4 h, the glucose sensitivities of NO-releasing and control sensors were comparable and remained constant (1.3–2.3 nA mM–1) over 10 d in PBS at 37 °C for all membrane formulations. In the absence of preconditioning, sensors exhibited poorer dynamic range and longer response times to changes in glucose concentration during the first several hours of testing (data not shown). Both NO-releasing and control sensors exhibited acceptable response times (<40 s) to an increase in glucose concentration of 5.6 mM. All sensors responded linearly to glucose between 1–12 mM after preconditioning in PBS. Furthermore, the amperometric selectivity coefficients for glucose over acetaminophen, ascorbic acid, and urea were 0.82 ± 0.30, 0.49 ± 0.11, and 0.03 ± 0.01, respectively, for blank sensors (i.e., sensors that were coated solely with polyurethane). As expected, selectivity for glucose was sufficient. The analytical performance merits of the sensors are provided in the SI (Table S1).
In Vivo Biosensor Run-In Time, Glucose Sensitivity, and Clarke Error Grid
Following implantation, both NO-releasing and control biosensors displayed a run-in period (i.e., the time required to achieve a stable baseline current) during which the sensor response was erratic (Figure S3 in the SI). While Gifford et al. reported a reduced run-in time for NO-releasing sensors versus control sensors in rodents,26 we observed no significant differences in run-in time between NO-releasing sensors and controls, with all four sensor configurations requiring ∼3–6 h to achieve a steady background current. The source of this discrepancy is unclear, but a number of variables (e.g., different animal model, implant method, and extended sensor hydration time) may have contributed to this result.
The potential analytical performance benefits of NO-releasing amperometric glucose biosensors were evaluated in a healthy swine model. The use of digital noise filters was required to achieve stable current traces due to swine motion and intermittent potentiostat RF transmitter dropout. The filtering algorithms were restricted to those compatible with real-time continuous glucose monitoring.32 As expected, the FIR and median filters sufficiently improved signal quality without introducing an undesirable artificial time delay (>20%) between sensor and reference signals. Subsequently, sensors were calibrated by comparison to corresponding reference blood glucose measurements using a two-point retrospective calibration. While a one-point calibration (which assumes a negligible background current) has been suggested to be superior to the two-point calibration,33,34 the in vivo background in our study was substantial (6–10 nA) compared to the in vitro baseline (1–3 nA), necessitating the use of a two-point calibration. Other researchers have also reported disparities between in vitro and in vivo sensor baseline currents.50 Despite minimizing the artificial delay caused by filtering, a physiological lag between the sensor signal and reference BG measurements was still observed. This delay arises from the slow mass transfer of glucose from the vasculature to the tissue and ultimately the sensor.35−37 An analysis of sensor performance on day 0 via the method of Poincaré36 indicated a ∼5 min lag between the reference signal and calibrated sensor signal. This lag time was thus accounted for in all remaining data sets (days 1, 3, 7, and 10) by shifting the reference signal in time relative to the sensor signal.
