
Figure 2.
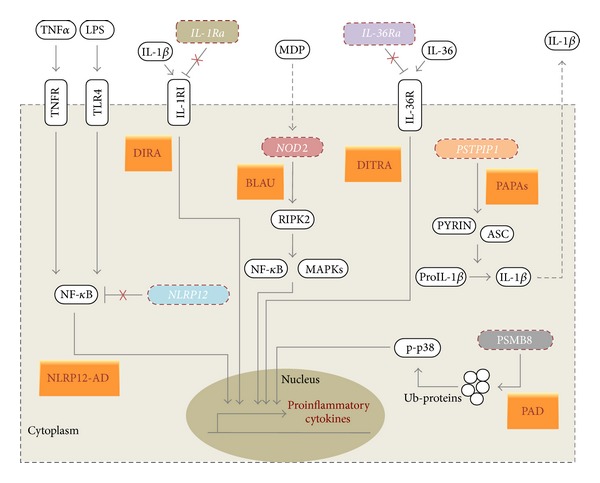
Schematic representation of the main pathophysiologic mechanisms involved in the less frequently diagnosed systemic autoinflammatory diseases. Blau syndrome (BLAU) and NLRP12-associated autoinflammatory disorder (NLRP12-AD) are caused by altered nuclear factor-κB (NF-κB) regulation, caused, respectively, by NOD2 and NLRP12 gene mutations. Deficiency of the interleukin-1 (IL-1) receptor antagonist (DIRA) and deficiency of interleukin-36 (IL-36) receptor antagonist (DITRA) are linked to mutations of genes coding, respectively, for IL-1 receptor antagonist (IL-1Ra) and IL-36 receptor antagonist (IL-36Ra). These mutations lead to the loss of IL-1β and IL-36 natural inhibition, resulting in uncontrolled proinflammatory responses. PAPA syndrome (PAPAs) is associated with increased IL-1β processing and secretion, as a consequence of proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1) gene mutations. Proteasome-associated diseases (PAD) are caused by mutations in the proteasome subunit beta type 8 (PSMB8), which lead to the accumulation of ubiquitinated proteins (Ub-proteins) and in turn to p38 phosphorylation, resulting in enhanced proinflammatory responses.