
Figure 4.
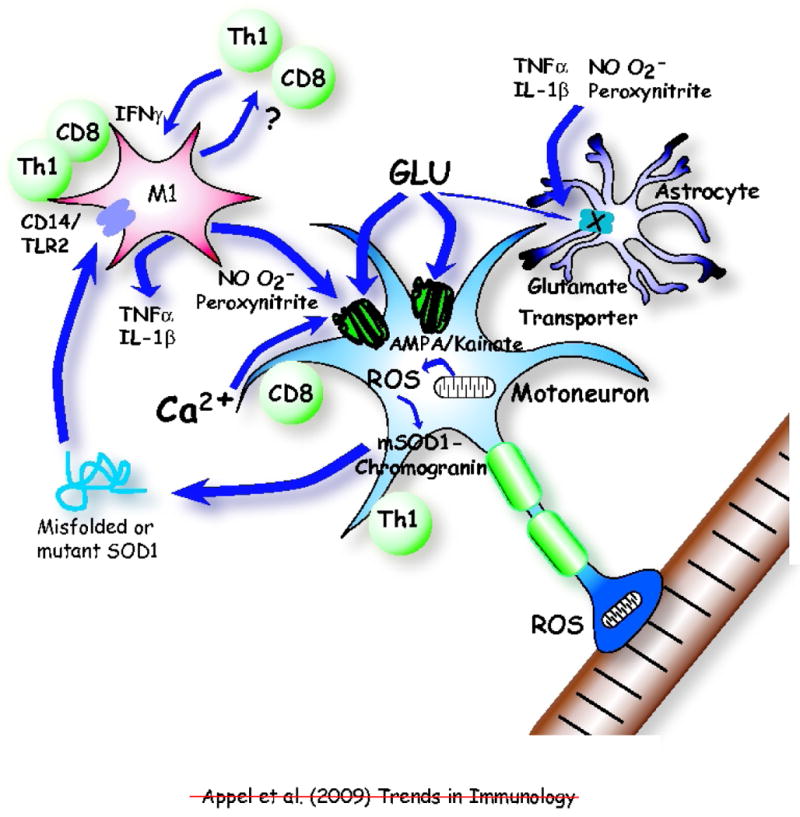
An immunological shift from protection to neurotoxicity accelerates disease in amyotrophic lateral sclerosis (ALS). Initial presentation of mutant superoxide dismutase (mSOD1) or oxidized and misfolded wild-type (WT) SOD1 to T cells might occur in the periphery. Later in disease, as the concentration of extracellular mutant or misfolded SOD1 accumulates, this now rogue protein activates the CD14/Toll-like receptors (TLRs) on microglia transforming them from a protective M2 phenotype to a neurotoxic M1 phenotype. Microglia may also re-present the rogue protein to T cells (Th1 and CD8). In vitro, the M1 microglia release the pro-inflammatory TNF-α and IL-1β cytokines and the reactive oxygen species (ROS) nitric oxide (NO) and superoxide (O2•−), which may form the highly reactive and toxic peroxynitrite. These ROS alter the motoneuron AMPA/kainate channel, resulting in increased glutamate (GLU) susceptibility and subsequent calcium (Ca2+) influx cytotoxicity. Extracellular glutamate concentrations may increase due to reduced numbers and abilities of the glutamate transporters expressed by astrocytes to remove glutamate resulting in further glutamate toxicity. The pro-inflammatory cytokines and ROS may also render the astroglial glutamate transporters ineffective further compounding the Ca2+-induced neurotoxicity. The M1 microglia also influence Th1 and CD8 T cells, which in turn maintain M1 microglial phenotype through the release of IFN-γ and may also exert direct toxic effects on motoneurons.