The clinical accuracies of NO-releasing and control in vivo glucose biosensors were first assessed via the Clarke error grid.39 The percentage of BG measurements falling in zones A and B (clinically accurate and clinically benign determinations, respectively) of the error grid are shown in Table 2. On the day of implantation (day 0), the MAP3/NO-based sensors performed slightly worse than control sensors, with a 2% difference in the percentage of determinations in zones A and B. However, the performance of MAP3/NO sensors on days 1 and 3 was superior to that of controls, with >7% difference in the percentage of clinically accurate and acceptable determinations. Concomitant with improved clinical performance, sensors that rapidly released NO were characterized as having greater glucose sensitivity on days 1 and 3 (0.59 ± 0.54 and 0.59 ± 0.40 nA mM–1, respectively) versus controls (0.14 ± 0.09 and 0.18 ± 0.04 nA mM–1, respectively). However, the MAP3/NO sensors exhibited similar clinical accuracy and glucose sensitivity to control sensors at implant periods beyond 3 days (i.e., days 7 and 10), suggesting that sensor performance is only improved during periods of active NO release. The trends in sensor clinical performance and glucose sensitivity correlate well with the NO-release kinetics from the sensors, with clear benefits to sensor performance early during in vivo use (i.e., days 1 and 3) but no improvements after the NO supply was exhausted. Of note, Nichols et al. noted no decrease in the FBR (>1 w) for implants with rapid NO release, suggesting that inflammation may be the primary culprit for decreased sensor performance beyond 3 d.25 Unexpectedly, the MPTMS-RSNO-based sensors exhibited clinical accuracy similar to that of MPTMS control sensors throughout the 10 d in vivo study. The sensitivity of the MPTMS-RSNO sensors to glucose appeared slightly greater than that of MPTMS controls beyond day 0, but these differences were not significant (p > 0.05). This result may be due to the low, sustained NO fluxes released from sensors when compared to those from the MAP3/NO-based sensors (Table 1).
Table 2. Clinical Performance and Apparent in Vivo Sensitivity of Glucose Biosensors.
| day | MAP3 control | MAP3/NO | MPTMS control | MPTMS-RSNO | |
|---|---|---|---|---|---|
| 0 | % points in zones A/B | 89.6 | 87.6 | 91.0 | 94.7 |
| Na | 183 | 105 | 311 | 321 | |
| sensitivity (nA mM–1) | 0.90 ± 0.87 | 0.72 ± 0.40 | 0.74 ± 0.47 | 0.60 ± 0.30 | |
| 1 | % points in zones A/B | 78.6 | 86.2 | 90.6 | 89.1 |
| Na | 168 | 174 | 224 | 347 | |
| sensitivity (nA mM–1) | 0.14 ± 0.09 | 0.59 ± 0.54b | 0.29 ± 0.18 | 0.39 ± 0.17 | |
| 3 | % points in zones A/B | 84.8 | 92.0 | 81.7 | 83.9 |
| Na | 169 | 173 | 180 | 124 | |
| sensitivity (nA mM–1) | 0.18 ± 0.04 | 0.59 ± 0.40b | 0.24 ± 0.16 | 0.49 ± 0.18 | |
| 7 | % points in zones A/B | 93.2 | 94.2 | 88.3 | 88.1 |
| Na | 115 | 87 | 157 | 69 | |
| sensitivity (nA mM–1) | 0.23 ± 0.15 | 0.39 ± 0.26 | 0.20 ± 0.07 | 0.45 ± 0.19 | |
| 10 | % points in zones A/B | 84.8 | 81.4 | 91.8 | 84.9 |
| Na | 138 | 97 | 135 | 66 | |
| sensitivity (nA mM–1) | 0.16 ± 0.06 | 0.20 ± 0.13 | 0.09 ± 0.02 | 1.3 ± 1.1 |
Total number of measurements.
Significantly different at p < 0.05.
Of importance, the majority (∼70%) of BG determinations were obtained in the 50–100 mg dL–1 range, as shown in the representative Clarke error grid analysis in Figure S4 of the SI. In addition to the similarities between swine and humans (e.g., skin, vasculature, subcutaneous tissue composition) which render the pig an appropriate model for evaluating in vivo biosensors, baseline blood glucose concentrations obtained in this study were comparable to human euglycemic levels.26,50−52 As maintenance of euglycemia increases the propensity of diabetic individuals to enter the hypoglycemic BG range,53 the Clarke error grid presents austere requirements for sensor accuracy in this region. Thus, the error grid analysis presented herein is at BG levels clinically and physiologically pertinent to humans.
