

Abstract
Mitochondrial ROS have emerged as an important mechanism of disease and redox signaling in the cardiovascular system. Under basal or pathological conditions, electron leakage for ROS production is primarily mediated by the electron transport chain and proton motive force consisting of a membrane potential (ΔΨ) and a proton gradient (ΔpH). Several factors controlling ROS production in mitochondria include FMN and the FMN-binding domain of complex I, ubisemiquinone and quinone-binding domain of complex I, FAD binding moiety and quinone-binding pocket (Qp) of complex II, and unstable semiquinone •Qo− mediated by the Q cycle of complex III. In mitochondrial complex I, specific cysteinyl redox domains modulate ROS production from the FMN moiety and iron sulfur clusters. In the cardiovascular system, mitochondrial ROS have been linked to mediating physiological effects of metabolic dilation and preconditioning-like mKATP channel activation. Furthermore, oxidative post-translational modification by glutathione in complex I and complex II has been shown to affect enzymatic catalysis, protein-protein interactions, and enzyme-mediated ROS production. Conditions associated with oxidative or nitrosative stress, such as myocardial ischemia and reperfusion, increase mitochondrial ROS production via oxidative injury of complexes I and II, and •O2−-induced hydroxyl radical production by aconitase. Further insight into cellular mechanisms by which specific redox post-translational modifications regulate ROS production in mitochondria will enrich our understanding of redox signal transduction and identify new therapeutic targets for cardiovascular diseases in which oxidative stress perturbs normal redox signaling.
Keywords: Mitochondria, Reactive Oxygen Species, Electron Transport Chain, Myocardial Ischemia and Reperfusion, Metabolic Dilation
1. How Mitochondria Generate ROS
1.1 Energy Transduction Mediated by Mitochondria
Mitochondria are the powerhouses of the living cell, producing most of the cell’s energy by oxidative phosphorylation. The process of energy transduction requires the coordinated action of four major respiratory enzyme complexes and ATP synthase (F0F1ATPase). High resolution structures are now available for the bacterial complex I, complete structures of mammalian complexes II−IV, and F1-ATPase (peripheral or headpiece domain of F0F1ATPase). In addition, mitochondria play a central role in the regulation of programmed cell death. Mitochondria trigger apoptosis by impairing electron transport and energy metabolism, by releasing cytochrome c and activating caspases that mediate apoptosis, and by altering cellular redox potential via reactive oxygen species (ROS) production(1, 2). The aforementioned mechanism can help to explain a variety of cardiovascular disease caused by mitochondrial dysfunction. In this review article, we focus on the mechanism of ROS production by the mitochondrial electron transport chain (ETC), the related physiological implications, and how these mechanisms control the disease process of myocardial infarction.
Mitochondrial energy transduction for ATP synthesis is driven by oxidation of NADH and FADH2, which is carried out by the ETC located within the inner membrane (Figure 1). The ETC mediates a stepwise electron flow from NADH or succinate to molecular oxygen through a series of electron carriers including complex I, complex II, ubiquinone, complex III, cytochrome c, and complex IV. Electron flow mediated by the ETC can drive proton translocation from the matrix side to the cytoplasmic side. ATP synthesis is then catalyzed by the F0F1ATPase driven by the flow of protons back across the membrane. This process is called oxidative phosphorylation.
Figure 1.
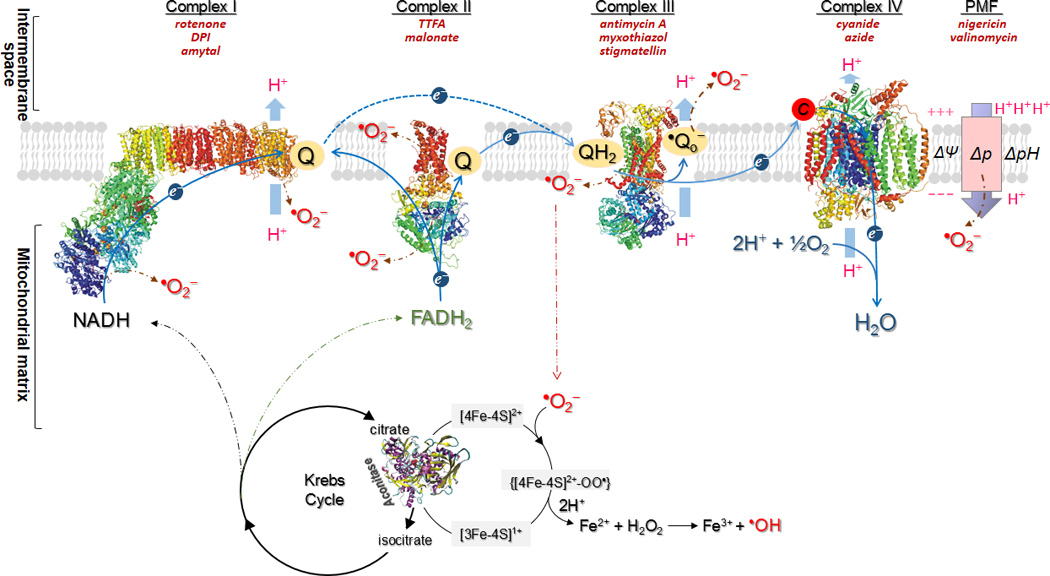
Schematic representation illustrating the mechanism of oxygen free radical(s) generation mediated by electron transport chain, the proton motive force (PMF, Δp), and the aconitase of Krebs cycle in mitochondria. Blue arrows show the path of electron transport from NADH or FADH2 to O2, or reverse electron flow from FADH2-linked succinate to complex I. Brown dashed arrows indicate the sites mediating •O2− generation in mitochondria. As electrons pass through the chain, protons are pumped from the mitochondrial matrix to the inter-membrane space, thereby establishing an electrochemical potential gradient or called proton motive force (Δp) across the inner membrane. The positive and negative charges on the membrane denote the membrane potential (ΔΨ). A proton gradient is denoted by ΔpH for the difference of pH across the membrane. Δp can contribute to •O2− generation in the respiratory conditions of state 2 and state 4. Black circles show aconitase of the Krebs Cycle that generates NADH and FADH2 as the substrates of the ETC that are the source of hydroxyl radical production induced by •O2−. The common inhibitors used for studying the ETC components, ΔpH, and ΔΨ are indicated in brick red italics.
1.2. Oxidative Phosphorylation is an Endogenous Source of ROS
Chance and Williams have proposed a convention following the typical order of addition of agents during an experiment using a Clark oxygen electrode, which allows definition of so called respiratory “states” and determination of the respiratory control index (RCI) (3, 4). In this experiment, mitochondria were added to an oxygen electrode chamber, followed by a mixture of glutamate and malate (NADH-linked, explained in Online Figure 1) or succinate (FADH2-linked) as substrate. Respiration is slow (state 2 respiration) due to a low amount of ADP and that the proton circuit is not completed by H+ re-entry through F0F1ATPase. A limited amount of ADP is added, allowing F0F1ATPase to synthesize ATP coupled to proton re-entry across the membrane, which is defined as “state 3 respiration”. Oxygen uptake is accelerated during state 3 respiration, and the total oxygen uptake is effectively used for ATP synthesis. When ADP is exhausted, respiration slows and finally anoxia is attained, which is state 4 respiration. State 4 respiration is not ADP-dependent, and technically can be attained by the F0F1ATPase inhibitor, oligomycin.
In myocytes, mitochondria comprise 30–40% of the volume, and generate about 90% of the ATP(5). Mitochondria are also the major source of ROS in the cardiovascular system(5). Under most physiological conditions, electron transport to O2 is tightly coupled to oxidative phosphorylation for ATP synthesis. However, oxidative phosphorylation is the major endogenous source of ROS such as •O2− (superoxide anion radical), H2O2 (hydrogen peroxide), and •OH (hydroxyl radical), which are toxic by-products of respiration(1, 2). The •O2− generation by mitochondria is primarily generated by electron leakage from the ETC (Figure 1). Under the physiological conditions of state 4 respiration, the oxygen tension in mitochondria is low; O2 consumption by the ETC does not meet the needs of oxidative phosphorylation. A decrease in the rate of mitochondrial phosphorylation increases the electron leakage from the ETC and subsequent production of •O2−. •O2− is converted to H2O2 by mitochondrial manganese superoxide dismutase (Mn SOD or SOD2 in Eq. 1), and H2O2 is further converted to H2O by glutathione peroxidase (GPx) in the presence of glutathione (GSH in Eq. 2). It was estimated that mitochondria produce up to 0.5 nmol of H2O2/min/mg protein, accounting for 2% of the oxygen uptake, under the conditions of state 4 respiration(6).
The metabolic H2O2of mitochondria under state 3 and 4 respiration may modestly induce mitochondrial oxidative stress, or diffuse to the cytosol, acting as a signaling molecule to trigger important physiological responses, such as endothelium-derived hyperpolarizing factor (EDHF)that mediates shear or acetylcholine-induced smooth muscle relaxation in small arteries(7, 8). Therefore, the H2O2 generated at low to moderate concentrations under normal physiological conditions has been recognized as a non-toxic by-product of cellular metabolism that can mediate physiological signaling (7).
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
| (Eq. 4) |
The mitochondrial ETC proteins are rich in metal cofactors such as hemes (complexes II, III, and IV) and iron sulfur clusters (complexes I, II, and III). Oxidative stress in mitochondria can be greatly enhanced in the presence of reduced transition metals because H2O2 can be converted to the highly reactive hydroxyl radical (HO•) via the Fe2+-dependent Fenton reaction (Eq. 3) orvia the Fe3+−catalyzed Haber-Weiss mechanism (Eq. 4). Under pathophysiological conditions of cardiovascular disease, decreasing or impairing the function of the ETC and oxidative phosphorylation increases mitochondrial ROS production. Acute ROS exposure can inactivate the iron-sulfur (Fe-S)centers of complexes I, II, and III, and Krebs cycle aconitase, resulting in shutdown of mitochondrial energy production, and chronic ROS exposure can result in oxidative damage to mitochondrial and cellular proteins, lipids, and nucleic acids. Therefore, oxidative stress induced by ROS overproduction in mitochondria is closely associated with disease pathogenesis.
1.3. Membrane Potential (ΔΨ) and Proton Gradient (ΔpH) as the Sources of ROS
Proton motive force (Δp or electrochemical gradient) across inner mitochondrial membrane is generated from proton pumping, and represents the potential energy driving proton re-entry to the matrix for ATP synthesis. The Δp consists of a membrane potential (ΔΨ) and a proton gradient (ΔpH), which partially contribute to the source of •O2−mediated by mitochondria (Figure 1). In vitro studies have firmly established the effect of membrane potential on the •O2− production by complex III (9–11).
