
Figure 4.
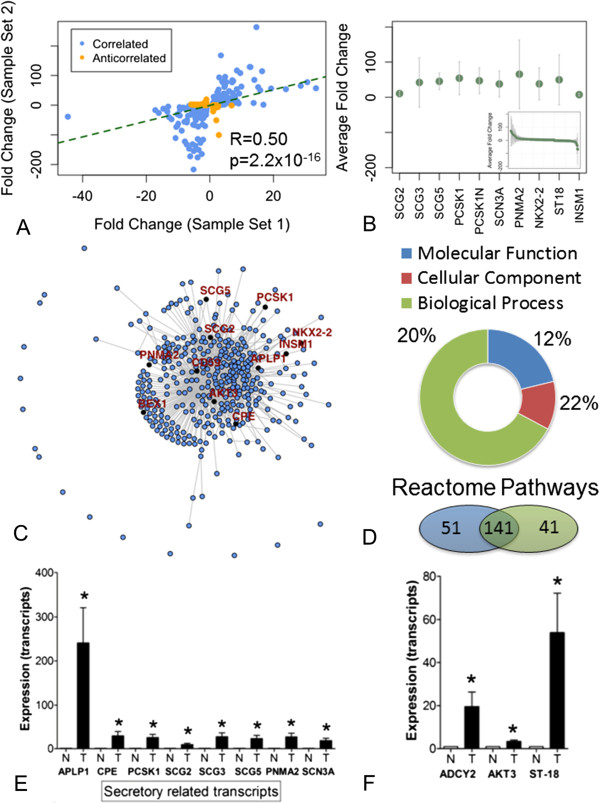
Co-analyses of the two small intestinal NET sets. A. Correlation profile of transcript alterations in each of the tumor sets. Both tissue databases were marginally correlated (R = 0.50). B. Commonly elevated transcripts in both datasets predominantly include genes involved in neuroendocrine secretion and regulation thereof. Error bars indicate the range of fold changes across the two datasets, while green points reflect average gene expression. C. Network analysis of the top ranked genes (see B) identified the most densely connected module to be related to secretion (interactome identified by multiple links). D. Gene-ontology and Reactome pathway demonstrating overlap between the two tumor sets; common pathways included secretion and xenobiotic responses (toxic environmental chemicals) as well as neurodevelopmental gene expression and alternative metabolic cycling (urea and TCA) consistent with a hypoxic phenotype (see Additional file 5: Table S1 and Additional file 6: Table S2). E. QPCR analysis of secretome-related transcripts in the independent set identified significant over-expression of all eight genes (ranging from APLP1 to SCN3A). *p <0.05 vs. normal mucosa. 3F. QPCR analysis of highly expressed transcripts in the independent set identified significant over-expression of ADCY2, AKT3 and ST18. Mean ± SEM, *p < 0.05 vs. normal mucosa. Tumors n = 13, normal mucosa n = 8.