Biosensor Numerical Accuracy and Adherence to ISO Criteria
To evaluate in vivo biosensor performance in more detail, the sensor numerical accuracy was represented using the MARD of each sensor from corresponding reference values.38 While the Clarke error grid measures sensor accuracy based on the clinical implications of a given BG measurement, the MARD represents a statistical entity that exemplifies the average percent deviation of the sensor from a reference. Additionally, ISO criteria for in vivo glucose biosensor performance was considered as a metric for numerical accuracy because it can be used to assess sensor accuracy in both hypoglycemic (≤70 mg dL–1) and euglycemic/hyperglycemic (>70 mg dL–1) BG ranges separately.38 A comparison of the numerical accuracies for control and NO-releasing sensors is shown in Figure 1. As anticipated, the analytical performance of MAP3/NO-based sensors on days 1 and 3 was superior to that of MAP3 (control) sensors. The improvements in numerical accuracy agree with the increased clinical accuracy and greater glucose sensitivity for the more rapid NO-releasing sensors. Furthermore, the performance of the MAP3/NO-based sensors was observed to worsen beyond 3 d implantation. The desirably lower MARD for rapid NO-releasing glucose sensors is attributed to the improved accuracy in both the hypoglycemic and euglycemic/hyperglycemic ranges, as shown in Table 3. Indeed, >55% of the total BG determinations obtained by MAP3/NO-based sensors agreed well with corresponding reference measurements in both BG ranges on days 1 and 3. Unexpectedly, the MARD for control (MAP3) sensors was lowest at 7 d implantation (21.9 ± 13.1%). Despite the inconsistent numerical accuracy for control sensors, the analytical performance was comparable to NO-releasing sensors at both 7 and 10 d.
Figure 1.

Comparison of MARD for (A) MAP3/NO (red circle) and control (MAP3) sensors (black, square) and (B) MPTMS-RSNO (red circle) and control (MPTMS) (black, square) sensors. Significant differences (p < 0.05) in the median value for the MARD are indicated with an asterisk.
Table 3. ISO Criteria for NO-Releasing and Control Sensors.
| Day | MAP3 Control (%) | MAP3/NO (%) | MPTMS Control (%) | MPTMS-RSNO (%) |
|---|---|---|---|---|
| 0 | 58.0a/50.0b | 51.9/61.5 | 55.7/60.0 | 60.2/67.0 |
| 1 | 37.9/39.2 | 55.6/56.7 | 45.9/59.7 | 55.5/59.4 |
| 3 | 52.9/47.7 | 65.6/57.3 | 39.5/57.3 | 58.5/74.7 |
| 7 | 62.5/62.7 | 42.1/57.8 | 35.5/45.2 | 42.1/52.0 |
| 10 | 55.6/54.9 | 30.6/45.9 | 15.0/34.8 | 63.6/45.5 |
Calculated as the percentage of determinations within 15 mg dL–1 of the reference measurement when BG ≤ 70 mg dL–1
Calculated as the percentage of determinations within 20% of the reference measurement when BG > 70 mg dL–1
Although the clinical accuracy of the MPTMS-RSNO-based sensors was comparable to that of controls, the numerical accuracy of NO-releasing sensors remained constant (MARD range 22.2–26.0%) throughout the experiment. Furthermore, the sensors that released NO for extended durations exhibited a significantly lower MARD on days 1 and 3 (26.0 ± 5.1 and 23.9 ± 8.6%, respectively) versus controls (34.3 ± 10.9 and 38.8 ± 10.4%, respectively). We attribute the good agreement between MPTMS-RSNO sensors and reference measurements to the increased accuracy of the NO-releasing biosensors in both the hypoglycemic and euglycemic/hyperglycemic BG ranges. The percentage of determinations for MPTMS-RSNO-based sensors that adhered to ISO criteria was typically >50% throughout implantation, while control sensor performance worsened with implant duration, particularly in the hypoglycemic range. The stable biosensor response provided by the sustained NO-releasing sensor membranes highlights the utility of NO release for continuous glucose monitoring.