The production of •O2−mediated by isolated mitochondria under the conditions of state 2 respiration can be induced by glutamate plus malate, and measured by EPR spin-trapping with 5’, 5’-dimethylpyrroline N-oxide (DMPO). Addition of ADP dissipates the proton gradient, initiates the conditions of state 3 respiration, and diminishes mitochondria-mediated •O2− generation, indicating that coupling of enhanced O2 consumption with oxidative phosphorylation decreased electron leakage, and a decrease of ΔpH is correlated with decreasing •O2− generation by mitochondria. Further addition of oligomycin A inhibits the F0F1ATPase activity, and initiates the conditions of state 4 respiration, which gradually restores ΔpH and ΔΨ. Due to restored Δp, the •O2−generation induced by glutamate plus malate at state 4 respiration is enhanced to the level of state 2 respiration. Addition of 4-(trifluoromethoxy) phenylhydrazone (FCCP, a proton ionophore) to mitochondria uncouples oxidative phosphorylation, which dissipates the ΔΨ and ΔpH. EPR analysis indicates that proton ionophore reduces the mitochondria-mediated •O2− production to the level of state 3 respiration. Analysis of H2O2 production by isolated mitochondria indicates the rate of •O2− generation by the ETC is decreased by un couplers or inhibitors of Δp (12, 13). Therefore, increasing ΔΨ and ΔpH-supported proton back pressure induce electron leakage from the normal pathway for •O2− production in mitochondria.
2. Factors Involved in the •O2− Generation Mediated by the Electron Transport Chain
Under normal physiological conditions, adecrease in the rate of oxidative phosphorylation can increase oxygen free radical production, in the form of •O2−, from the early stages of the ETC. Two segments of the ETC have been widely hypothesized to be responsible for •O2− generation. One, on the NADH dehydrogenase (or flavin subcomplex) of complex I, operates via electron leakage from the reduced flavin mononucleotide(14, 15). The other, on complex III, mediates •O2− production through a Q-cycle mechanism, in which electron leakage results from auto-oxidation of ubisemiquinone(16)and reduced cytochrome b (low potential b566 or bL) (11). Furthermore, there is an increasing body of evidence that links •O2−overproduction with complex II (17, 18)or a defect in complex II(19, 20). Under ischemic conditions, phosphorylation of complex IV may also increase electron leakage and •O2− production(21). Therefore, mediation of •O2− production by complex II or complex IV is relevant in disease conditions.
2.1. Mediation of •O2−Generation by Complex I
Mitochondrial complex I [EC 1.6.5.3. NADH: ubiquinone reductase (NQR)] is the first energy-conserving segment of the ETC. Purified bovine heart complex I contains 45 different polypeptides with a total molecular mass approaching 980 kDa(22). Complex I can be resolved into three subcomplexesusing achaotropic agent: a flavoprotein fraction (Fp), an iron sulfur protein fraction (Ip), and a hydrophobic fraction (Hp). The Fp contains the catalytic activity of NADH dehydrogenase (NDH) or NADH ferricyanide reductase (NFR). The redox centers, apart from the FMN, are iron-sulfur centers, which cannot be studied by optical spectroscopy, but instead require the more labor intensive technique of low temperature EPR(23). Complex I catalyzes the transfer of two electrons from NADH to ubiquinone in a reaction that is coupled with the translocation of four protons across the membrane, and it is inhibited by rotenone and piericidine A (Table I). The redox centers of mammalian complex I that are involved in mediation of two-electron transfer include a non-covalent FMN, 8 iron-sulfur clusters, and ubiquinone. Current evidence suggests that the proton translocation stoichiometry is 4H+/2e−. In addition to electron-transfer and energy transduction, the catalysis of complex I provides the major source of ROS generation in mitochondria with detectable •O2− formed under the conditions of enzyme turnover(24–26). Studies using isolated mitochondria indicate •O2− and H2O2 production by complex I are mainly controlled by NADH/NAD+ redox coupling and succinate-induced reverse electron transfer(13, 27). Two segments of complex I are widely recognized to be responsible for enzyme-mediated •O2− generation. One is involved in the cofactor of FMN and FMN-binding moiety at the 51 kDa polypeptide (FMN-binding subunit) (28–31) and the other is located on the ubiquinone-binding site that mediates ubiquinone reduction.
Table I.
Summary of the Inhibitors Used to Study the Components of Mitochondrial Electron Transport Chain (ETC) and Proton Motive Force (PMF) In Vitro and In Vivo
| ETC/PMF Components | Inhibitors | Experimentation | Mechanism of Inhibition |
|---|---|---|---|
| Complex I | DPI | In vitro | Inhibiting electron leakage at FMN- binding subunit. |
| Complex I | rotenone, piericidine A | In vitro and in vivo | Blocking the oxidation of Fe-S clusters of complex I. |
| Complex I | amytal | In vitro and in vivo | Inhibiting Fp activity, and subsequently block oxidation of Fe- S clusters. |
| Complex II | TTFA | In vitro | Blocking ubiquinone reduction by occupying ubiquinone-binding site. |
| Complex II | malonate oxaloacetate | In vitro | Competitive inhibitor for succinate- binding and blocking FADH2-linked ROS production. |
| Complex II | diazoxide | In vitro and in vivo | Inhibiting complex II and weaken SCR supercomplex assembly, increasing ROS mediated by complex III. |
| Complex III | antimycin A | In vitro | Blocking the electron transfer between bH to semiquinone bound at the Qi site, increasing ROS production. |
| Complex III | myxothiazol | In vitro | Blocking the electron transfer between ubiquinol and Reiske iron- sulfur cluster, inhibiting •Qo− and ROS production by complex III. |
| Complex III | stigmatellin | In vitro | Inhibiting the electron transfer to cytochrome c1, inhibiting •Qo− and ROS production by complex III. |
| Complex IV | cyanide, azide, CO | In vitro | Inhibiting the electron transfer to O2 by binding tightly with the iron coordinated in cytochrome a3. |
| F0F1ATPase | oligomycin A | In vitro | Blocking the flow of protons through the channel by binding to the F0portion of F0F1ATPase. |
| ΔpH | nigericin | In vitro and in vivo | H+/K+ antiporter, collapsing proton gradient and increasing ΔΨ. |
| ΔΨ | valinomycin | In vitro and in vivo | K+ mobile carrier ionophore, collapsing mitochondrial ΔΨ. |
| Δp | DNP, FCCP | In vitro and in vivo | Protonophore, mediating the net electrical uniport of protons and uncoupling the process of Δp. |
2.1.a. ROS Generation by the FMN moiety of Complex I
The mechanism of •O2− generation is likely derived from one-electron transfer of reduced FMN (FMN H2) to molecular oxygen(30). Based on investigations using models of intact complex I and its Fp subcomplex (NDH) along with EPR spin-trapping with 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide (DEPMPO), enzyme-mediated •O2− generation by intact complex I or NDH isolated from bovine heart can be inhibited by the general flavoprotein inhibitor, diphenyleneiodinium chloride (DPI). Addition of free FMN can increase enzyme-mediated •O2− generation by intact complex I or NDH(28). However, FMN addition could not reverse the inhibition by either DPI treatment or heat denaturation of either intact complex I or NDH. These results support the involvement of FMN and the FMN-binding protein moiety in the mediation of •O2− generation by the Fp of complex I. Further evidence has been provided by redox titration of mitochondrial •O2− generation using submitochondrial particles(SMP) (29). The redox potential of •O2− production at the site of complex I was determined to be ~ −295 mV. The midpoint potential of the •O2− producing site at complex I resembles the value of free FMN (~ −310 mV for the redox couple of FMN/FMN H•−), also supporting the FMN moiety as a potential site of •O2− generation.
2.1.b. ROS Generation by the Ubiquinone-binding Domain of Complex I
The second site associated with complex I-mediated •O2− generation has been proposed to be the site of ubiquinone reduction due to the formation of an unstable ubisemiquinone radical (SQ) that is the source of •O2−(13, 27, 32, 33). The ubisemiquinone radical is formed via incomplete reduction of ubiquinone under the conditions of enzyme turnover. Evidence of SQ involvement has been investigated using EPR detection of enzymatic systems in the presence or absence of spin trap(25). SQ can be categorized as stable ubisemiquinone radical that is EPR detectable, and unstable ubisemiquinone radical that is not detected by EPR due to a short half-life at room temperature. Unstable ubisemiquinone radical is thus a source of •O2− under physiological conditions. Studies using isolated complex I and EPR spin-trapping indicated that enzyme-mediated •O2− generation driven by NADH was enhanced 2-fold in the presence of ubiquinone-1 (Q1) compared with that in the absence of Q1. More than 80% of the enhanced •O2− generation can be inhibited by rotenone, thus supporting a •O2− generation mechanism involving the reduction of ubiquinone to an unstable semiquinone radical(25). The formation of an unstable semiquinone radical under enzyme turnover conditions is likely mediated through the ubiquinone-binding site of the Hp subcomplex in complex I. Ohnishi et al. have reported the presence of two distinct semiquinone species with different spin relaxation times in complex I in situ based on a study using the SMP from rat heart(34) and proteoliposomes of isolated complex I(35). The unstable semiquinone as a source of •O2− is likely to be SQNf(semiquinone with fast relaxing time), which is highly sensitive to rotenone or piericidin A.
Studies from Brand et al. using isolated mitochondria suggest •O2− production from the ubiquinone moiety of complex I is large under conditions of reverse electron transport mediated by respiring to succinate(13),or the conditions of forward electron transport in the presence of ATP and piericidin A (or rotenone) (27). The •O2− generation by complex I during reverse/forward electron transport can be inhibited by uncoupler and nigericin, suggesting •O2− production by complex I is more dependent on the ΔpH than ΔΨ(13, 27). Note that nigericin is an ionophore functioning as a specific carrier of protons and catalyzing a K+/H+ exchange that abolishes ΔpH, thus allowing ΔΨ to increase to the full magnitude of the proton motive force. Whereas, an uncoupler (e.g. FCCP) functions as a carrier of protons and charge that collapses both ΔpH and ΔΨ.
2.1.c. The Role of Iron Sulfur Clusters in ROS Generation by Complex I
Mammalian complex I hosts as many as eight iron-sulfur clusters, which are essential redox centers controlling electron transfer during enzyme turnover. Iron-sulfur (Fe-S) clusters identified with the use of EPR include the binuclear clusters N1a and N1b and the tetranuclear clusters N2, N3, N4, and N5. Two more tetranuclear clusters, N6a and N6b were identified in the subunit TYKY. Based on the x-ray structure of complex I from Thermus Thermophilus, organization of iron sulfur clusters in mammalian complex I has been inferred. The main route for electron transfer within the enzyme is likely to be NADH→FMN→N3→N1b→N4→N5→N6a→N6b→N2→ubiquinone(36). The iron sulfur clusters in complex I should play a secondary role in the generation of •O2−by complex I. Studies using intact mitochondria from rat brain indicates rotenone-insensitive •O2− generation by complex I is far less sensitive to p-chloromercuriobenzoate(an iron sulfur cluster blocker) treatment than DPI treatment(29). However, several research groups have proposed that in intact mitochondria and SMP, most of the •O2− may be generated around the N2/ubiquinone site, suggesting that electron leakage at the N2 center is coupled to incomplete reduction of ubiquinone(32).
