

Abstract
Background
Surfactant protein D (SP-D) is a collection that plays important roles in modulating host defense functions and maintaining phospholipid homeostasis in the lung. The aim of current study was to characterize comparatively the SP-D response in bronchoalveolar lavage (BAL) and serum in three murine models of lung injury, using a validated ELISA technology for estimation of SP-D levels.
Methods
Mice were exposed to lipopolysaccharide, bleomycin, or Pneumocystis carinii (Pc) and sacrificed at different time points.
Results
In lipopolysaccharide-challenged mice, the level of SP-D in BAL increased within 6 h, peaked at 51 h (4,518 ng/ml), and returned to base level at 99 h (612 ng/ml). Serum levels of SP-D increased immediately (8.6 ng/ml), peaked at 51 h (16 ng/ml), and returned to base levels at 99 h (3.8 ng/ml). In a subacute bleomycin inflammation model, SP-D levels were 4,625 and 367 ng/ml in BAL and serum, respectively, 8 days after exposure. In a chronic Pc inflammation model, the highest level of SP-D was observed 6 weeks after inoculation, with BAL and serum levels of 1,868 and 335 ng/ml, respectively.
Conclusions
We conclude that serum levels of SP-D increase during lung injury, with a sustained increment during chronic inflammation compared with acute inflammation. A quick upregulation of SP-D in serum in response to acute airway inflammation supports the notion that SP-D translocates from the airways into the vascular system, in favor of being synthesized systemically. The study also confirms the concept of using increased SP-D serum levels as a biomarker of especially chronic airway inflammation.
Keywords: Surfactant protein D, Lung injury Lipopolysaccharide, Bronchoalveolar lavage, Bleomycin, Pneumocystis carinii
Introduction
The hydrophilic lung surfactant protein D (SP-D) is a collection that plays important roles in our innate immune defense system against inhaled microorganisms, in the immunomodulation of our adaptive immune response, and in the homeostasis of surfactant phospholipids [1]. There have been several studies showing the association of regulation of SP-D and human diseases such as infections [2, 3], acute respiratory distress syndrome (ARDS) [4], acute lung injury (ALI) [5], chronic obstructive pulmonary disease (COPD) [6], asthma [7], and cystic fibrosis [8, 9]. Studies have often aimed to refine diagnosis and monitoring of disease activity using measurements of SP-D levels in serum and bronchoalveolar lavage (BAL) [10–12]. It has also been demonstrated that lung injury and subsequent inflammation lead to decomposition of SP-D oligomers via S-nitrosylation of cysteine residues in the N-termini of SP-D oligomers, and hence to decreased binding and decreased antimicrobial activities [13, 14]. In humans, cigarette smoking induces an increase in SP-D serum levels but a decrease in SP-D BAL levels [11, 15, 16]. In order to characterize disease mechanisms, several mouse studies have applied semiquantitative methods to estimate the regulation of SP-D. Studies that examined mice exposed to cigarette smoke showed an increased expression of SP-D mRNA and protein in the lungs but not in the circulation [17]. Further studies with mice have shown that SP-D regulation is under the influence of the proinflammatory cytokines IL-4, IL-13, and TNF-α [18, 19]. The source of plasma SP-D has not been fully clarified. Pulmonary intravascular leakage or extrapulmonary synthesis has been suggested to cause the increase of SP-D in the circulation during lung injury [20]. Recent studies indicate also that post-translational modifications of SP-D resulting in smaller subunits may play a role in the intravascular leakage during inflammation [15, 21]. To fully characterize the kinetics of the regulation of SP-D during inflammation in murine lung injury models and provide a basis for further regulatory and association studies, we have characterized the regulation of SP-D in both BAL and serum during acute and chronic lung injury by means of a validated SP-D ELISA [22]. As a model of acute inflammation, lipopolysaccharide (LPS), which activates macrophages and induces inflammatory mediators via Toll-like receptor 4, was used to study SP-D regulation during a rapid and transient lung injury [23]. As a model of subacute inflammation, the toxic effect of bleomycin was used to study severe acute lung injury and remodeling, including pulmonary fibrosis, over the course of a month [24]. Finally, as model of chronic inflammation, Pneumocystis carinii (Pc) infection was used to study long-term lung damage spanning several weeks [25]. We found that SP-D was strongly upregulated in all three models of inflammation.
Methods
Reagents, Solutions, Buffers, and Mice
Unless otherwise stated, reagents were obtained from Sigma-Aldrich (Broendby, Denmark). The LPS solution was 0.125 mg LPS/ml HBSS (Hanks’ Balanced Salt Solution, Lonza, Taastrup, Denmark). Tris-Buffered Saline (TBS) was made of 10 mM Tris and 150 mM NaCl (pH 7.4). Female virgin Balb/C mice (Taconic, Denmark), C57/BL6 mice (Charles River Laboratories, Wilmington, MA, USA), and Swiss Black wild-type mice (Taconic, Inc., Germantown, NY, USA) were housed at local animal facilities under controlled conditions, and the national animal welfare committees approved the studies ahead of performance.