Of importance, the NO-release kinetics also correlated with the magnitude of the improvement in numerical accuracy for NO-releasing sensors versus controls. For example, MAP3/NO-based sensors showed vastly decreased MARD versus MAP3 (control) sensors on day 1 (22.0 ± 6.6 and 47.3 ± 8.1%, respectively), whereas sensors with longer NO-release durations (MPTMS-RSNO) exhibited more modest improvements relative to controls (28.4 ± 5.9 and 34.3 ± 10.9%, respectively). However, the differences in the MARD between MAP3/NO and MPTMS-RSNO sensors on days 1 and 3 were not statistically significant (p > 0.05). The enhanced numerical accuracy afforded by rapid NO release from sensor membranes indicates a possible advantage to greater NO fluxes, as MAP3/NO-based sensors delivered ∼3.1 μmol cm–2 NO in <24 h. While MPTMS-RSNO sensors had a near constant MARD throughout the experiment duration, the improvements in numerical accuracy provided by lower, more sustained NO release may not have been large enough to result in improved clinical performance. Collectively, these results suggest that sensor performance benefits to a greater extent with prolonged NO release and that these gains are dependent on the fluxes at which NO is liberated.
Biosensor Lag Time
While poor glucose sensitivity often contributes to undesirable sensor performance in vivo, diminished accuracy also results from sluggish response of the sensor to changes in BG levels.54 In addition to an inherent blood–tissue glucose lag, progression of the FBR increases the difficulty of glucose diffusion to the sensor. Distinct properties of the collagen capsule (i.e., thickness, density, and avascularity) produced upon resolution of the foreign body response have been shown to affect the transport properties of small molecules in the subcutaneous tissue.12−14 Even in the absence of a mature fibrotic capsule, biofouling and inflammation at the sensor–tissue interface may create a diffusion barrier to glucose.55 As amperometric glucose biosensors are diffusion-limited with respect to glucose, a longer response time may hinder the competence of the sensor to track rapid changes in BG levels, resulting in decreased accuracy. Since tissue surrounding NO-releasing implants exhibits less inflammation,23−25 reduced collagen encapsulation,23,25 and low impedance to glucose transport,24 NO-releasing sensors may show more rapid response to changes in BG. While time-shifting methods (i.e., Poincaré dynamical analysis) have been used to correct CGM data for time-lag effects,35,36 calibration of the sensor signal may corrupt a comparison of sensor lag times. Cross-correlation of the raw sensor signals and paired reference signals were thus used to estimate sensor delay, avoiding the requirement for sensor calibration.40,41
Initially (i.e., 0–1 d implant period), NO release had little effect on sensor lag times (Figure 2). However, NO release did impact sensor lag times on days 3, 7, and 10. The MPTMS-RSNO-based sensors yielded a faster response to changing glucose concentrations during the IVGTT (<4.2 min) compared with both control (MPTMS) and MAP3/NO-based sensors (>5.8 min). As well, the response time of the MAP3/NO-based sensors worsened with implantation time analogous to control sensors, suggesting that the benefit of reduced response time is only attained when sensors are still releasing NO. Despite similar NO payloads, the difference in lag time between the two types of NO-releasing sensors is corroborated by the work of Nichols et al.,25 in which rapid NO release at 3 and 7 d yielded no reduction in FBR, while extended NO release provided a lessened FBR at both 3 and 7 d. Likewise, sustained NO release from percutaneously implanted microdialysis probes reduced tissue impedance to glucose transport,24 which may explain the reduced sensor lag time observed in the present study.
Figure 2.