The 75 kDa subunit hosts three iron sulfur clusters (N1b, N4, N5) of mammalian complex I. Residues 100–120 of the 75 k Daprotein precursor,100WNILTNSEKTKKAREGVMEFL120(peptide of p75), exhibit a β-sheet-α-turn-helix conformation (blue region in the homolog model of 75 kDa subunit in the Figure 2B). The peptide of p75 was designed as a B cell epitope to make an antibody that has been used to probe structure and function of complex I(25, 37). The polyclonal antibody generated against p75 is named Ab75. Binding of Ab75 antibody to mammalian complex I resulted in the inhibition of •O2−generation by 35% as measured by EPR spin-trapping(25). Therefore, (i)binding of Ab75 is likely to stabilize the conformation of the protein matrix surrounding the iron-sulfur clusters, thus enhancing electron transfer efficiency; and (ii)Ab75 binding may protect the protein matrix from molecular oxygen’s accessing the iron-sulfur clusters, including N1b, N4, and N5 in bovine complex I (Figure 2B), minimizing the electron leakage from iron-sulfur clusters.
Figure 2.
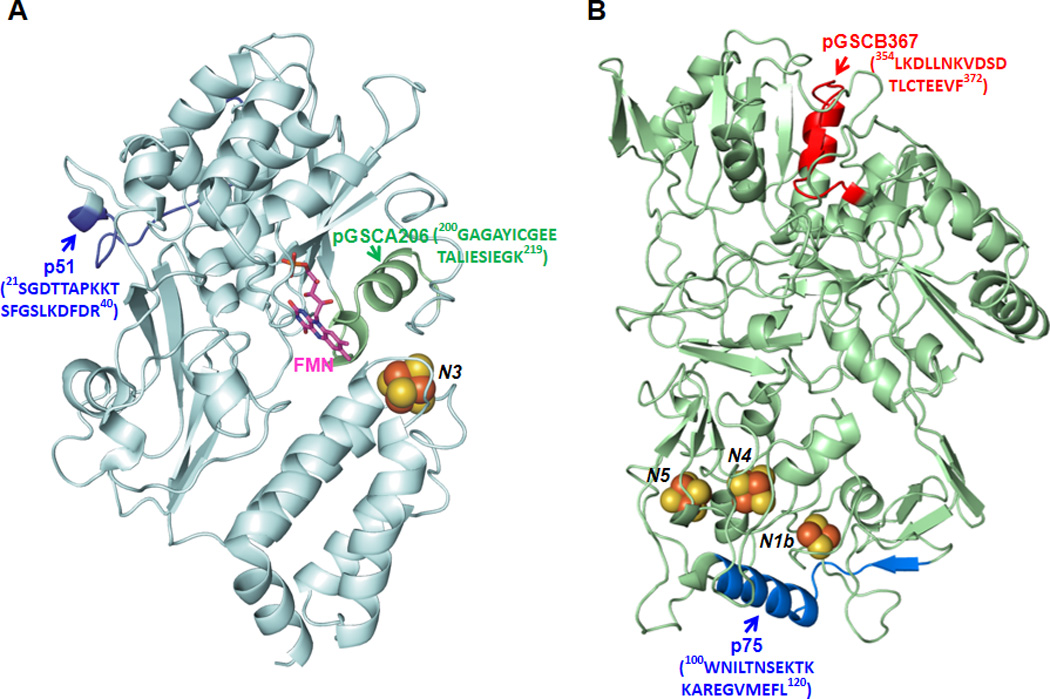
A, Homology model of the 51 kDa subunit of bovine heart complex I using the crystal structure of respiratory complex I from T. thermophilius with a protein data bank accession code, 3I9V (pdb 3I9V), as a template (36). Arrows show the domains of peptide pGSCA206 and peptide p51, denoted by green and blue ribbons. B, Model of the 75 kDa subunit of bovine heart complex I using 3I9V as a template. Arrows show the domains of peptide pGSCB367 and peptide p75, denoted by red and blue ribbons.
2.1.d. The role of cysteinyl redox domains
The mitochondrial redox pool contains a high physiological concentration of GSH. Overproduction of •O2− and •O2−-derived oxidants increases the ratio of GSSG to GSH. Moreover, the proteins of the mitochondrial ETC are rich in protein thiols. Mammalian complex I is the major component of the ETC to host the reactive/regulatory thiols that are thought to function in antioxidant defense and redox signaling. Physiologically, the complex I-derived regulatory thiols have been implicated in the regulation of respiration, nitric oxide utilization, and redox status of mitochondria. It is documented that the 51 kDa (FMN-binding protein) and 75 kDa (iron-sulfur protein) subunits of the complex I are two major polypeptides hosting redoxthiols(24–26, 28, 38–40).
The C206 (Cys206) moiety of the 51 kDa subunit plays a unique role as a redoxthiol in oxidative damage to complex I. The C206 of the 51 kDa subunit is also involved in site-specific S-glutathionylation (via binding of GSH) (24, 26). The peptide identified to form protein thiyl radical and GS-binding is 200GAGAYIC206GEETALIESIEGK219, which is highly conserved in the bacterial, fungal, and mammalian enzymes (28). An X-ray crystal structure of the hydrophilic domain of complex I from T. Thermophilus reveals that this conserved cysteine (Cys182 in T. Thermophilus) is only 6 Å from the FMN, which is consistent with the role of C206 as a redox-sensitive thiol and FMN’s serving as a source of •O2−(36).
The polypeptide of 75 kDa is the other subunit of complex I to be involved in redox modification via S-glutathionylation or protein thiyl radical formation(24, 26, 40). Based on the LC/MS/MS analysis, S-glutathionylation of C367 can be induced by oxidized GSSG through protein thiol disulfide exchange. C554 and C727 were S-glutathionylated when complex I of ratheart mitochondria was oxidatively stressed by diamide(40). The GS-binding peptides identified include 361VDSDTLC367TEEVFPTAGAGTDLR382, 544MLFLLGADGGC554ITR557, and713AVTEGAHAVEEPSIC727.
Based on EPR spin-trapping studies, GSSG-induced S-glutathionylation of complex I at the 51 kDa and the 75 kDa subunits affects the •O2− generation activity of complex I by marginally decreasing electron leakage and increasing electron transfer efficiency(26). High dosage of GSSG or diamide-induced S-glutathionylation tends to decrease the catalytic function of complex I and increase enzyme-mediated •O2− generation(38, 40). Binding of antibodies against the peptide of 200GAGAYIC206GEETALIESIEGK219(pGSCA206 in Figure 2A) decreases complex I-mediated •O2− generation by 37%(25). Since FMN serves as a source of •O2−, binding of antibodies may prevent molecular oxygen from accessing FMN resulting in a subsequent decrease in •O2− production. In addition, binding of the antibodies against the peptide of 354LKDLLNKVDSDTLC367TEEVF372(pGSCB367 in Figure 2B) inhibits •O2− production by complex I by 57%. The distance of pGSCB367 to iron sulfur clusters is relatively long, thus binding of antibodies to the 75 kDa subunit of complex I likely triggers long range conformational changes in the 75 kDa polypeptide to reduce O2 interactions. Binding of either antibody does not affect the electron transfer activity of complex I. Therefore, the redox domains involved in the S-glutathionylation are responsible for regulating electron leakage for •O2− production by complex I.
2.2 Mediation of •O2− Generation by Complex II
Mitochondrial Complex II (EC 1.3.5.1. succinate:ubiquinone reductase or SQR) is a key membrane complex in the Krebs cycle that catalyzes the oxidation of succinate to fumarate in the mitochondrial matrix. Succinate oxidation is coupled to reduction of ubiquinone at the mitochondrial inner membrane as one part of the ETC. Complex II mediates electron transfer from succinate to ubiquinone through the prosthetic groups of flavin adenine nucleotide (FAD); [2Fe-2S] (S1), [4Fe-4S] (S2), and [3Fe-4S] (S3); and heme b. The enzyme is composed of two parts: a soluble succinate dehydrogenase (SDH) and a membrane-anchoring protein fraction. SDH contains two subunits, a 70 kDa protein with a covalently bound FAD, and a 30 kDa iron-sulfur protein hosting S1, S2, and S3 iron-sulfur clusters. The membrane anchoring protein fraction contains two hydrophobic polypeptides (14 kDa and 9 kDa) with heme b binding.
The catalysis of complex II is believed to contribute to ROS generation in mitochondria. Two regions of the enzyme complex are responsible for generating the •O2−. One is located on the FAD cofactor, while the other is located on the ubiquinone-binding site which acts in the mediation of ubiquinone reduction(17, 41).
2.2.a. ROS Generation by the FAD Moiety of Mammalian Complex II
The generation of •O2− by the FAD moiety of complex II may arise from FADH2autooxidation or FADH•−semiquinone autooxidation. Evidence has been provided from the mammalian enzyme via use of the inhibitor, 2-thenoyltrifluoroacetone (TTFA) (42, 43). TTFA is a classical inhibitor for the ubiquinone reduction of complex II by occupying its ubiquinone binding sites(20). Therefore, binding of TTFA to the enzyme induces electron accumulation at the early stage of complex II. Based on EPR spin-trapping studies using isolated complex II and a super complex (SCR) hosting complex II and complex III, the inhibitory effect of TTFA on the •O2− generation by complex II or SCR indicates that FADH2autooxidation mediated by FAD binding moiety partially contributes to the •O2− production(43). The production of •O2− by complex II or SCR is minimized under the conditions of enzyme turnover(42, 43).
2.2.b. ROS Generation by the Ubiquinone-binding Site of Complex II
Mammalian complex II contains at least two ubiquinone-binding sites (namely, Qp and Qd) (20). Qp is near the matrix side and Qd is near the inter-membrane space site(20). Because the ubiquinone-binding site in complex II is close to the TTFA-binding site, EPR signal derived from ubisemiquinone of complex II is sensitive to TTFA(44). The location of the Qp site and its quinone-binding pocket has been revealed by the X-ray structure of porcine heart complex II (pdb 1ZOY). The amino acid residues involved in the quinone-binding pocket at the Qp site form the quinone-binding environment (Figure 3) (20, 41). Under physiological conditions, ubiquinone reduction mediated by complex II is well controlled via the quinone-binding environment. Alteration of the quinone-binding environment results in incomplete reduction of ubiquinone, leading to increased ROS production.
Figure 3.