LPS Preparation and Aerosol Exposures of Mice
LPS exposures were carried out as previously described, with minor modifications [23]. In brief, 65 ml of LPS solution was loaded into the nebulizer (BGI Inc., Waltham MA, USA), to which a 25-psi pressure was applied, resulting in a flow rate of approximately 0.40 ml/min. After 1.5 h of exposure, the nebulizer was refilled with 35 ml LPS. After a total exposure of 3 h, female virgin Balb/C mice were sacrificed at 3, 6, 15, 27, 51, or 99 h after LPS exposure (n = 4–6). To serve as a control and a measurement of steady state, five mice not exposed to LPS were sacrificed.
Mouse Model of Bleomycin-Induced Lung Injury
Swiss Black mice were anesthetized; then, with the use of a syringe, 50 μl of either saline or bleomycin sulfate (3.0 U/kg; Bristol-Myers Squibb, New York, NY, USA) was injected directly into the surgically exposed trachea, as previously described [26]. Eight days after bleomycin exposure, the mice were sacrificed and serum samples and BAL were collected.
Mouse Model of Chronic Infection with P. carinii
Before and during the experiment, C57/BL6 mice were subjected to selective depletion of CD4-positive cells via an intraperitoneal (i.p.) injection of the monoclonal antibody GK 1.5 twice a week [25]. Control mice received an i.p. injection of equal volumes of phosphate-buffered saline (PBS). Pc was obtained from the lungs of athymic mice (nu/nu on a BALB/c background) in which Pc was propagated by serial passages, as previously described [27]. Four or 6 weeks after inoculation, infected and uninfected mice were euthanized and samples of serum and BAL were collected.
Serum and Bronchoalveolar Lavage
Mice were euthanized via i.p. injection of pentobarbital (150 mg/kg body weight). Approximately 500 μl of blood from the heart chambers was collected, and serum was isolated and stored at −80 °C. BAL was collected from LPS-exposed mice by gently lavaging three times with 1 ml saline each time. The BAL, approximately 2–3 ml, was centrifuged at 2,500 rpm (1,125 g) for 15 min at 4 °C and stored at −80 °C. BAL was obtained from Pc-inoculated and bleomycin-exposed mice as above, but their lungs were lavaged with 0.5-ml aliquots of sterile saline to a total of 5 ml.
Estimation of the Total Cell Number in BAL Fluid and Cytospin Preparation
Cell densities were estimated by microscopy using Trypan blue staining and hemacytometers. Cytospins of 200,000 cells per slide were prepared and stained manually. Differential cell counts of neutrophils, macrophages, and dendritic cells were obtained for ten areas with approximately 200 cells per each area per mouse.
ELISA for Quantification of Mouse SP-D
Mouse SP-D ELISA was carried out as described previously [22]. Samples of BAL and serum were diluted 1:64 and 1:2, respectively, and measured in duplicate.
Immunohistochemistry
Nine in-house monoclonal and two commercial polyclonal anti-mouse SP-D antibodies were tested by immunohistochemistry on cryo- or formalin-fixed/paraffin-embedded sections using general procedures for optimization and antigen retrieval, as described previously [28, 29]. None of the tested antibodies worked on formalin-fixed sections, but a polyclonal antibody gave specific staining on cryo-sections using the following protocol. Snap-frozen and Tissue-Tek (Sakura, Finetek Europe, Zoeterwoude, The Netherlands)-preserved cryosamples of mouse lungs were cut into sections of 5 μm and immobilized on polylysine L-coated glass slides. The sections were fixed for 10 min at room temperature in acetone and washed in TBS twice. Subsequently, the sections were blocked in TBS with 2 % bovine serum albumin (BSA) for 10 min and incubated for 2 h at room temperature with the primary antibody, rabbit anti-mouse SP-D polyclonal antibody (Chemicon International, Temecula, CA, Cat. AB3434), diluted 1:300 in TBS with 1 % BSA. In negative controls, the use of the primary antibody was omitted and substituted for with a subclass-matched nonsense antibody. The sections were washed three times with TBS and incubated for 30 min with EnVision ? System Labelled Polymer-HRP anti-rabbit (Dako, Glostrup, Denmark). After washing, the color reaction was developed with aminoethyl carbazol for 20 min. The sections were then counterstained with Mayer’s hematoxylin and mounted with Aquatex.
Data Processing
Figure preparation was performed using GraphPad Prism5 and Adobe Illustrator CS5.
Results
LPS-Induced Acute Lung Injury
Five mice were used for replica measurements and the average cell count of each group of mice for each post-LPS-exposure time point (Fig. 1c). The total number of cells, represented by the adjusted averaged cell density in BAL from nonexposed mice, was estimated to be 1.5 × 106 (SEM ± 4.9 × 105) cells/ml, and during the 6 h of LPS exposure, the density increased to 7.4 × 106 (±3.1 × 106) cells/ml. The cell density continued to increase and peaked at 1.3 × 107 (±3.5 × 106) cells/ml at 9 h post LPS exposure. The cell density slowly declined and nearly reached the base levels after 99 h. Cytological analyses were done by cytospin preparations (Fig. 1a, b) that allowed for differential cell counts (Fig. 1d). Macrophage and dendritic cells could not be distinguished by their microscopic morphology. At pre-exposure (time = 0), BAL consisted entirely of cells with the appearance of macrophages or dendritic cells. Immediately after LPS exposure (time = 3), 87 % of the BAL cells were neutrophils and 13 % were macrophages and dendritic cells. From 6 to 27 h after exposure, the relative composition was 95 % neutrophils and 5 % macrophages and dendritic cells. At 99 h the composition had returned to base levels.
