

Summary
Although the basic mechanisms of prokaryotic transcription are conserved, it has become evident that some bacteria require additional factors to allow for efficient gene transcription. CarD is an RNA polymerase (RNAP) binding protein conserved in numerous bacterial species and essential in mycobacteria. Despite the importance of CarD, its function at transcription complexes remains unclear. We have generated a panel of mutations that individually target three independent functional modules of CarD: the RNAP interaction domain, the DNA binding domain, and a conserved tryptophan residue. We have dissected the roles of each functional module in CarD activity and built a model where each module contributes to stabilizing RNAP-promoter complexes. Our work highlights the requirement of all three modules of CarD in the obligate pathogen Mycobacterium tuberculosis, but not in Mycobacterium smegmatis. We also report divergent use of the CarD functional modules in resisting oxidative stress and pigmentation. These studies provide new information regarding the functional domains involved in transcriptional regulation by CarD while also improving understanding of the physiology of M. tuberculosis.
Keywords: Infection, Mycobacteria, Stress Response, Transcription, Tuberculosis, rRNA
Introduction
The World Health Organization reported 8.6 million new cases of tuberculosis (TB) in 2012, contributing to the 2 billion people infected with Mycobacterium tuberculosis worldwide and 1.3 million TB-related deaths that year (WHO, 2013). Control of the M. tuberculosis epidemic is further complicated by the alarming rise in multidrug-resistant tuberculosis cases, which accounted for 450,000 new infections in 2012 (WHO, 2013). There is growing concern that, without the development of new antibiotics, we are not equipped with the tools to battle this epidemic. Insight into the pathways involved in mycobacterial pathogenesis is required to develop new chemotherapeutic strategies for TB. Previous studies have shown that the transcriptional regulator CarD is required for M. tuberculosis survival during oxidative stress, treatment with certain antibiotics, and acute and persistent infection of mice (Stallings et al., 2009; Weiss et al., 2012). As CarD is not conserved in eukaryotes, targeting CarD with small molecules is an attractive treatment strategy. Notably, CarD proteins are ubiquitous in mycobacteria and conserved in many other eubacteria (Stallings et al., 2009; Srivastava et al., 2013), indicating that new therapeutic strategies involving CarD will not only be relevant to mycobacterial infections, but may also be efficacious for diverse bacterial pathogens that encode CarD homologs. However, developing inhibitors of CarD requires understanding of which of CarD’s activities are essential.
CarD interacts directly with the β1 region of the RNA polymerase (RNAP) β subunit through its N-terminal RNAP interaction domain (RID) (Stallings et al., 2009; García-Moreno et al., 2010; Weiss et al., 2012). Chromatin immunoprecipitation-sequencing (ChIP-seq) experiments demonstrated that CarD is localized to promoters throughout the genome, indicating that CarD is a global regulator of transcription initiation (Srivastava et al., 2013). In addition, mycobacterial strains expressing mutants of CarD with weakened affinity for the RNAP βsubunit display lower levels of promoter activation in promoter-lacZ fusion experiments as compared to strains expressing wild-type (WT) CarD (Srivastava et al., 2013). Recently, we have also shown that CarD stabilizes RNAP-promoter complexes during in vitro transcription experiments and its association with the RNAP β subunit is required for this activity (Srivastava et al., 2013). Most of our knowledge regarding the mechanisms of transcription comes from studies performed in model organisms, including studies in Escherichia coli to understand prokaryotic transcription. Recently, it has become clear that mycobacteria differ substantially from the model organisms traditionally used to study transcription. For instance, many of the sequences and proteins required to regulate transcription initiation in E. coli are absent in mycobacteria, including Fis (Johnson and Simon, 1985; Kahmann et al., 1985; Johnson et al., 1986; Koch and Kahmann, 1986), DksA (Kang and Craig, 1990), AT-rich upstream activating elements (Newlands et al., 1992; Rao et al., 1994), and GC-rich discriminator sequences (Travers, 1980). The identification of CarD as a protein that is not present in E. coli but is essential for transcription regulation in mycobacteria is an example of the importance of investigating prokaryotic transcription in diverse bacterial species.
Understanding of CarD has hitherto been limited to the RNAP binding activity encoded by the N-terminus of the protein since the role of the C-terminus of CarD had remained elusive. Recently, crystal structures of both the Thermus thermophilus CarD and the M. tuberculosis CarD have been solved (Srivastava et al., 2013; Gulten and Sacchettini, 2013; Kaur et al., 2014) as well as a crystal structure of the C-terminus of CarD in isolation of the first 82 amino acids (Gangwar et al., 2014). The crystal structures of full length CarD confirmed the predicted structure of the N-terminal RID and revealed a compact helical C-terminal domain. Since ChIP-seq experiments showed CarD was present at promoter sequences (Srivastava et al., 2013), we modeled CarD onto the structure of the RNAP promoter initiation complex based on the well- characterized interaction between the CarD-RID and RNAP-β (Weiss et al., 2012). This model predicted that the C-terminus of CarD may act as a DNA binding domain, specifically interacting with DNA via a conserved tryptophan residue and a surrounding basic patch at the distal tip of the C-terminal alpha helices (Srivastava et al., 2013). Electrophoretic mobility shift assays (EMSAs) confirmed that CarD is capable of binding and shifting double stranded DNA (dsDNA) in a sequence-independent manner and the C-terminal residues 83–162 are sufficient for this activity (Srivastava et al., 2013; Gulten and Sacchettini, 2013). These studies still left many open questions including what residues within CarD were necessary for its interaction with DNA, what the roles of this interaction are in CarD-mediated transcriptional regulation and pathogenesis, and how do the CarD/RNAP and CarD/DNA interactions affect one another. We have addressed each of these open questions in this manuscript by isolating single point mutants in the CarD C-terminus and using these mutants to parse out the role of the CarD C-terminus in DNA binding, bacterial stress responses, pathogenesis, and regulation of transcription. In addition, we have begun to clarify the relationship between the RNAP binding and DNA binding activities of CarD. Our studies demonstrate that the conserved tryptophan residue in CarD that was previously implicated in binding DNA does not phenotypically group with the mutants that abolish the dsDNA interaction and performs a function that is important for CarD activity but is distinct from the dsDNA and RNAP binding activities. Therefore, this is the first report of an in vivo analysis of the CarD C-terminal mutants and has revealed a functional distinction between the conserved tryptophan and the surrounding basic patch in the C-terminus of CarD. Together our studies dissect three functional regions of CarD, elucidate their interplay as it relates to CarD activity, and propose a model of how CarD modifies transcription initiation complexes. These studies represent a step in understanding the mechanism of transcriptional regulation by CarD.