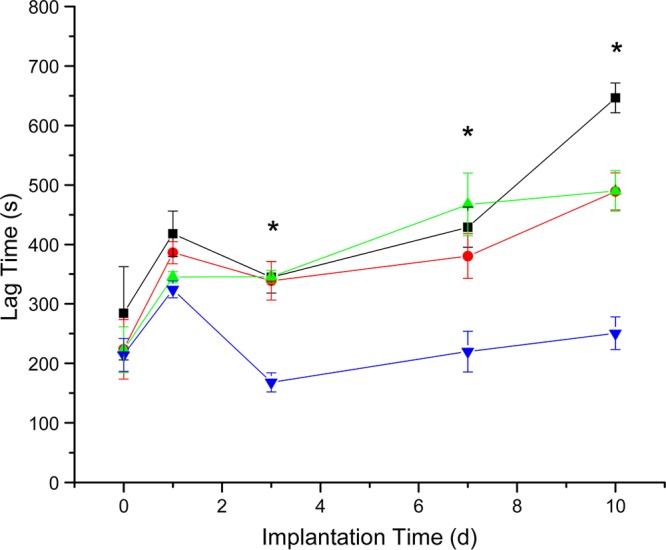
Estimation of sensor lag time via cross-correlation. MPTMS-RSNO biosensors (blue inverted triangle) exhibited significantly reduced lag times on days 3, 7, and 10 versus MAP3/NO sensors (red circle), and MAP3 and MPTMS controls (black square and green triangle, respectively). Asterisks denote significant differences (p < 0.05) in the median values for lag time between the MPTMS-RSNO sensors and all other sensor types.
Postexplantation Analysis
Approximately 40% of implanted NO-releasing and control sensors functioned beyond 3 d, indicating a limitation in this study. Following sensor explantation, the sensors were imaged via environmental scanning electron microscopy to investigate the implant surfaces and perhaps understand the potential sources of sensor failure. Representative scanning electron micrographs (SEMs) of the surface of the sensors are provided in SI (Figure S5). Electrical failure via membrane delamination or cracking contributed considerably to in vivo sensor failure. Most likely, mechanical stresses to the percutaneous sensor were at fault for damage to the sensor membranes.50 Koschwanez et al. previously reported that micromotion and the associated stress for percutaneous implants yielded unanticipated results for studies evaluating glucose sensor coatings.56 Indeed, the significant mechanical stress to percutaneous implants convolutes the interpretation of sensor failure. Nonetheless, percutaneous glucose sensors remain the most realistic method for implementing continuous glucose monitoring, due to their low cost and facile implantation, and serve as a suitable model for evaluating candidate biomaterials.50,57 Furthermore, NO has been shown to provide benefits to percutaneous implants even in the presence of such physical factors.24
Conclusions
Nitric oxide was shown to clearly enhance the analytical performance of in vivo glucose biosensors with the associated benefits being dependent on the NO-release kinetics from the outer sensor membranes. Both rapid and extended NO-releasing sensors exhibited improved numerical accuracy versus controls. Rapid NO release from sensors resulted in positive differences in both clinical accuracy and glucose sensitivity, while sustained NO release from MPTMS-RSNO biosensors provided constant numerical accuracy over the entire 10 d implant period. The MPTMS-RSNO sensors were characterized by a quicker response to the IVGTT than both the MPTMS control and MAP3-based sensors, which we attribute to the sustained generation of NO. It is hypothesized that shorter lag times for the MPTMS-RSNO sensors are the result of improved glucose transport to the sensor from the tissue surrounding the implants. The predictable performance of MPTMS-RSNO glucose biosensors suggests that materials that are capable of releasing large NO payloads for even longer durations (i.e., several weeks) represent the ultimate NO-release strategy for long-term glucose sensing technologies (i.e., months), rather than the short-term (i.e., ∼10 d) period that was the focus of this study. However, the effects of NO on diabetic tissue may be dissimilar. Indeed, diabetic tissue is characterized by numerous deficiencies including altered wound repair,58,59 lessened inflammation and pro-inflammatory cytokine production at wound sites,59 disrupted blood flow,60 and susceptibility to infection.61 Work characterizing the response of diabetic tissue to implantation has been limited thus far. The disparities between diabetic and healthy tissue motivate the need for understanding the diabetic response to sensor implantation and warrant a careful investigation of the role of NO on the diabetic FBR. Planning of these studies in a diabetic swine model is ongoing.