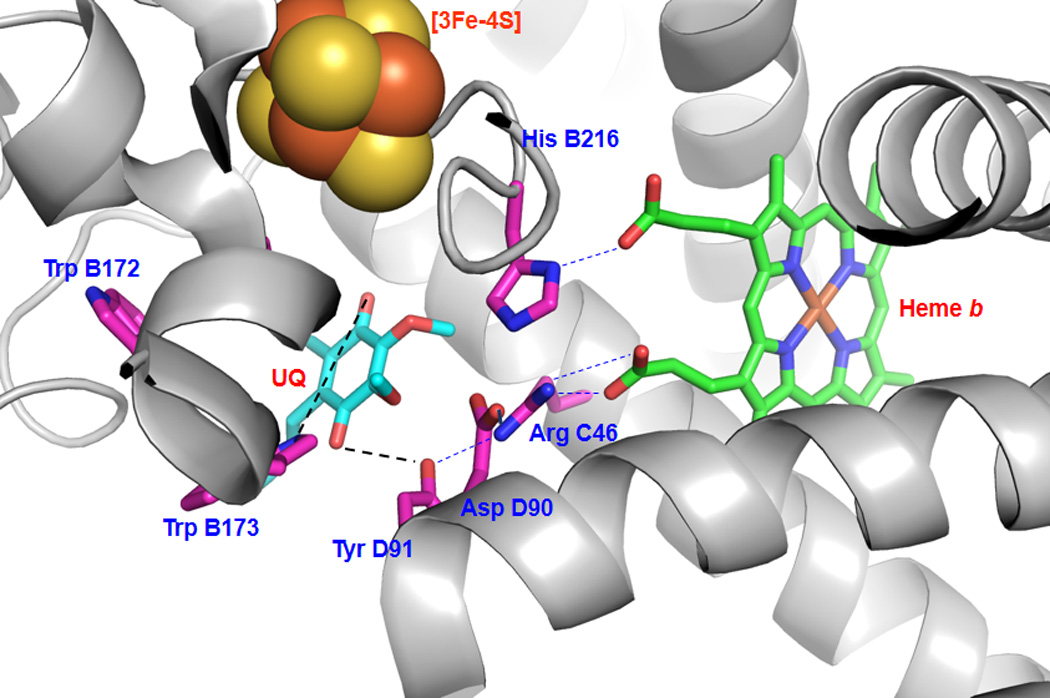
Hydrophobic residues and polar interactions (dashed lines) in the ubiquinone-binding site (Qp site) of mammalian complex II. The quinone-binding pocket is revealed by the X-ray structure (pdb 1ZOY), and involves the residues Trp B172, Trp B173, His B216, Arg C46, Asp D90, Tyr D91. The residues of the ubiquinone-binding site are determined by X-ray structure to be Trp B173 and Tyr D91. UQ denotes ubiquinone.
2.3. Mediation of •O2− Generation by Complex III
2.3.a. The Q-cycle Mediated by Complex III
Mitochondrial complex III catalyzes the electron transfer from ubiquinol (QH2) to ferricytochrome c, which is coupled to proton translocation for ATP synthesis. The redox centers of complex III consist of QH2, hemes bL (low potential b or b566), bH (high potential b or b562), and c1, and the Rieske iron-sulfur cluster (RISP). The electron transfer from QH2 to cytochrome c follows the Q-cycle mechanism.
In the Q-cycle mechanism, there are two semiquinones formed in different parts of the cycle (Figure 4A). An unstable semiquinone (•Qo−) is formed near the cytoplasmic site (outer site or positive site). The other semiquinone (•Qi−), formed near the matrix side (inner site or negative site), is a stable semiquinone and EPR detectable. During the enzyme turnover of complex III, one electron from QH2 is sequentially transferred to RISP, then cytochrome c1, and then ferricytochrome c. QH2 contains two electrons, but cytochrome c only accepts one electron. This leaves an unstable semiquinone(•Qo−)formed at the cytoplasmic site. One electron from the unstable semiquinone is transferred to the low potential heme b (bL), and then transferred to the high potential heme b (bH). One electron from bH is then transferred to ubiquinone (Q) to form a stable semiquinone at the matrix side. The stable semiquinone can accept one electron from a second single turnover to complete the cycle, forming QH2.
Figure 4.
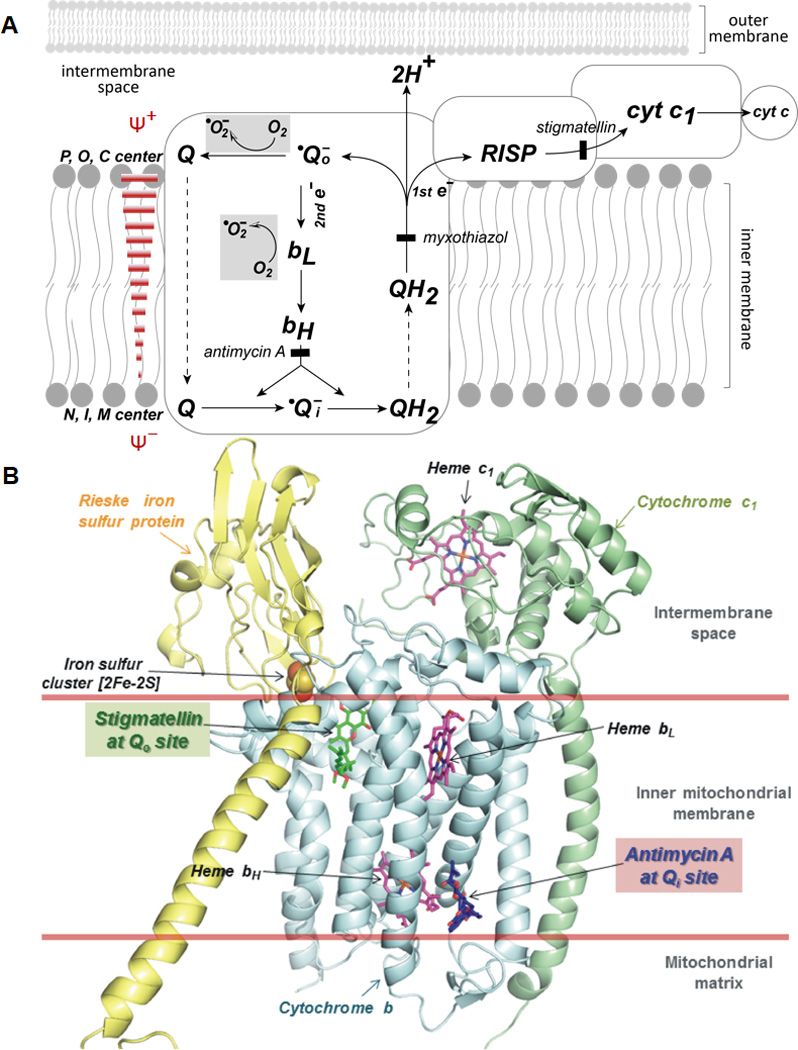
(A) Superoxide generation mediated by the Q-cycle mechanism in complex III. The scheme is adapted from Ref. (43) with modification. Gray areas symbolize the reactions involved in •O2− production. P, O, and C represent positive, outside, and cytoplasmic side respectively. N, I, and M stands for negative, inside, and matrix side, respectively. Solid bars in brick red show the opposition of electron transfer from the bL to bH by the membrane potential (ΔΨ) and inhibition of bH reoxidation by antimycin A. (B) X-ray structure (pdb 1PPJ) of complex III shows Qo site (occupied by Qo site inhibitor, stigmatellin in green color) is located immediately next to the inter-membrane space. Structure also shows the Qi site (occupied by Qi site inhibitor, antimycin A in blue color) is located next to the matrix site. The subunits of cytochrome b, cytochrome c1, and Rieske iron sulfur protein (RISP) are denoted by cyan, pale green, and yellow ribbons, respectively.
2.3.b. •O2−Generation by the Q-cycle
The Q-cycle mechanism provides an important theoretical basis to explain why mammalian complex III is an endogenous source of ROS in mitochondria(9, 43, 45, 46). The •Qo− formed near the cytoplasmic space is a source of •O2− due to its poor stability. Antimycin A, myxothiazol, and stigmatellin are inhibitors of complex III (Figure 4A). The mechanism of inhibition by these three inhibitors strongly supports the Qo site as the source of •O2− generation. Antimycin Ainhibits electron transfer from bL to bH, preventing the formation of the relatively stable •Qi−, increasing •Qo−formation and subsequent •O2− production. Myxothiazolinhibits electron transfer to the RISP and blocks the formation of •Qo−; while stigmatellin inhibits electron transfer to the cytochrome c1, raising the midpoint potential of the Rieske iron sulfur cluster from +290 mV to +540 mV, thus deceasing •Qo−formation and •O2− production.
2.3.c. The Role of Cytochrome bL and Membrane Potential (ΔΨ) in •O2− Generation by Complex III
As indicated in Figure 4A of the Q-cycle mechanism, low potential heme b (bL) accepts one electron from the unstable semiquinone. The mid-point potential of bL is −50 mV which tends to donate an electron. In vitro evidence has supported the notion of ferrocytochrome bL to be a source of •O2−(43, 47). Experimental evidence has shown the involvement of •O2− generation during intra-molecular electron transfer from ferrocytochrome b to ferricytochrome c1 (43).
In vitro studies using the vesicles of phospholipid (PL)-reconstituted complex III indicated the rate of •O2− generation can be controlled by modulation of the ΔΨ(10). Experimental evidence has revealed that the rate of •O2− generation by PL-reconstituted complex III is enhanced exponentially by increased ΔΨ and nigericin treatment(10), suggesting •O2− generation by complex III is more dependent on ΔΨ than ΔpH. Opposing membrane potential (ΔΨ) inhibits electron transfer from heme bL to heme bH, thereby promoting •Qo− formation and stimulating •O2− generation from reduced bL or •Qo− (Figure 4A).
2.3.d. Bi-directionality of Superoxide Release as Mediated by Complex III
Since the anionic form of •O2− is too strongly negatively charged to readily pass the inner membrane, •O2− production mediated by complex III can exhibit a distinct membrane sidedness or “topology.” The Qo site, one of the sources of •O2−, is located near the inter-membrane space (Figure 4B). The close proximity of the Qo site to the inter-membrane space can result in invariable release of a fraction of complex III-derived •O2− to the cytoplasmic side of the inner membrane(48, 49). Experimental evidence has supported that ~50% of total electron leakage in mitochondria lacking CuZn-SOD accounts for extra-mitochondrial •O2− release(50). The remaining ~50% of electron leakage is due to •O2− released to the matrix. Hydrogen peroxide derived from dismutation of •O2− in the mitochondrial matrix can induce aconitase inhibition. The inhibition of aconitase by H2O2 in the matrix can be relieved by the Qo site inhibitor stigmatellin. These results indicate that •O2− is released to both sides of the inner mitochondrial membrane by the semiquinone at the Qo site(49, 50).