Fig. 1.
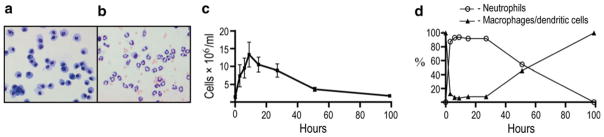
Validation of acute inflammation induced by LPS aerosol challenge. a Cytological studies of BAL cells isolated from nonexposed mice. b Same as a but 6 h after ending LPS exposure. c Total cell counts of BAL for each group of mice (n = 5) killed at 3, 6, 9, 15, 27, 51, and 99 h, respectively, after ending exposure to LPS aerosols. d Differential cell counts obtained from cytological staining. The data set at 0 h represents the control group of mice not exposed to LPS (a measure for steady-state). The error bars represent the standard error of mean (SEM)
Immunohistochemical Analyses of SP-D During Acute Lung Injury
The lung tissue from mice exposed to LPS was dominated by inflammatory cell infiltration with neutrophils, lymphocytes, and macrophages (Fig. 2). In mice not exposed to LPS, SP-D immunoreactivity in the lung tissue was associated with moderate to weak staining. Upon LPS challenge, increasing immunoreactivity was already observed at 3, 6, and 9 h after LPS exposure, and very potent immunoreactivity was observed at 15, 27, and 51 h after LPS exposure. As found in previous studies, strong immunoreactivity for SP-D was associated with macrophages (Fig. 2e) and type II pneumocytes (Fig. 2f) [30]. To allow for visualization of upregulation, the applied concentration of antibody was adjusted to yield moderate staining during steady state. This resulted in only weak to absent immunoreactivity of Clara cells (Fig. 2e), which have been associated with SP-D synthesis. No immunoreactivity was found in the controls using subclass-matched nonsense antibodies.
Fig. 2.
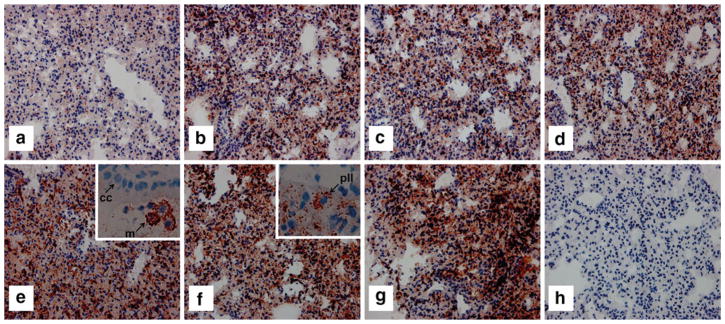
Immunohistochemical analysis of SP-D expression and localization in lung tissues from mice exposed to LPS. The mice were sacrificed at different time points: b 3 h, c 6 h, d 9 h, e 15 h, f 51 h, and g 99 h after LPS exposure. a Control mice not exposed to LPS. h Control with subclass-matched nonsense antibody. Clara cells (cc) were identified by their location in the small airways and the appearance of dome-shaped cells. Macrophages (mp) were identified by their high content of granules and their location in the lumen. Type II pneumocytes (pII) were identified by the appearance of rounded cells in the alveoli. e, f Original magnification ×200 and ×400 for the inserted close-up images
SP-D Regulation During LPS-Induced Acute Lung Injury
Levels of SP-D in BAL and serum were estimated by means of ELISA (Figs. 3 and 4). In nonexposed mice and at 99 h post exposure the average level was 554 (±81) ng SP-D/ml BAL. Immediately after exposure (time = 3) there was no change in the level, but within 6 h the level increased and reached a maximum of 4,518 (±426) ng SP-D/ml BAL at 51 h post exposure. In serum from non-exposed mice, the level was 3.90 (±0.36) ng SP-D/ml serum, and immediately after LPS exposure (time = 3) the level increased to 8.55 (±0.81) ng SP-D/ml serum. The level continued to rise to a maximum of 16 (±1.29) ng SP-D/ml serum at 9–51 h post exposure. At 99 h the serum level of SP-D had returned to base level.
Fig. 3.

SP-D BAL levels at different time points after LPS exposure. Each square represents the average concentration of SP-D in BAL for each group of mice sacrificed at 3, 6, 9, 15, 27, 51, and 99 h after LPS exposure. The data set at 0 h represents the control group of mice not exposed to LPS (a measure for steady-state). The error bars represent the standard error of mean (SEM)
Fig. 4.