Acknowledgments
The National Institutes of Health supported this research (R01 EB000708 and R43DK093119). R.J.S. gratefully acknowledges a John Motley Morehead Fellowship and a Francis Preston Venable Fellowship. We express our gratitude to Jaime Davis, Jessica Fuller, Dr. Alice Will, and the staff at Synchrony, LLC, for their expertise in the swine model and assistance in characterizing in vivo biosensor performance. The authors also thank Wallace Ambrose for assistance with the SEM and Collin McKinney for modifications to the wireless bipotentiostats. Additionally, we thank Justin Johnson and Dr. Yuan Lu for synthesizing the NO-releasing MAP3 nanoparticles used in this study.
Supporting Information Available
Real-time NO-release profiles of glucose sensor outer membranes, glucose sensor sensitivity as a function of storage time in PBS, nanoparticle characterization, representative EGA, sensor run-in times, experimental protocols, and representative SEMs of sensor surfaces postexplantation. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Mark Schoenfisch is a co-founder, a member of the board of directors, and maintains a financial interest in Novan Therapeutics, Inc. Novan is commercializing macromolecular nitric oxide storage and release vehicles for dermatological indications.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Koh A.; Nichols S. P.; Schoenfisch M. H. J. Diabetes Sci. Technol. 2011, 5, 1052–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson G. S.; Zhang Y.. In Vivo Glucose Sensing; John Wiley & Sons: Hoboken, 2009. [Google Scholar]
- Anderson J. M.; Rodriguez A.; Chang D. T. Sem. Immunol. 2008, 20, 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford R.; Kehoe J. J.; Barnes S. L.; Kornilayev B. A.; Alterman M. A.; Wilson G. S. Biomaterials 2006, 27, 2587–2598. [DOI] [PubMed] [Google Scholar]
- Vroman L.; Adams A. L.; Fischer G. C.; Munoz P. C. Blood 1980, 55, 156–159. [PubMed] [Google Scholar]
- Thomé-Duret V.; Gangnerau M. N.; Zhang Y.; Wilson G. S.; Reach G. Diabetes Metab. 1996, 22, 174–178. [PubMed] [Google Scholar]
- Wilson G. S.; Ammam M. FEBS J. 2007, 274, 5452–5461. [DOI] [PubMed] [Google Scholar]
- Nichols S. P.; Koh A.; Storm W. L.; Shin J. H.; Schoenfisch M. H. Chem. Rev. 2013, 113, 2528–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K.; Nomura H.; Mihara T.; Kurita K.; Iwasaki Y.; Nakabayashi N. J. Biomed. Mater. Res. 1998, 39, 323–330. [DOI] [PubMed] [Google Scholar]
- Ju Y. M.; Yu B. Z.; West L.; Moussy Y.; Moussy F. J. Biomed. Mater. Res. A 2010, 92, 650–658. [DOI] [PubMed] [Google Scholar]
- Massia S. P.; Stark J.; Letbetter D. S. Biomaterials 2000, 21, 2253–2261. [DOI] [PubMed] [Google Scholar]
- Sharkawy A. A.; Klitzman B.; Truskey G. A.; Reichert W. M. J. Biomed. Mater. Res. 1997, 37, 401–412. [DOI] [PubMed] [Google Scholar]
- Sharkawy A. A.; Klitzman B.; Truskey G. A.; Reichert W. M. J. Biomed. Mater. Res. 1998, 40, 586–597. [DOI] [PubMed] [Google Scholar]