2.3.e. Protein-Protein Interaction with Complex I Minimizes the •O2− Generation by Complex III
NADH cytochrome c reductase(NCR) is the supercomplex hosting complex I and complex III, mediating electron transfer from NADH to ferricytochrome c.Direct evidence from EPR measurements shows that NCR mediated •O2− generation is controlled by the enzyme turnover of complex I, whereas •O2−generation mediated by the enzyme turnover of complex III is minimized in the intact supercomplex of NCR; indicating the protein-protein interaction with complex I modulates •O2− production by complex III. Impairment of protein-protein interaction via oxidative injury of complex I may amplify the complex III-mediated •O2− generation(51).
2.4. Hyperphosphorylation of Complex IV Increases Enzyme-mediated ROS Generation
Mitochondrial complex IV is cytochrome c oxidase that catalyzes the sequential transfer of four electrons from the ferrocytochrome c to O2, forming 2H2O. The reduction of O2 to H2O provides the driving force for proton translocation and ATP synthesis. The catalytic function of complex IV is mainly controlled by subunit I and subunit II with both subunits encoded by mitochondrial DNA(52, 53). The redox centers of complex IV include 2 CuA (in a cluster with sulfur atoms in subunit II), 2 a-type hemes (heme a and heme a3 are located in subunit I), and CuB (in subunit I). Subunit II is the location of two CuAsites that undergo a one-electron oxidation-reduction reaction. The binuclear CuAcenter receives electrons, one at a time, from ferrocytochrome c.
One axial coordination position to the iron of heme a3 is not occupied by an amino acid ligand. This is the position where oxygen binds before its reduction to water. Heme a3 is also the site of binding of several inhibitors, including cyanide, azide, NO, and CO. CuB, the third copper, is immediately adjacent to heme a3; it has three histidine ligands, suggesting a fourth coordination position may be occupied by a reaction product during a specific stage of the oxygen reduction reaction. One of three ligands, histidine-240 (H240 in bovine heart complex IV), is cross-linked to a nearby specific tyrosine residue (Y244 in bovine heart complex IV) through a covalent bond. This specific tyrosine is proved to participate in the formation of a protein-tyrosyl radical intermediate during enzyme turnover or under oxidant stress induced by H2O2(54–56). CuBand nearby heme a3 form the binuclear center which tightly controls oxygen binding, the formation of intermediates (oxy, compound P, and compound F), and reduction of oxygen to H2O. Electron leakage for •O2− generation is not likely occurring during enzyme turnover of complex IV or under normal physiological conditions. However, •O2− production can be mediated by hyperphosphorylated complex IV under ischemic conditions(21).
2.5. Aconitase Is a Source of Hydroxyl Radical
Mitochondrial aconitase hosts a [4Fe-4S] cluster in its active site which catalyzes the conversion of citrate to isocitrate (a reaction of stereo specific hydration and rehydration) in the Krebs cycle. Aconitase activity in mitochondria was reported to be a redox sensor of ROS(57–59). The reaction of aconitase with superoxide (k ~ 107 M−1s−1) rapidly inactivates its enzymatic activity by producing an inactive [3Fe-4S] cluster (g=2.018), free iron (II), and H2O2.Consequently, inactivation of mitochondrial aconitase by superoxide facilitates the formation of hydroxyl radical via a Fenton-type mechanism (Figure 1) (60).
3. The Effects of Mitochondrial ROS in the Heart and Role in Disease
3.1. The Effects of Mitochondrial ROS on Myocytes
The heart requires a constant supply of energy in order to support contractile activity. This obligation is met by the daily synthesis of ATP via oxidative phosphorylation. In normal heart, about 70% of the ATP requirement is met by the catabolism of free fatty acids via β-oxidation, giving rise to a greater ATP yield than with glucose. Oxidative phosphorylation is the endogenous source of mitochondrial ROS production, and overproduction of ROS can establish a vicious cycle of oxidative stress in mitochondria and MPT (mitochondrial permeability transition) induction with ROS induced ROS release(61), which has important pathophysiological effects. Excess ROS production under pathophysiological conditions executes the detrimental effects on myocytes via mitochondrial dysfunction and bioenergtic decline. Chronic exposure of myocytes to ROS leads to impairment of excitation-contraction coupling, causing arrhythmias, and contributing to cardiac remodeling by inducing cardiac hypertrophy, apoptosis, necrosis and fibrosis(62–64).
3.2. The Effects of Mitochondrial ROS on Endothelial Cells
In the vasculature, excess production of ROS from mitochondria is responsible for the inflammatory vascular reaction that has been implicated in the pathogenesis of atherosclerosis, hypertension, diabetes(65), and corruption of coronary collateral growth (66). However, basal low levels of mitochondrial ROS released under normal metabolic activity may exert beneficial effects on cardiovascular physiology and function(7). Myocardial work or cardiac work is tightly controlled by coronary blood flow and heart rate. In the endothelium of coronary arterioles, generation of H2O2 in mitochondria is induced by shear stress. Mitochondrial H2O2subsequently mediates flow-induced dilation of vessel via activation of large-conductance Ca2+−activated potassium channel(Kca channel) coupled with intracellular signaling of PKG-1α activation in the smooth muscle cells(8, 67). H2O2produced in response to flow was demonstrated to be a byproduct of complex I and complex III from the endothelial ETC(8). Therefore, H2O2 originating from mitochondrial •O2−in the vasculature has been described as an EDHF that acts as an intracellular signaling molecule to mediate shear- or acetylcholine-induced smooth muscle relaxation(8) (Figure 5).
Figure 5.
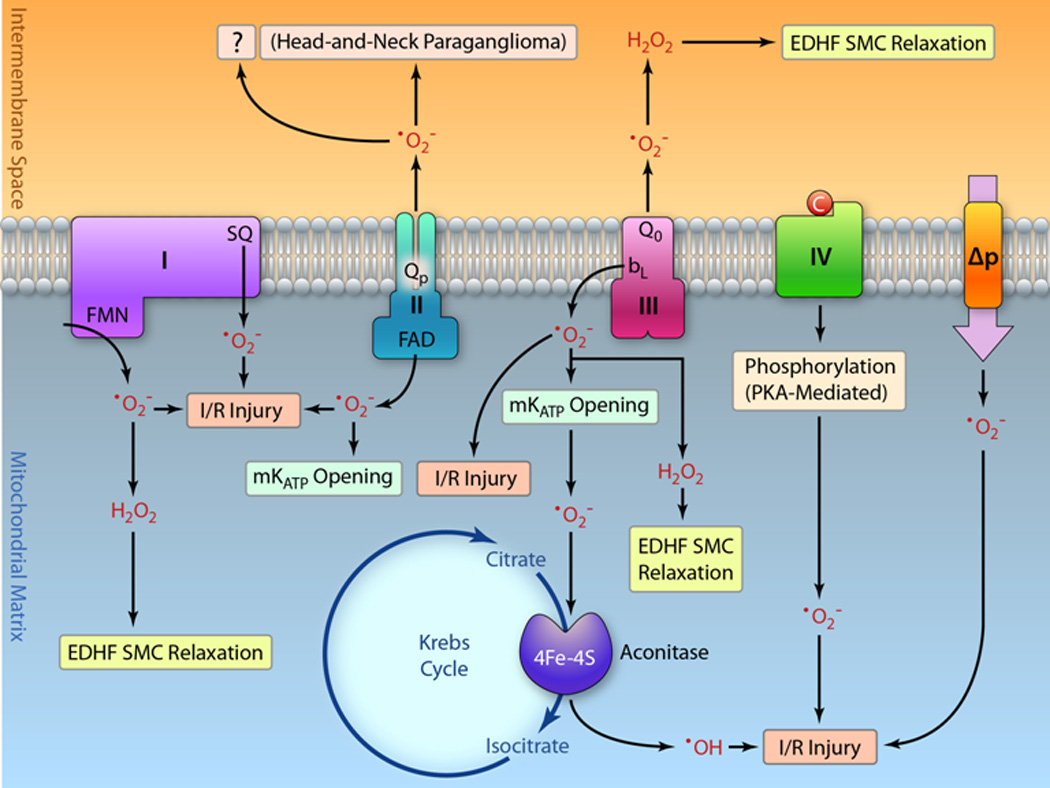
Schematic depiction showing the proposed physiological (highlighted with green boxes) and pathophysiological (highlighted with red boxes and bold) roles of •O2− generation by each respiratory electron transport complex, the proton motive force (Δp), and the aconitase of Krebs cycle. The H2O2 derived from •O2− by complex I and complex III can function as EDHF to mediate the intracellular signaling of metabolic dilation via smooth cell (SMC) relaxation. Intra-mitochondrial H2O2 formed at the matrix site and extra-mitochondrial H2O2 formed at the Qo site can diffuse to the cytosol to trigger the physiological responses. Unlike complex I, complex II-mediated •O2− may modulate pre-conditioning via mKATP channel activation. However, it remains unclear the physiological role of •O2− mediated by the Qp site of complex II. In addition to the physiological role of EDHF, complex III-mediated •O2− also has been linked to diazoxide-induced transient opening of mKATP. Under the pathophysiological conditions of myocardial ischemia and reperfusion (I/R), the •O2− generation by complexes I, III, and flavin protein of complex II all mediate I/R injury and increase pro-oxidant activity of aconitase, thus further augmenting I/R injury. PKA-mediated phosphorylation of complex IV under ischemic conditions predisposes complex IV to generate •O2− and augment I/R injury. Proton motive force is proposed as a source of •O2− for I/R injury because reperfusion partially restores the membrane potential. Overproduction of •O2− from the Qp site of complex II is associated with the disease of head-to-neck paraganglioma, however, it’s pathological role in I/R injury remains unexplored.
In the cardiac myocytes, H2O2 derived from mitochondrial •O2− has also been proposed to regulate collateral blood flow via underlying diffusion to the micro-vascular system of the heart (68). This has important physiological implications providing a link between mitochondrial H2O2 and metabolic flow regulation, and couples the production of H2O2 as EDHF to myocardial metabolism. Evidence has indicated the mitochondrial H2O2is the vasoactive product of metabolically active cardiac myocytes, functioning as a coronary metabolic dilator coupling myocardial oxygen consumption to coronary blood flow in a redox-dependent manner. The above coronary metabolic dilation is mediated by activation of voltage-dependent potassium channels (Kv channels) and thiol redox-dependent signaling(69, 70). Therefore, it is expected that metabolic H2O2 produced by increased perfusion during intense exercise likely contributes to endothelium-dependent hyperpolarization.