SP-D serum levels at different time points after LPS exposure. Each square represents the average concentration of SP-D in serum for each group of mice sacrificed at 3, 6, 9, 15, 27, 51, and 99 h after LPS exposure. The data set at 0 h represents the control group of mice not exposed to LPS (a measure for steady-state). The error bars represent the standard error of mean (SEM)
SP-D Regulation During Bleomycin-Induced Lung Injury
SP-D levels in BAL and serum were estimated by ELISA (Fig. 5a). Eight days after bleomycin exposure, the average levels were 4,625 (±1,205) ng SP-D/ml BAL and 367 (±161) ng SP-D/ml serum. In the controls the average levels were 305 (±32) ng SP-D/ml BAL and 6 (±0) ng SP-D/ml serum. The inflammatory properties of the applied model of bleomycin-induced lung injury were characterized previously [24, 26].
Fig. 5.

Regulation of SP-D during subacute and chronic infection and inflammation. a Subacute inflammation was induced by intratracheal instillation of bleomycin in Swiss Black mice. SP-D levels were assessed 8 days post exposure in control mice exposed to saline (n = 7, open bars) and in bleomycin-challenged mice (n = 7, black bars). b Chronic infection with P. carinii in CD4-depleted C57BL/6 mice. Uninfected mice (n = 5, open bars), mice sacrificed 4 weeks post challenge (n = 5, gray bars), and mice sacrificed 6 weeks post challenge (n = 6, black bars). Error bars represent the standard error of mean (SEM)
SP-D Regulation During Chronic Lung Injury Induced by P. carinii
Four weeks after Pc inoculation, the average levels were 1,789 (±470) ng SP-D/ml BAL and 94 (±33) ng SP-D/ml serum (Fig. 5b). Six weeks post Pc inoculation, the average levels of SP-D in BAL and serum increased to 1,868 (±387) ng/ml and 335 (±102) ng/ml, respectively. In the controls average levels of 253 (±22) ng SP-D/ml BAL and 4.70 (±0.70) ng SP-D/ml serum were observed. The inflammatory properties of the applied model of Pc infection were characterized previously [27, 31].
Discussion
In the present study, we demonstrated a multifold upregulation of SP-D in serum in response to either acute or chronic lung injury. It was evident that chronic injury, not acute injury, induced the highest upregulation of SP-D in serum, 71-fold vs. 4.0-fold, respectively. To our knowledge this is the first study that compares regulation of SP-D in three models of lung injury: acute, subacute, and chronic inflammation.
Our model of acute lung injury by inhalation of LPS aerosols worked well. The maximum total number of cells in BAL was observed 9 h after exposure. Inflammation declined from 15 h post exposure and reached base levels at 99 h. Throughout the study, the immunohistochemical analysis agreed with the degree of inflammation (Fig. 2). Thus, the results confirmed that the inhalation of LPS aerosols is a good model of acute lung injury and allows the study of the regulation of SP-D. In BAL, the level of SP-D was upregulated 6 h post LPS exposure, and it continued to increase, peaking at 51 h at a level 9.1-fold higher than the base level (Table 1). At 99 h SP-D had returned to base level. In serum, the level of SP-D was upregulated 2.2-fold immediately after exposure and peaked at 51 h at 4.0-fold the base level. As with the situation in the BAL, serum levels were normalized at 99 h. These data demonstrated that levels of SP-D in both BAL and serum responded to proinflammatory stimuli and increased during acute inflammation. According to the LPS inhalation protocol [23], the mice were exposed to LPS 6 h before the first measurements. Thus, it was unknown how SP-D in BAL and serum was regulated during this period (0–3 h). The immediate increase of SP-D in serum at time = 3, ahead of the upregulation in BAL at 6 h post exposure, showed that SP-D nearly immediately translocated from the lungs and into circulation during the 3 h of LPS exposure. Increased endothelial permeability, potentially augmented by disruption of SP-D multimeric structure into relatively low-molecular-weight single subunits, is likely the cause of the immediate intravascular leakage. It is unlikely that extrapulmonary synthesis of SP-D is the source since it requires that extrapulmonary synthesis respond faster to inflammatory stimuli than pulmonary cells, which otherwise are a known to be the primary source of SP-D synthesis and are located at the site of inflammation. The BAL:serum ratio of SP-D was calculated to obtain a more straightforward comparison of the changes that occur during inflammation (Table 1). A high ratio was interpreted as a pronounced rise in BAL SP-D, whereas a low ratio was interpreted as a pronounced rise in serum SP-D. In nonexposed mice, the ratio of BAL SP-D to serum SP-D was 130, and during inflammation, when the SP-D concentration peaked in BAL, the same ratio was 290 in favor of BAL. Our results for BAL and serum levels agree well with those of our previous pilot study [22]. However, in a recent similar study by Suda et al. [32], the steady-state BAL/arterial ratio was estimated to be only 34.0 compared with 130 in the current study [32]. Besides the great difference in blood and BAL sampling, we have no explanation for this difference. In a study of lung tumors in mice, Zhang et al. [33] found via a polyclonal antibody–based ELISA that the base levels in serum were 4–8 ng/ml, which corresponds well with the average base level of 3.90 (±0.36) ng/ml observed in the current study. Experiments with Aspergillus fumigatus have shown that allergic airway inflammation alters surfactant homeostasis of SP-D in BALB/c mice after intranasal treatment [34]. Using densiometry of Western blots, Haczku et al. [34] observed a 9-fold increase in SP-D BAL levels 24 h after the last treatment. We observed a similar increase of 7.3-fold 27 h post exposure. Another study performed in ovalbumin-sensitized mice showed an increase in SP-D serum level to 24 ng/ml immediately after challenge with aerosolized ovalbumin [33]. In our study, the highest level of SP-D in serum was 16 ng/ml after 51 h. The divergence may be caused by unevaluated SP-D measurements or a pronounced Th2-mediated lung injury in ovalbumin-sensitized mice, leading to sustained increased leakage of SP-D into the vascular system.