- Sharkawy A. A.; Klitzman B.; Truskey G. A.; Reichert W. M. J. Biomed. Mater. Res. 1998, 40, 598–605. [DOI] [PubMed] [Google Scholar]
- Cao H.; McHugh K.; Chew S. Y.; Anderson J. M. J. Biomed. Mater. Res. 2010, 93, 1151–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton L. W.; Tegnell E.; Toporek S. S.; Reichert W. M. Biomaterials 2005, 26, 3285–3297. [DOI] [PubMed] [Google Scholar]
- Ward W. K.; Hansen J. C.; Massoud R. J.; Engle J. M.; Takeno M. M.; Hauch K. D. J. Biomed. Mater. Res. 2010, 94, 280–287. [DOI] [PubMed] [Google Scholar]
- Ziesche E.; Scheiermann P.; Bachmann M.; Sadik C. D.; Hofstetter C.; Zwissler B.; Pfeilschifter J.; Muhl H. Clin. Exp. Immunol. 2009, 157, 370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klueh U.; Dorsky D. I.; Kreutzer D. L. Biomaterials 2005, 26, 1155–1163. [DOI] [PubMed] [Google Scholar]
- Cooke J. P. Atheroscler. Suppl. 2003, 4, 53–60. [DOI] [PubMed] [Google Scholar]
- Schwentker A.; Vodovotz Y.; Weller R.; Billiar T. R. Nitric Oxide 2002, 7, 1–10. [DOI] [PubMed] [Google Scholar]
- Riccio D. A.; Schoenfisch M. H. Chem. Soc. Rev. 2012, 41, 3731–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetrick E. M.; Prichard H. L.; Klitzman B.; Schoenfisch M. H. Biomaterials 2007, 28, 4571–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols S. P.; Le N. N.; Klitzman B.; Schoenfisch M. H. Anal. Chem. 2011, 83, 1180–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols S. P.; Koh A.; Brown N. L.; Rose M. B.; Sun B.; Slomberg D. L.; Riccio D. A.; Klitzman B.; Schoenfisch M. H. Biomaterials 2012, 33, 6305–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford R.; Batchelor M. M.; Lee Y.; Gokulrangan G.; Meyerhoff M. E.; Wilson G. S. J. Biomed. Mater. Res. 2005, 75, 755–766. [DOI] [PubMed] [Google Scholar]
- Frost M. C.; Batchelor M. M.; Lee Y.; Zhang H.; Kang Y.; Oh B.; Wilson G. S.; Gifford R.; Rudich S. M.; Meyerhoff M. E. Microchem. J. 2003, 74, 277–288. [Google Scholar]
- Riccio D. A.; Nugent J.; Schoenfisch M. H. Chem. Mater. 2011, 23, 1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A.; Lu Y.; Schoenfisch M. H. Anal. Chem. 2013, 85, 10488–10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A.; Riccio D. A.; Sun B.; Carpenter A. W.; Nichols S. P.; Schoenfisch M. H. Biosens. Bioelectron. 2011, 28, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J. H.; Marxer S. M.; Schoenfisch M. H. Anal. Chem. 2004, 76, 4543–4549. [DOI] [PubMed] [Google Scholar]
- Bequette B. W. J. Diabetes Sci. Technol. 2010, 4, 404–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choleau C.; Klein J. C.; Reach G.; Aussedat B.; Demaria-Pesce V.; Wilson G. S.; Gifford R.; Ward W. K. Biosens. Bioelectron. 2002, 17, 641–646. [DOI] [PubMed] [Google Scholar]
- Choleau C.; Klein J. C.; Reach G.; Aussedat B.; Demaria-Pesce V.; Wilson G. S.; Gifford R.; Ward W. K. Biosens. Bioelectron. 2002, 17, 647–654. [DOI] [PubMed] [Google Scholar]
- Garg S. K.; Voelmle M.; Gottlieb P. A. Diabetes Res. Clin. Pract. 2010, 87, 348–353. [DOI] [PubMed] [Google Scholar]