3.3. Role of Mitochondrial ROS in Myocardial Infarction
Mitochondrial dysfunction is a critical consequence of myocardial infarction. Impairment of mitochondrial function during myocardial ischemia-reperfusion injury is caused by oxidative stress(71–75). In the ischemic heart, oxygen delivery to the myocardium is not sufficient to meet the need for substrate oxidation in mitochondria during the physiological conditions of hypoxia, leaving the mitochondrial ETC in a highly reduced state. This results in increased electron leakage from the ETC that in turn reacts with residual O2 to give •O2−. Ischemia converts most ATP to ADP and AMP. On reperfusion, most ADP is converted back to ATP and AMP as catalyzed by adenylate kinase. Because of the significant depletion of ADP in the early stage of reperfusion(76), re-introduction of O2with reperfusion will greatly increase electron leakage along with a decrease in scavenging capacity, leading to marked overproduction of •O2− and •O2−−derived ROS in the mitochondria. Specifically, an increased hyperoxygenation induced by reperfusion in the post-ischemic heart has been detected by in vivo EPR oximetry(77). The overproduction of mitochondrial ROS also initiates oxidative inactivation of the ETC proteins, antioxidant enzymes, lipid peroxidation, and mt DNA damage. Mitochondrial dysfunction caused by this injury to the ETC was marked in the post-ischemic rat heart. Pre-treatment of the heart with the radical trap, DMPO, protected the ETC from post-ischemic injury, preventing the oxidative damage to the ETC that occurred following myocardial ischemia and reperfusion(78).
The activity of the mitochondrial aconitase is highly susceptible to oxidative stress, which further predisposes the enzyme as a source of hydroxyl radical (section 2.5). Physiological conditions of myocardial ischemia have been reported to impair the aconitase activity in mitochondria(79). Post-ischemic reperfusion increased mitochondrial •O2− production, and further enhanced oxidative protein damage of aconitase(79). Therefore, oxidative inactivation of mitochondrial aconitase activity may serve as an additional disease marker of myocardial infarction (Figure 5).
Myocardial ischemia and reperfusion provide a stimulus to alter NO metabolism, including involvement of eNOS(77, 80), increased nitrite disproportionation(81–83), and increased iNOS expression in chronic reperfusion injury(80, 84). Therefore, myocardial ischemia and reperfusion indirectly changes the balance between NO and •O2− in mitochondria. Increased NO production, subsequent excess peroxynitrite (ONOO−) formation, and related protein tyrosine nitration was greatly increased in the mitochondria of the post-ischemic heart(84, 85).
3.3.a. Mitochondrial Complex I in the Post-ischemic Heart
Complex I is viewed as the major contributor of mitochondrial ROS metabolism in cells(86). ROS generated by complex I in the cardiovascular system are thus proposed to mediate the physiological effects related to vascular signaling as discussed in section 3.2 (vide supra).
However, pathophysiological production of excess ROS by a leak at mitochondrial complex I has been implicated in myocardial infarction (Figure 5). Oxidative impairment of mitochondrial complex I is detected in isolated rat hearts subjected to global ischemia and reperfusion(51, 87), and in animal disease models of myocardial ischemia and reperfusion(25, 77). In mitochondria of post-ischemic hearts, more severe impairment of ETC in mitochondria was observed in complex I, which correlated with mitochondrial dysfunction, diminished NADH-linked state 3 oxygen consumption, and enhanced NADH-linked •O2− generation and the capacity of mitochondria to produce H2O2(51, 87). Furthermore, the activity of intact complex I was impaired with no significant loss of NADH-ferricyanide reductase activity (the enzymatic activity of NADH dehydrogenase or Fp subcomplex) due to the restoration of the Fp function during reperfusion. The functional recovery of Fp likely plays a role in •O2− generation during reperfusion, since a functional Fp is one of the major sources for complex I-mediated •O2− production(51). In the post-ischemic heart, ROS-induced damage of mitochondrial cardiolip in and respiratory supercomplex assemblies also has been attributed to a defect in complex I activity, increasing electron leakage for complex I-mediated •O2− generation, perpetuating a cycle of oxidative damage, and ultimately leading to mitochondrial dysfunction(87, 88). Furthermore, protein tyrosine nitration of complex I at the subcomplexes Fp and Ip was marked in the post-ischemic heart, implicating that peroxynitrite-mediated complex I inactivation occurs during myocardial infarction(85).
3.3.b. Mitochondrial Complex II in the Post-ischemic Heart
The role of complex II in mitochondrial ROS production in vivo remains uncertain and further investigation is required to determine its role in the cardiovascular system. To date, there is no evidence indicating that complex II-mediated ROS metabolism is involved in vascular signaling (unlike complex I). However, specific complex II inhibitors diazoxide or at pen in A5 have been employed as pharmacologic agents to induce preconditioning-like cardio protective effects by activating mitochondrial ATP-sensitive potassium (mKATP) channels(89–92). Basal levels of ROS generated by complex II are likely physiologically relevant in enabling mitochondria to modulate cardiac function and exert cardio protection via mKATP channels (Figure 5).
Defects in mammalian complex II leading to •O2− overproduction have been linked with the disease pathogenesis of pheochromocytoma and head-to-neck paraganglioma as well as myocardial infarction (42, 84, 93–97). The disease phenotype attributed to a point mutation of complex II is associated with pheochromocytoma and head-to-neck paraganglioma, causing electron leakage from the prosthetic groups or the Qp site in complex II, producing excess ROS and leading to tumor formation (Figure 3 and Figure 5).
In animal models of myocardial ischemia and reperfusion injury, oxidative damage of the electron transfer activity of complex II is marked in regions of myocardial infarction(42, 84, 98). Damage of complex II is associated with loss of FADH2-linked oxygen consumption in the post-ischemic heart. Further evaluation of the redox biochemistry of complex II indicated that alterations of oxidative post-translational modification are present in post-ischemic myocardium, including deglutathionylation of the 70 kDa FAD-binding subunit (loss of GSH binding) and an increase in the level of protein tyrosine nitration of the 70 kDa subunit. Since S-glutathionylation of complex II at the FAD-binding subunit decreases the electron leakage that leads to •O2− generation in vitro, it is thus expected that the complex II deglutathionylation that occurs during myocardial ischemia and reperfusion enhances complex II-mediated •O2−generation(42). Overproduction of •O2−and NO results in subsequent oxidative inactivation and protein nitration of complex II, which can serve as a disease marker of myocardial infarction(84).
3.3.c. Mitochondrial Complex III in the Post-ischemic Heart
Uninhibited complex III (versus complex I) is considered a more minor source of mitochondrial ROS with a physiological role in the regulation of metabolic dilation(7). Despite this minor role, the extra-mitochondrial role and effect of •O2− produced at the Qo site is expected to be relevant. Furthermore, electron leakage at complex III has been linked to pharmacological preconditioning by diazoxide via mKATP channel opening through inhibition of complex II and promoting transient generation of ROS for signaling at complex III(91) (Figure 5).
Complex III is proposed as one of the major sources for mitochondrial •O2− production in the post-ischemic heart (Figure 5). Increased unstable semiqunone radical, •Qo−, is attributed to be the source of •O2−(99, 100). Evidence includes increased lipid peroxidation of cardiolip in required for complex III activity(101)and increased protein tyrosine nitration at the polypeptide of the Rieske iron-sulfur protein (85), which may increase •Qo− formation. Oxidative injury or protein nitration of complex I and complex II weakens the supercomplex interaction with complex III, which can subsequently enhance •O2− generation by complex III in the mitochondria of the post-ischemic heart(51).
3.3.d. Mitochondrial Complex IV in the Ischemic Heart
•O2− production can be mediated by complex IV under ischemic conditions. The activity of complex IV is modulated in response to O2 tension. A marked decrease in complex IV activity has been measured in the mitochondria of ischemic hearts(21, 51), and submitochondrial particles exposed to hypoxic conditions in vitro (102). Hypoxic exposure of murine monocyte macrophages results in cAMP-mediated inhibition of complex IV activity(21). The inhibition of complex IV is accompanied by phosphorylation mediated by protein kinase A (PKA) and markedly increased •O2− generation by phosphorylated complex IV. Conditions of myocardial ischemia also activate mitochondrial PKA, enhancing hyperphosphorylation of complex IV, vastly increasing ROS production by complex IV, and augmenting ischemic and subsequent reperfusion injuries(21) (Figure 5). Therefore, specific inhibitors of PKA have been proposed to render cardiac protection against ischemia/reperfusion injury via suppression of the pro-oxidant activity of complex IV. Furthermore, blocking β1-adreno receptor activation during ischemia/reperfusion reversed PKA-mediated depression of the complex IV activity, and reduced myocardium at risk for post-ischemic injury(79), implying that β1-adrenergic stimulation contributes to myocardial ischemic and reperfusion damage via the mechanism of PKA/cAMP-mediated complex IV inactivation(79).
4. Conclusions and Perspective
Mitochondrial ROS play critical roles in disease pathogenesis and redox signal transduction. The diagram of Figure 5s ummarizes the proposed physiological role and I/R-related pathophysiological role of mitochondrial ROS generated by complexes I-IV, Δp, and aconitase. Complexes I and III are the most extensively characterized enzyme complexes mediating ROS generation in mitochondria, and appear to be responsible for the majority of mitochondrial ROS in cardiovascular physiology and disease. The primary physiological role of mitochondrial ROS is to mediate intracellular signaling of vascular metabolic dilation in the cardiovascular system. In the disease setting of myocardial ischemia/reperfusion, oxidative injury of complex I with increased tyrosine nitration or protein thiylradical-dependent S-glutathionylation can weaken supercomplex interaction, and enhance ROS production by complex III. Recently, deglutathionylated complex II and phosphorylated complex IV have also been demonstrated to be involved in mitochondrial ROS generation in the disease progression of myocardial infarction. Increased peroxynitrite-mediated complex II nitration in the post-ischemic heart further supports the role of complex II in ROS generation and disease. ROS mediated by the ETC may also synergistically enhance oxidative stress in mitochondria via induction of the pro-oxidant activity of aconitase. The redox domains of S-glutathionylation in complex Imodulate the electron leakage that leads to ROS production from FMN and iron-sulfur clusters. Ongoing further evaluation of the role of the aforementioned redox domains in the normal and post-ischemic heart will enable increased understanding of how mitochondrial ROS mediate cellular signaling in cardiovascular physiology and disease.
Further identification of specific redox domains that regulate mitochondrial ROS generation under physiological or pathological conditions is a major frontier in the fields of redox regulation and signaling. Currently, specific S-glutathionylated domains of complex I and complex II have been identified. In addition to S-glutathionylation, specific redox modifications such ascysteine nitrosylation(103, 104) and tyrosine nitration have been detected in complexes I−III, but it is currently unknown whether any of these modifications regulates mitochondrial ROS production. Future studies are likely to reveal new layers of complexity, including more specific oxidative post-translational modifications and more diverse mechanisms of mitochondrial ROS production and regulation, including how oxidative post-translational modifications regulate ROS generation by the ETC in vivo, topology of •O2− production by the ETC, and how •O2− production by the ETC provokes the pro-oxidant activity of Krebs cycle enzymes such as aconitase. As this frontier of knowledge advances, continued attention to the fundamental processes of mitochondrial ROS production will be needed to understand cardiovascular disease pathogenesis with the goal of developing improved treatments to ameliorate disease.