Table 1.
Summary of SP-D levels
| LPS (51 h) | Bleomycin (8 days) | P. carinii (6 weeks) | |
|---|---|---|---|
| BAL | 4,518 ± 426 (n = 5) | 4,625 ± 1205 (n = 4) | 1,868 ± 387 (n = 6) |
| Controls | 497 ± 92 (n = 4) | 305 ± 32 (n = 2) | 253 ± 22 (n = 4) |
| Serum | 16 ± 1.3 (n = 5) | 367 ± 161 (n = 3) | 335 ± 102 (n = 4) |
| Controls | 4.0 ± 0.4 (n = 5) | <6.0 ± 0a (n = 2) | 4.7 ± 0.7 (n = 5) |
| BAL:serum ratio | 290 | 13 | 5.6 |
| Controls | 130 | >51a | 54 |
Concentrations are given in ng/ml, ± standard error of mean (SEM)
BAL bronchoalveolar lavage, LPS lipopolysaccharide
Due to a limited amount of serum, additional dilution made the practical detection limit 6.0 ng/ml
Instillation of bleomycin induces an early inflammatory response followed by fibrosis and finally resolution of inflammation [24]. In the current study, the SP-D levels at day 8 were 15- and 51-fold upregulated in BAL and serum, respectively, with levels at 4,625 and 367 ng/ml. This resulted in a BAL:serum ratio of 12.6. The level of serum SP-D corresponds relatively well with that of a previous work on bleomycin-induced lung injury in mice, where the authors found a maximum serum level of 425 ng/ml [24]. In a study performed with rats, the SP-D level in serum was measured by ELISA after instillation of 2 units of bleomycin [35]. In the rats, the BAL and serum levels of SP-D peaked at 550 and 470 ng/ml, respectively, 3 and 7 days after instillation. The serum level corresponds to that measured in the current study. In contrast, the SP-D level in BAL differed considerably between the studies, with a substantially higher level in the current study. Incomparable sampling may have caused the divergence of BAL levels between the different species used in the studies. The protective role of SP-D was confirmed in a study on bleomycin-induced lung injury in SP-D-deficient mice, leading to increased mortality, enhanced lung inflammation, and alterations in nitric oxide metabolism. Moreover, resistance against bleomycin-induced mortality and morbidity was demonstrated in transgene mice overexpressing SP-D [26]. Although bleomycin-induced lung injury induces fibrosis, the supply of bleomycin generates subacute inflammation which resolves after 4 weeks [24].
Inoculation with Pc leads to long-term damage with chronic inflammation. Previous works have studied the kinetics of Pc infection in immunocompromized mice and major changes in surfactant composition at 4–6 weeks after inoculation have been observed [31]. Therefore, the present study focused on regulation of SP-D at these time points and found a 7.1–7.4-fold increase in SP-D concentration in BAL corresponding to 1,789 and 1,868 ng/ml. In serum, the level of SP-D increased with time as well, with 20- and 71-fold increases corresponding to 94 and 335 ng/ml at 4 and 6 weeks, respectively. This resulted in BAL:serum ratios of 19 and 5.6 at 4 and 6 weeks after instillation, respectively. Pc infection is associated with hypoxemic respiratory insufficiency, and it affects predominantly immunocompromised patients. The pathogenesis is unclear, but several studies have suggested that dysfunction of the pulmonary surfactant system may contribute to impaired gas exchange [31, 36]. Previous studies have also measured the SP-D level in BAL due to Pc infection in mice. Focusing on expression of surfactant composition 4–6 weeks after Pc inoculation, a 3.3–3.5-fold increase in the level of SP-D in BAL was observed by means of Western blotting and densitometry [27, 36]. Our observation falls relatively in line with those observations of previous studies, and the divergence likely reflects the different techniques used for estimating SP-D levels. Compared with previous work, the strength of the current study is the simultaneous assessment of SP-D regulation and response in both serum and alveolar compartments. We found a pronounced increase in serum SP-D during chronic inflammation in comparison with acute inflammation.
The exact mechanism for the increase in serum SP-D associated with lung injury has not been clarified, likely because of several factors. The pronounced increase in serum SP-D associated with chronic and subacute inflammation may be due to sustained lung inflammation. In an attempt to repair the lungs, remodeling takes place, which leads to structural changes, including damaged epithelium and endothelium and angiogenesis. The changes contribute to permeability defects and allow for intravascular sieving of relatively large pulmonary molecules. Also during inflammation, the multimeric form of SP-D will be modified by S-nitrosylation and disassembled into single subunits made of three polypeptide chains that intravasculate easier than multimeric SP-D. SP-D upregulation by type II cells results in a large concentration gradient between the pulmonary environment and the blood, which augments the intravascular sieving. It is likely that these mechanisms allow the organisms to either take systemic advantage of the protective effects of SP-D, which include the antimicrobial properties, or allow for a systemic response to lung injury [37]. However, this study did not assess the physical state of SP-D or the barrier dysfunction. We find it likely that increased SP-D synthesis, and hence increasing concentrations of nonmicrobial complex SP-D in both BAL and blood, may favor anti-inflammatory properties via SIRP-alpha on phagocytes [13, 38]. Several human studies have investigated the regulation of SP-D in lung injury and they support the idea that a similar translocation of SP-D to the vascular system also applies to humans [3, 4].