- Kovatchev B. P.; Shields D. S.; Breton M. Diabetes Technol. Ther. 2009, 11, 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan D. B.; Mastrototaro J. J.; Weinzimer S. A.; Steil G. M. Biomed. Signal Process. 2013, 8, 81–89. [Google Scholar]
- Clarke W. L.; Kovatchev B. P. J. Diabetes Sci. Technol. 2007, 1, 669–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke W. L. Diabetes Technol. Ther. 2005, 7, 776–779. [DOI] [PubMed] [Google Scholar]
- Venton B. J.; Michael D. J.; Wightman R. M. J. Neurochem. 2003, 84, 373–381. [DOI] [PubMed] [Google Scholar]
- Jacovitti G.; Scarano G. IEEE Trans. Signal Process. 1993, 41, 525–533. [Google Scholar]
- McDonald J. H.Handbook of Biological Statistics, 2nd ed.; Sparky House Publishing: Baltimore, Maryland, U.S.A., 2009. [Google Scholar]
- Chen S.; Jones J. A.; Xu Y.; Low H. Y.; Anderson J. M.; Leong K. W. Biomaterials 2010, 31, 3479–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbe C.; Bartlett J.; Kong L. G.; Finnie K.; Lin H. Q.; Larkin M.; Calleja S.; Bush A.; Calleja G. Adv. Mater. 2004, 16, 1959–1966. [Google Scholar]
- Shin J. H.; Metzger S. K.; Schoenfisch M. H. J. Am. Chem. Soc. 2007, 129, 4612–4619. [DOI] [PubMed] [Google Scholar]
- Al-Sádoni H. H.; Ferro A. Curr. Med. Chem. 2004, 11, 2679–2690. [DOI] [PubMed] [Google Scholar]
- Vaughn M. W.; Kuo L.; Liao J. C. Am. J. Physiol. 1998, 274, 2163–2176. [DOI] [PubMed] [Google Scholar]
- Hetrick E. M.; Schoenfisch M. H. Biomaterials 2007, 28, 1948–1956. [DOI] [PubMed] [Google Scholar]
- Storm W. L.; Schoenfisch M. H. ACS Appl. Mater. Interfaces 2013, 5, 4904–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschwanez H. E.; Reichert W. M. Biomaterials 2007, 28, 3687–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M. O.; Rolin B. ILAR Journal 2004, 45, 303–313. [DOI] [PubMed] [Google Scholar]
- Swindle M. M.; Makin A.; Herron A. J.; Clubb F. J.; Frazier K. S. Vet. Pathol. 2012, 49, 344–356. [DOI] [PubMed] [Google Scholar]
- The Diabetes Control and Complications Trial Group N. Engl. J. Med. 1993, 329, 977–986. [DOI] [PubMed] [Google Scholar]
- Wilson G. S.; Gifford R. Biosens. Bioelectron. 2005, 20, 2388–2403. [DOI] [PubMed] [Google Scholar]
- Rebrin K.; Fischer U.; Hahn von Dorsche H.; von Woetke T.; Abel P.; Brunstein E. J. Biomed. Eng. 1992, 14, 33–40. [DOI] [PubMed] [Google Scholar]
- Koschwanez H. E.; Yap F. Y.; Klitzman B.; Reichert W. M. J. Biomed. Mater. Res. 2008, 87, 792–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry C. Anal. Chem. 1998, 70, 594A–598A. [PubMed] [Google Scholar]
- Schaper N. C.; Havekes B. Diabetalogia 2012, 55, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey T. J.; Sadaty A.; Jones W. G.; Barber A.; Smoller B.; Shires G. T. J. Surg. Res. 1991, 50, 308–313. [DOI] [PubMed] [Google Scholar]
- Vinik A. I.; Maser R. E.; Mitchell B. D.; Freeman R. Diabetes Care 2003, 26, 1553–1579. [DOI] [PubMed] [Google Scholar]
- Wheat L. J. Diabetes Care 1980, 3, 187–197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.