Supplementary Material
Acknowledgments
We thank Dr. Patrick T. Kang (Northeast Ohio Medical University) for helping the Figures 2–4
Sources of Funding
This work was supported in part by National Institutes of Health Grants HL83237, HL65608, HL63744, and HL38324.
Abbreviations
- ROS
reactive oxygen species
- •O2−
superoxide anion radical
- Qp
quinone-binding pocket or quinone binding site in complex II
- •Qo−
unstable ubisemiquinone formed at the Qo site of complex III
- •Qi−
stable ubisemiquinone formed at the Qi site of complex III
- pdb
protein data bank
- ATPase
F1F0-ATP synthase
- ETC
mitochondrial electron transport chain
- RCI
respiratory control index
- MnSOD
manganese containing superoxide dismutase
- GPx
glutathione peroxidase
- GSH
glutathione
- GSSG
oxidized glutathione, EDHF, endothelium-derived hyperpolarizing factor
- HO•
hydroxyl radical
- Δp or PMF
proton motive force or electrochemical gradient
- ΔΨ
membrane potential
- ΔpH
proton gradient
- DMPO
5’, 5’-dimethyl pyrroline N-oxide
- DEPMPO
5-(Diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide
- FCCP
carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- DNP
2,4-dintrophenol
- EPR
electron paramagnetic resonance
- NDH
NADH dehydrogenase or Fp subcomplex of complex I
- SMP
submitochondrial particles
- SQ
ubisemiquinone radical of complex I
- Q1
ubiquinone-1
- Q2
ubiquinone-2
- DPI
diphenyleneiodonium chloride
- TTFA
2-thenoyltrifluoroacetone
- SCR
succinate cytochrome c reductase
- NCR
NADH cytochrome c reductase
- QH2
ubiquinol or reduced ubiquinone
- RISP
Rieske iron-sulfur protein/or cluster of complex III
- bL
low potential heme b or heme b566 of complex III
- bH
high potential heme b or heme b562 of complex III
- mtDNA
mitochondrial DNA
- NOS
nitric oxide synthase
- ONOO−
peroxynitrite
- I/R
ischemia and reperfusion
- SMC
smooth muscle cell
- mKATP
mitochondrial ATP-sensitive potassium channel
Footnotes
Disclosures
None
References
- 1.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem. 1955;217:383–393. [PubMed] [Google Scholar]
- 4.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- 5.Murphy E, Steenbergen C. Preconditioning: the mitochondrial connection. Annu Rev Physiol. 2007;69:51–67. doi: 10.1146/annurev.physiol.69.031905.163645. [DOI] [PubMed] [Google Scholar]
- 6.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H2023–H2031. doi: 10.1152/ajpheart.01283.2006. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- 9.Zhang L, Yu L, Yu CA. Generation of superoxide anion by succinate-cytochrome c reductase from bovine heart mitochondria. J Biol Chem. 1998;273:33972–33976. doi: 10.1074/jbc.273.51.33972. [DOI] [PubMed] [Google Scholar]
- 10.Rottenberg H, Covian R, Trumpower BL. Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. J Biol Chem. 2009;284:19203–19210. doi: 10.1074/jbc.M109.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nohl H, Jordan W. The mitochondrial site of superoxide formation. Biochem Biophys Res Commun. 1986;138:533–539. doi: 10.1016/s0006-291x(86)80529-0. [DOI] [PubMed] [Google Scholar]
- 12.Votyakova TV, Reynolds IJ. Delta Psi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 13.Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cadenas E, Boveris A, Ragan CI, Stoppani AO. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys. 1977;180:248–257. doi: 10.1016/0003-9861(77)90035-2. [DOI] [PubMed] [Google Scholar]
- 15.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 17.Guo J, Lemire BD. The ubiquinone-binding site of the Saccharomyces cerevisiae succinate-ubiquinone oxidoreductase is a source of superoxide. Journal of Biological Chemistry. 2003;278:47629–47635. doi: 10.1074/jbc.M306312200. [DOI] [PubMed] [Google Scholar]
- 18.Messner KR, Imlay JA. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J Biol Chem. 2002;277:42563–42571. doi: 10.1074/jbc.M204958200. [DOI] [PubMed] [Google Scholar]
- 19.Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature. 1998;394:694–697. doi: 10.1038/29331. [DOI] [PubMed] [Google Scholar]
- 20.Sun F, Huo X, Zhai Y, Wang A, Xu J, Su D, Bartlam M, Rao Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121:1043–1057. doi: 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 21.Prabu SK, Anandatheerthavarada HK, Raza H, Srinivasan S, Spear JF, Avadhani NG. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J Biol Chem. 2006;281:2061–2070. doi: 10.1074/jbc.M507741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem. 2006;281:32724–32727. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- 23.Ohnishi T. Iron-sulfur clusters/semiquinones in complex I. Biochim Biophys Acta. 1998;1364:186–206. doi: 10.1016/s0005-2728(98)00027-9. [DOI] [PubMed] [Google Scholar]
- 24.Kang P, Zhang L, Chen C, Chen J, Green K, Chen Y. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radical in Biology and Medicine. 2012;53:962–973. doi: 10.1016/j.freeradbiomed.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Chen CL, Rawale S, Chen CA, Zweier JL, Kaumaya PT, Chen YR. Peptide-based antibodies against glutathione-binding domains suppress superoxide production mediated by mitochondrial complex I. J Biol Chem. 2010;285:3168–3180. doi: 10.1074/jbc.M109.056846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen CL, Zhang L, Yeh A, Chen CA, Green-Church KB, Zweier JL, Chen YR. Site-specific S-glutathiolation of mitochondrial NADH ubiquinone reductase. Biochemistry. 2007;46:5754–5765. doi: 10.1021/bi602580c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I) J Biol Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 28.Chen YR, Chen CL, Zhang L, Green-Church KB, Zweier JL. Superoxide generation from mitochondrial NADH dehydrogenase induces self-inactivation with specific protein radical formation. J Biol Chem. 2005;280:37339–37348. doi: 10.1074/jbc.M503936200. [DOI] [PubMed] [Google Scholar]
- 29.Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J Biol Chem. 2004;279:4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- 30.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci U S A. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 32.Ohnishi ST, Ohnishi T, Muranaka S, Fujita H, Kimura H, Uemura K, Yoshida K, Utsumi K. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. J Bioenerg Biomembr. 2005;37:1–15. doi: 10.1007/s10863-005-4117-y. [DOI] [PubMed] [Google Scholar]
- 33.Ohnishi ST, Shinzawa-Itoh K, Ohta K, Yoshikawa S, Ohnishi T. New insights into the superoxide generation sites in bovine heart NADH-ubiquinone oxidoreductase (Complex I): the significance of protein-associated ubiquinone and the dynamic shifting of generation sites between semiflavin and semiquinone radicals. Biochim Biophys Acta. 2010;1797:1901–1909. doi: 10.1016/j.bbabio.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 34.Magnitsky S, Toulokhonova L, Yano T, Sled VD, Hagerhall C, Grivennikova VG, Burbaev DS, Vinogradov AD, Ohnishi T. EPR characterization of ubisemiquinones and iron-sulfur cluster N2, central components of the energy coupling in the NADH-ubiquinone oxidoreductase (complex I) in situ. J Bioenerg Biomembr. 2002;34:193–208. doi: 10.1023/a:1016083419979. [DOI] [PubMed] [Google Scholar]
- 35.Ohnishi T, Ohnishi ST, Shinzawa-Itoh K, Yoshikawa S, Weber RT. EPR detection of two protein-associated ubiquinone components (SQ(Nf) and SQ(Ns)) in the membrane in situ and in proteoliposomes of isolated bovine heart complex I. Biochim Biophys Acta. 2012;1817:1803–1809. doi: 10.1016/j.bbabio.2012.03.032. [DOI] [PubMed] [Google Scholar]
- 36.Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 37.Kang PT, Yun J, Kaumaya PP, Chen YR. Design and use of peptide-based antibodies decreasing superoxide production by mitochondrial complex I and complex II. 2010 doi: 10.1002/bip.21457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor ER, Hurrell F, Shannon RJ, Lin TK, Hirst J, Murphy MP. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J Biol Chem. 2003;278:19603–19610. doi: 10.1074/jbc.M209359200. [DOI] [PubMed] [Google Scholar]
- 39.Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 40.Hurd TR, Requejo R, Filipovska A, Brown S, Prime TA, Robinson AJ, Fearnley IM, Murphy MP. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: potential role of CYS residues in decreasing oxidative damage. J Biol Chem. 2008;283:24801–24815. doi: 10.1074/jbc.M803432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, Byrne B, Cecchini G, Iwata S. Architecture of succinate dehydrogenase and reactive oxygen species generation.[see comment] Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 42.Chen YR, Chen CL, Pfeiffer DR, Zweier JL. Mitochondrial Complex II in the Post-ischemic Heart: OXIDATIVE INJURY AND THE ROLE OF PROTEIN S-GLUTATHIONYLATION. J Biol Chem. 2007;282:32640–32654. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 43.Chen YR, Chen CL, Yeh A, Liu X, Zweier JL. Direct and indirect roles of cytochrome b in the mediation of superoxide generation and NO catabolism by mitochondrial succinate-cytochrome c reductase. J Biol Chem. 2006;281:13159–13168. doi: 10.1074/jbc.M513627200. [DOI] [PubMed] [Google Scholar]
- 44.Miki T, Yu L, Yu CA. Characterization of ubisemiquinone radicals in succinate-ubiquinone reductase. Archives of Biochemistry & Biophysics. 1992;293:61–66. doi: 10.1016/0003-9861(92)90365-4. [DOI] [PubMed] [Google Scholar]
- 45.Cape JL, Bowman MK, Kramer DM. A semiquinone intermediate generated at the Qo site of the cytochrome bc1 complex: importance for the Q-cycle and superoxide production. Proc Natl Acad Sci U S A. 2007;104:7887–7892. doi: 10.1073/pnas.0702621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muller FL, Roberts AG, Bowman MK, Kramer DM. Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry. 2003;42:6493–6499. doi: 10.1021/bi0342160. [DOI] [PubMed] [Google Scholar]
- 47.Gong X, Yu L, Xia D, Yu CA. Evidence for electron equilibrium between the two hemes bL in the dimeric cytochrome bc1 complex. J Biol Chem. 2005;280:9251–9257. doi: 10.1074/jbc.M409994200. [DOI] [PubMed] [Google Scholar]
- 48.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 49.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. 2002;277:44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 50.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 51.Lee H, Chen C, Yeh S, Zweier J, Chen Y. Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2012;302:H1410–H1422. doi: 10.1152/ajpheart.00731.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 53.Schultz BE, Chan SI. Structures and proton-pumping strategies of mitochondrial respiratory enzymes. Annu Rev Biophys Biomol Struct. 2001;30:23–65. doi: 10.1146/annurev.biophys.30.1.23. [DOI] [PubMed] [Google Scholar]