Conclusions
In many human diseases there is a focus on finding a plasma biomarker of lung injury, and SP-D seems to be a highly valuable candidate [5]. The current study is the first to provide a detailed description of the SP-D response and regulation in both BAL and serum during different acute and chronic lung injury models in the same study. Unlike previous mouse studies that often applied relative measurements of SP-D levels, the strength of current study is the simultaneous quantitative ELISA measurements of both BAL and serum levels of SP-D. We conclude that serum levels of SP-D increase especially during chronic lung injury than in acute injury. The quick upregulation of serum SP-D in response to acute airway inflammation, which occurs immediately at the end of LPS challenge at 3 h, supports the notion that SP-D translocates from the airways into the vascular system, in favor of being synthesized systemically. At this time point, upregulation of SP-D synthesis and secretion has not even been effectuated. The study also confirms the concept of using increased SP-D serum levels as a biomarker of chronic airway inflammation.
Acknowledgments
We thank technicians Jette Brandt (University of Southern Denmark) and Vivi Schmidt (University of Southern Denmark) for help with establishing and continuously validating the SP-D ELISA. We thank professors W. Michael Foster and Jo Rae Wright (Duke University Medical Center) for help with establishing the Hinner exposure chamber and protocol for LPS exposure. This study was part of a Ph.D. study supported financially by the Department of Otorhinolaryngology, Odense University Hospital, University of Southern Denmark and the Region of Southern Denmark. We thank the Hoejbjerg Foundation and the A.P. Moeller Foundation for financial support to obtain materials and reagents. This work was supported by NIH ES P30-013508. The supporting organizations had no part in the protocol or conductance of the study and had no financial interest in the outcome.
Footnotes
Ethical Standards
The Institutional Animal Care and Use Committees of the University of Pennsylvania and the Danish National Council for Animal Testing have approved the procedures for the use of mice. Thus, the authors declare that the experiments comply with the current laws of the country in which they were performed.
Conflict of interest The authors have no conflicts of interest to disclose.
Contributor Information
Maria Quisgaard Gaunsbaek, Email: maria.quisgaard@ouh.regionsyddanmark.dk, Department of Otorhinolaryngology, Odense University Hospital, Sdr. Boulevard 29, 5000 Odense C, Denmark.
Karina Juhl Rasmussen, Department of Cancer and Inflammation Research, Institute of Molecular Medicine, University of Southern Denmark, Winsloevsparken 21, 1, 5000 Odense C, Denmark.
Michael F. Beers, Pulmonary and Critical Care Division, University of Pennsylvania, Surfactant Biology Laboratories, Translational Research Center, Room 11-104, 3400 Civic Center Blvd., Philadelphia, PA 19104-5159, USA
Elena N. Atochina-Vasserman, Pulmonary and Critical Care Division, University of Pennsylvania, Surfactant Biology Laboratories, Translational Research Center, Room 11-104, 3400 Civic Center Blvd., Philadelphia, PA 19104-5159, USA
Soren Hansen, Department of Cancer and Inflammation Research, Institute of Molecular Medicine, University of Southern Denmark, Winsloevsparken 21, 1, 5000 Odense C, Denmark.