- 54.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 55.Chen YR, Gunther MR, Mason RP. An electron spin resonance spin-trapping investigation of the free radicals formed by the reaction of mitochondrial cytochrome c oxidase with H2O2. J Biol Chem. 1999;274:3308–3314. doi: 10.1074/jbc.274.6.3308. [DOI] [PubMed] [Google Scholar]
- 56.MacMillan F, Kannt A, Behr J, Prisner T, Michel H. Direct evidence for a tyrosine radical in the reaction of cytochrome c oxidase with hydrogen peroxide. Biochemistry. 1999;38:9179–9184. doi: 10.1021/bi9911987. [DOI] [PubMed] [Google Scholar]
- 57.Gardner PR, Fridovich I. Superoxide sensitivity of the Escherichia coli aconitase. J Biol Chem. 1991;266:19328–19333. [PubMed] [Google Scholar]
- 58.Gardner PR, Fridovich I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J Biol Chem. 1992;267:8757–8763. [PubMed] [Google Scholar]
- 59.Gardner PR, Nguyen DD, White CW. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc Natl Acad Sci U S A. 1994;91:12248–12252. doi: 10.1073/pnas.91.25.12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vasquez-Vivar J, Kalyanaraman B, Kennedy MC. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J Biol Chem. 2000;275:14064–14069. doi: 10.1074/jbc.275.19.14064. [DOI] [PubMed] [Google Scholar]
- 61.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maack C, Dabew ER, Hohl M, Schafers HJ, Bohm M. Endogenous activation of mitochondrial KATP channels protects human failing myocardium from hydroxyl radical-induced stunning. Circ Res. 2009;105:811–817. doi: 10.1161/CIRCRESAHA.109.206359. [DOI] [PubMed] [Google Scholar]
- 64.Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–3535. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 66.Pung YF, Sam WJ, Stevanov K, Enrick M, Chen CL, Kolz C, Thakker P, Hardwick JP, Chen YR, Dyck JR, Yin L, Chilian WM. Mitochondrial oxidative stress corrupts coronary collateral growth by activating adenosine monophosphate activated kinase-alpha signaling. Arterioscler Thromb Vasc Biol. 2013;33:1911–1919. doi: 10.1161/ATVBAHA.113.301591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res. 2012;110:471–480. doi: 10.1161/CIRCRESAHA.111.258871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saitoh S, Zhang C, Tune JD, Potter B, Kiyooka T, Rogers PA, Knudson JD, Dick GM, Swafford A, Chilian WM. Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol. 2006;26:2614–2621. doi: 10.1161/01.ATV.0000249408.55796.da. [DOI] [PubMed] [Google Scholar]
- 69.Saitoh S, Kiyooka T, Rocic P, Rogers PA, Zhang C, Swafford A, Dick GM, Viswanathan C, Park Y, Chilian WM. Redox-dependent coronary metabolic dilation. Am J Physiol Heart Circ Physiol. 2007;293:H3720–H3725. doi: 10.1152/ajpheart.00436.2007. [DOI] [PubMed] [Google Scholar]
- 70.Rogers PA, Chilian WM, Bratz IN, Bryan RM, Jr, Dick GM. H2O2 activates redox- and 4-aminopyridine-sensitive Kv channels in coronary vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2007;292:H1404–H1411. doi: 10.1152/ajpheart.00696.2006. [DOI] [PubMed] [Google Scholar]
- 71.Ferrari R. Importance of oxygen free radicals during ischemia and reperfusion in the experimental and clinical setting. Oxygen free radicals and the heart. Am J Cardiovasc Pathol. 1992;4:103–114. [PubMed] [Google Scholar]
- 72.Ferrari R. Oxygen-free radicals at myocardial level: effects of ischaemia and reperfusion. Advances in Experimental Medicine & Biology. 1994;366:99–111. doi: 10.1007/978-1-4615-1833-4_8. [DOI] [PubMed] [Google Scholar]
- 73.Ferrari R, Ceconi C, Curello S, Cargnoni A, Pasini E, De Giuli F, Albertini A. Role of oxygen free radicals in ischemic and reperfused myocardium. Am J Clin Nutr. 1991;53:215S–222S. doi: 10.1093/ajcn/53.1.215S. [DOI] [PubMed] [Google Scholar]
- 74.Watts JA, Kline JA. Bench to bedside: the role of mitochondrial medicine in the pathogenesis and treatment of cellular injury. Academic Emergency Medicine. 2003;10:985–997. doi: 10.1111/j.1553-2712.2003.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 75.Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70:181–190. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 76.Lasley RD, Ely SW, Berne RM, Mentzer RM., Jr Allopurinol enhanced adenine nucleotide repletion after myocardial ischemia in the isolated rat heart. J Clin Invest. 1988;81:16–20. doi: 10.1172/JCI113288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao X, He G, Chen YR, Pandian RP, Kuppusamy P, Zweier JL. Endothelium-derived nitric oxide regulates postischemic myocardial oxygenation and oxygen consumption by modulation of mitochondrial electron transport. Circulation. 2005;111:2966–2972. doi: 10.1161/CIRCULATIONAHA.104.527226. [DOI] [PubMed] [Google Scholar]
- 78.Zuo L, Chen Y, Reyes L, Lee H, Chen C, Villamena F, Zweier J. The radical trap 5,5-dimethyl-1-pyrroline N-oxide exerts dose-dependent protection against myocardial ischemia-reperfusion injury through preservation of mitochondrial electron transport. J Pharmacol Exp Ther. 2009;329:515–523. doi: 10.1124/jpet.108.143479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spear JF, Prabu SK, Galati D, Raza H, Anandatheerthavarada HK, Avadhani NG. beta1-Adrenoreceptor activation contributes to ischemia-reperfusion damage as well as playing a role in ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2007;292:H2459–H2466. doi: 10.1152/ajpheart.00459.2006. [DOI] [PubMed] [Google Scholar]
- 80.Zhao X, Chen YR, He G, Zhang A, Druhan LJ, Strauch AR, Zweier JL. Endothelial nitric oxide synthase (NOS3) knockout decreases NOS2 induction, limiting hyperoxygenation and conferring protection in the postischemic heart. Am J Physiol Heart Circ Physiol. 2007;292:H1541–H1550. doi: 10.1152/ajpheart.00264.2006. [DOI] [PubMed] [Google Scholar]
- 81.Zweier JL, Samouilov A, Kuppusamy P. Non-enzymatic nitric oxide synthesis in biological systems. Biochim Biophys Acta. 1999;1411:250–262. doi: 10.1016/s0005-2728(99)00018-3. [DOI] [PubMed] [Google Scholar]
- 82.Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat Med. 1995;1:804–809. doi: 10.1038/nm0895-804. [DOI] [PubMed] [Google Scholar]
- 83.Tiravanti E, Samouilov A, Zweier JL. Nitrosyl-heme complexes are formed in the ischemic heart: evidence of nitrite-derived nitric oxide formation, storage, and signaling in post-ischemic tissues. J Biol Chem. 2004;279:11065–11073. doi: 10.1074/jbc.M311908200. [DOI] [PubMed] [Google Scholar]
- 84.Chen CL, Chen J, Rawale S, Varadharaj S, Kaumaya PP, Zweier JL, Chen YR. Protein tyrosine nitration of the flavin subunit is associated with oxidative modification of mitochondrial complex II in the post-ischemic myocardium. J Biol Chem. 2008;283:27991–28003. doi: 10.1074/jbc.M802691200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu B, Tewari AK, Zhang L, Green-Church KB, Zweier JL, Chen YR, He G. Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: Mitochondria as the major target. Biochim Biophys Acta. 2009;1794:476–485. doi: 10.1016/j.bbapap.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. doi: 10.1042/bse0470053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res. 2004;94:53–59. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 88.Gadicherla AK, Stowe DF, Antholine WE, Yang M, Camara AK. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim Biophys Acta. 2012;1817:419–429. doi: 10.1016/j.bbabio.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu B, Zhu X, Chen CL, Hu K, Swartz HM, Chen YR, He G. Opening of the mitoKATP channel and decoupling of mitochondrial complex II and III contribute to the suppression of myocardial reperfusion hyperoxygenation. Mol Cell Biochem. 2010;337:25–38. doi: 10.1007/s11010-009-0283-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol. 2009;104:121–129. doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Drose S, Hanley PJ, Brandt U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochim Biophys Acta. 2009;1790:558–565. doi: 10.1016/j.bbagen.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 92.Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- 93.Astuti D, Douglas F, Lennard TW, Aligianis IA, Woodward ER, Evans DG, Eng C, Latif F, Maher ER. Germline SDHD mutation in familial phaeochromocytoma. Lancet. 2001;357:1181–1182. doi: 10.1016/S0140-6736(00)04378-6. [DOI] [PubMed] [Google Scholar]
- 94.Baysal BE. Hereditary paraganglioma targets diverse paraganglia. J Med Genet. 2002;39:617–622. doi: 10.1136/jmg.39.9.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, Cornelisse CJ, 3rd, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 96.Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 97.Taschner PE, Jansen JC, Baysal BE, Bosch A, Rosenberg EH, Brocker-Vriends AH, van Der Mey AG, van Ommen GJ, Cornelisse CJ, Devilee P. Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer. 2001;31:274–281. doi: 10.1002/gcc.1144. [DOI] [PubMed] [Google Scholar]
- 98.Zhang L, Chen CL, Kang PT, Garg V, Hu K, Green-Church KB, Chen YR. Peroxynitrite-mediated oxidative modifications of complex II: relevance in myocardial infarction. Biochemistry. 2010;49:2529–2539. doi: 10.1021/bi9018237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zweier JL, Flaherty JT, Weisfeldt ML. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci U S A. 1987;84:1404–1407. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zweier JL, Rayburn BK, Flaherty JT, Weisfeldt ML. Recombinant superoxide dismutase reduces oxygen free radical concentrations in reperfused myocardium. J Clin Invest. 1987;80:1728–1734. doi: 10.1172/JCI113264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J. 2003;17:714–716. doi: 10.1096/fj.02-0729fje. [DOI] [PubMed] [Google Scholar]
- 102.Chandel NS, Budinger GR, Choe SH, Schumacker PT. Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J Biol Chem. 1997;272:18808–18816. doi: 10.1074/jbc.272.30.18808. [DOI] [PubMed] [Google Scholar]
- 103.Kohr MJ, Aponte A, Sun J, Gucek M, Steenbergen C, Murphy E. Measurement of S-nitrosylation occupancy in the myocardium with cysteine-reactive tandem mass tags: short communication. Circ Res. 2012;111:1308–1312. doi: 10.1161/CIRCRESAHA.112.271320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M, Steenbergen C. Characterization of potential S-nitrosylation sites in the myocardium. Am J Physiol Heart Circ Physiol. 2011;300:H1327–H1335. doi: 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.