References
- 1.Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol. 2003;21:547–578. doi: 10.1146/annurev.immunol.21.120601.140954. [DOI] [PubMed] [Google Scholar]
- 2.Lahti M, Lofgren J, Marttila R, Renko M, Klaavuniemi T, Haataja R, et al. Surfactant protein D gene polymorphism associated with severe respiratory syncytial virus infection. Pediatr Res. 2002;51:696–699. doi: 10.1203/00006450-200206000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Leth-Larsen R, Nordenbaek C, Tornoe I, Moeller V, Schlosser A, Koch C, et al. Surfactant protein D (SP-D) serum levels in patients with community-acquired pneumonia small star, filled. Clin Immunol. 2003;108:29–37. doi: 10.1016/s1521-6616(03)00042-1. [DOI] [PubMed] [Google Scholar]
- 4.Greene KE, Wright JR, Steinberg KP, Ruzinski JT, Caldwell E, Wong WB, et al. Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am J Respir Crit Care Med. 1999;160:1843–1850. doi: 10.1164/ajrccm.160.6.9901117. [DOI] [PubMed] [Google Scholar]
- 5.Eisner MD, Parsons P, Matthay MA, Ware L, Greene K. Plasma surfactant protein levels and clinical outcomes in patients with acute lung injury. Thorax. 2003;58:983–988. doi: 10.1136/thorax.58.11.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betsuyaku T, Kuroki Y, Nagai K, Nasuhara Y, Nishimura M. Effects of ageing and smoking on SP-A and SP-D levels in bronchoalveolar lavage fluid. Eur Respir J. 2004;24:964–970. doi: 10.1183/09031936.04.00064004. [DOI] [PubMed] [Google Scholar]
- 7.Cheng G, Ueda T, Numao T, Kuroki Y, Nakajima H, Fukushima Y, et al. Increased levels of surfactant protein A and D in bronchoalveolar lavage fluids in patients with bronchial asthma. Eur Respir J. 2000;16:831–835. doi: 10.1183/09031936.00.16583100. [DOI] [PubMed] [Google Scholar]
- 8.Noah TL, Murphy PC, Alink JJ, Leigh MW, Hull WM, Stahlman MT, et al. Bronchoalveolar lavage fluid surfactant protein-A and surfactant protein-D are inversely related to inflammation in early cystic fibrosis. Am J Respir Crit Care Med. 2003;168:685–691. doi: 10.1164/rccm.200301-005OC. [DOI] [PubMed] [Google Scholar]
- 9.Postle AD, Mander A, Reid KB, Wang JY, Wright SM, Moustaki M, et al. Deficient hydrophilic lung surfactant proteins A and D with normal surfactant phospholipid molecular species in cystic fibrosis. Am J Respir Cell Mol Biol. 1999;20:90–98. doi: 10.1165/ajrcmb.20.1.3253. [DOI] [PubMed] [Google Scholar]
- 10.Honda Y, Kuroki Y, Matsuura E, Nagae H, Takahashi H, Akino T, et al. Pulmonary surfactant protein D in sera and bronchoalveolar lavage fluids. Am J Respir Crit Care Med. 1995;152:1860–1866. doi: 10.1164/ajrccm.152.6.8520747. [DOI] [PubMed] [Google Scholar]
- 11.Honda Y, Takahashi H, Kuroki Y, Akino T, Abe S. Decreased contents of surfactant proteins A and D in BAL fluids of healthy smokers. Chest. 1996;109:1006–1009. doi: 10.1378/chest.109.4.1006. [DOI] [PubMed] [Google Scholar]
- 12.Lomas DA, Silverman EK, Edwards LD, Locantore NW, Miller BE, Horstman DH, et al. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur Respir J. 2009;34:95–102. doi: 10.1183/09031936.00156508. [DOI] [PubMed] [Google Scholar]
- 13.Guo CJ, Atochina-Vasserman EN, Abramova E, Foley JP, Zaman A, Crouch E, et al. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol. 2008;6:e266. doi: 10.1371/journal.pbio.0060266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atochina-Vasserman EN. S-nitrosylation of surfactant protein D as a modulator of pulmonary inflammation. Biochim Biophys Acta. 2012;1820:763–769. doi: 10.1016/j.bbagen.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winkler C, Atochina-Vasserman EN, Holz O, Beers MF, Erpenbeck VJ, Krug N, et al. Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir Res. 2011;12:29. doi: 10.1186/1465-9921-12-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mutti A, Corradi M, Goldoni M, Vettori MV, Bernard A, Apostoli P. Exhaled metallic elements and serum pneumoproteins in asymptomatic smokers and patients with COPD or asthma. Chest. 2006;129:1288–1297. doi: 10.1378/chest.129.5.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirama N, Shibata Y, Otake K, Machiya J, Wada T, Inoue S, et al. Increased surfactant protein-D and foamy macrophages in smoking-induced mouse emphysema. Respirology. 2007;12:191–201. doi: 10.1111/j.1440-1843.2006.01009.x. [DOI] [PubMed] [Google Scholar]
- 18.Homer RJ, Zheng T, Chupp G, He S, Zhu Z, Chen Q, et al. Pulmonary type II cell hypertrophy and pulmonary lipoproteinosis are features of chronic IL-13 exposure. Am J Physiol Lung Cell Mol Physiol. 2002;283:52–59. doi: 10.1152/ajplung.00438.2001. [DOI] [PubMed] [Google Scholar]
- 19.Ikegami M, Whitsett JA, Chroneos ZC, Ross GF, Reed JA, Bachurski CJ, et al. IL-4 increases surfactant and regulates metabolism in vivo. Am J Physiol Lung Cell Mol Physiol. 2000;278:75–80. doi: 10.1152/ajplung.2000.278.1.L75. [DOI] [PubMed] [Google Scholar]
- 20.Sorensen GL, Husby S, Holmskov U. Surfactant protein A and surfactant protein D variation in pulmonary disease. Immunobiology. 2007;212:381–416. doi: 10.1016/j.imbio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Kingma PS, Zhang L, Ikegami M, Hartshorn K, McCormack FX, Whitsett JA. Correction of pulmonary abnormalities in Sftpd−/− mice requires the collagenous domain of surfactant protein D. J Biol Chem. 2006;281:24496–24505. doi: 10.1074/jbc.M600651200. [DOI] [PubMed] [Google Scholar]
- 22.Hansen S, Schmidt V, Steffensen MA, Jensen PH, Gjerstorff M, Thiel S, et al. An enzyme-linked immunosorbent assay (ELISA) for quantification of mouse surfactant protein D (SP-D) J Immunol Methods. 2008;330:75–85. doi: 10.1016/j.jim.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Hollingsworth JW, Cook DN, Brass DM, Walker JK, Morgan DL, Foster WM, et al. The role of Toll-like receptor 4 in environmental airway injury in mice. Am J Respir Crit Care Med. 2004;170:126–132. doi: 10.1164/rccm.200311-1499OC. [DOI] [PubMed] [Google Scholar]
- 24.Fujita M, Shannon JM, Ouchi H, Voelker DR, Nakanishi Y, Mason RJ. Serum surfactant protein D is increased in acute and chronic inflammation in mice. Cytokine. 2005;31:25–33. doi: 10.1016/j.cyto.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 25.Atochina-Vasserman EN, Gow AJ, Abramova H, Guo CJ, Tomer Y, Preston AM, et al. Immune reconstitution during Pneumocystis lung infection: disruption of surfactant component expression and function by S-nitrosylation. J Immunol. 2009;182:2277–2287. doi: 10.4049/jimmunol.0802775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Casey J, Kaplan J, Atochina-Vasserman EN, Gow AJ, Kadire H, Tomer Y, et al. Alveolar surfactant protein D content modulates bleomycin-induced lung injury. Am J Respir Crit Care Med. 2005;172:869–877. doi: 10.1164/rccm.200505-767OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atochina EN, Beck JM, Scanlon ST, Preston AM, Beers MF. Pneumocystis carinii pneumonia alters expression and distribution of lung collectins SP-A and SP-D. J Lab Clin Med. 2001;137:429–439. doi: 10.1067/mlc.2001.115220. [DOI] [PubMed] [Google Scholar]
- 28.Braber S, Verheijden KA, Henricks PA, Kraneveld AD, Folkerts G. A comparison of fixation methods on lung morphology in a murine model of emphysema. Am J Physiol Lung Cell Mol Physiol. 2010;299:843–851. doi: 10.1152/ajplung.00192.2010. [DOI] [PubMed] [Google Scholar]
- 29.Selman L, Skjodt K, Nielsen O, Floridon C, Holmskov U, Hansen S. Expression and tissue localization of collectin placenta 1 (CL-P1, SRCL) in human tissues. Mol Immunol. 2008;45:3278–3288. doi: 10.1016/j.molimm.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 30.Madsen J, Kliem A, Tornoe I, Skjodt K, Koch C, Holmskov U. Localization of lung surfactant protein D on mucosal surfaces in human tissues. J Immunol. 2000;164:5866–5870. doi: 10.4049/jimmunol.164.11.5866. [DOI] [PubMed] [Google Scholar]
- 31.Beers MF, Atochina EN, Preston AM, Beck JM. Inhibition of lung surfactant protein B expression during Pneumocystis carinii pneumonia in mice. J Lab Clin Med. 1999;133:423–433. doi: 10.1016/s0022-2143(99)90019-7. [DOI] [PubMed] [Google Scholar]
- 32.Suda K, Tsuruta M, Eom J, Or C, Mui T, Jaw JE, et al. Acute lung injury induces cardiovascular dysfunction: effects of IL-6 and budesonide/formoterol. Am J Respir Cell Mol Biol. 2011;45:510–516. doi: 10.1165/rcmb.2010-0169OC. [DOI] [PubMed] [Google Scholar]
- 33.Zhang F, Pao W, Umphress SM, Jakowlew SB, Meyer AM, Dwyer-Nield LD, et al. Serum levels of surfactant protein D are increased in mice with lung tumors. Cancer Res. 2003;63:5889–5894. [PubMed] [Google Scholar]
- 34.Haczku A, Atochina EN, Tomer Y, Chen H, Scanlon ST, Russo S, et al. Aspergillus fumigatus-induced allergic airway inflammation alters surfactant homeostasis and lung function in BALB/c mice. Am J Respir Cell Mol Biol. 2001;25:45–50. doi: 10.1165/ajrcmb.25.1.4391. [DOI] [PubMed] [Google Scholar]
- 35.Pan T, Nielsen LD, Allen MJ, Shannon KM, Shannon JM, Selman M, et al. Serum SP-D is a marker of lung injury in rats. Am J Physiol Lung Cell Mol Physiol. 2002;282:824–832. doi: 10.1152/ajplung.00421.2000. [DOI] [PubMed] [Google Scholar]
- 36.Atochina EN, Beers MF, Scanlon ST, Preston AM, Beck JM. P. carinii induces selective alterations in component expression and biophysical activity of lung surfactant. Am J Physiol Lung Cell Mol Physiol. 2000;278:599–609. doi: 10.1152/ajplung.2000.278.3.L599. [DOI] [PubMed] [Google Scholar]
- 37.Holmskov U. Lung surfactant proteins (SP-A and SP-D) in non-adaptive host responses to infection. J Leukoc Biol. 1999;66(5):747–752. doi: 10.1002/jlb.66.5.747. [DOI] [PubMed] [Google Scholar]
- 38.Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, et al. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell. 2003;115:13–23. doi: 10.1016/s0092-8674(03)00758-x. [DOI] [PubMed] [Google Scholar]