Results
Single point mutations in the CarD C-terminal basic patch abolish the interaction between CarD and DNA in vitro
To determine the residues in the CarD C-terminus that were responsible for the interaction with DNA, we purified CarD proteins harboring a single mutation in the tryptophan or lysine residues (W85, K90, and K125 (Fig. 1A)) that are highly conserved among CarD homologs (Srivastava et al., 2013). EMSAs were performed to assay the effect of mutating W85, K90, and K125 on the ability of CarD to bind the DNA fragment rrnAPL, which contains the promoters and leader sequences of the M. smegmatis ribosomal RNA (rRNA) rrnA operon. This is the same DNA fragment that was used in the experiments that identified the C-terminus of CarD as a DNA binding domain (Srivastava et al., 2013) and was originally chosen due to CarD’s described role in regulating rRNA transcription (Stallings et al., 2009; Weiss et al., 2012). The CarDWT, CarDW85A, and CarDK125A proteins were all able to bind dsDNA, as determined by a decrease in the unbound DNA band and the appearance of a slower migrating protein-bound DNA band in the EMSA (Fig. 1B). In contrast, the CarDK90A, CarDK90E, and CarDK125E mutants lost the ability to bind DNA, as determined by the dramatic decrease in the slower migrating protein-bound DNA band and the retention of the unbound DNA band in the EMSA (Fig. 1B). Therefore, the conserved lysine residues in the basic patch are essential for binding DNA and single point mutations in the basic patch compromise this activity.
Figure 1. CarD’s C-terminal basic patch is responsible for its interaction with DNA.
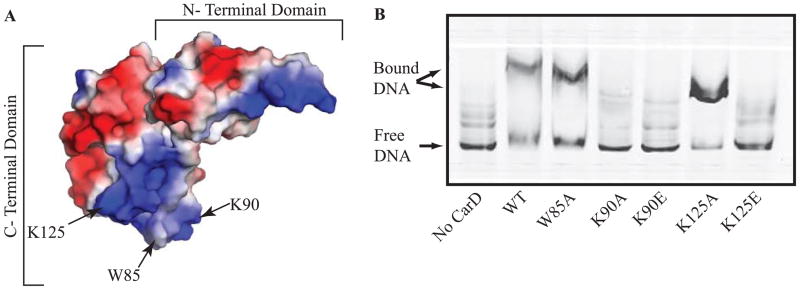
A. Electrostatic surface diagram of M. tuberculosis CarD modified from (Srivastava et al., 2013) illustrating the location of W85, K90, and K125 within the basic patch.
B. Image of a nondenaturing polyacrylamide gel from an EMSA with no protein, M. smegmatis CarDWT, CarDW85A, CarDK90A, CarDK90E, CarDK125A, or CarDK125E incubated with IRDye labeled M. smegmatis rrnAPL DNA. The reactions were separated on a nondenaturing polyacrylamide gel, which was then imaged using the Odyssey CLX imaging system (LI-COR).
Disrupting the interaction between CarD and DNA does not affect the association of CarD with the RNAP but is detrimental for growth and viability
To investigate the role of the interaction between CarD and DNA in vivo, we attempted to replace the carD gene in M. smegmatis and M. tuberculosis with alleles encoding CarDK90A, CarDK90E, CarDK125A, or CarDK125E using a gene switching technique (Pashley and Parish, 2003; Weiss et al., 2012). We successfully obtained all 4 mutants in M. smegmatis (Table 1). However, in M. tuberculosis we were only able to replace the carDWT gene with the carDK125A allele, which encodes the mutant that retains DNA binding activity in vitro during EMSA experiments (Fig. 1B and Table 1). In order to study the roles of the conserved tryptophan in CarD, we also attempted to replace the carD gene in M. smegmatis and M. tuberculosis with an allele encoding CarDW85A. We were able to engineer M. smegmatis strains expressing the CarDW85A mutant, but were unable to replace the M. tuberculosis carD allele with the carDW85A allele (Table 1). We had previously shown that M. tuberculosis was more sensitive to interfering with the RNAP binding activity of CarD than M. smegmatis (Weiss et al., 2012). These studies demonstrate that M. tuberculosis is also more sensitive to interfering with CarD’s DNA binding activity and the conserved tryptophan, supporting the observation that M. tuberculosis has more stringent requirements for CarD’s activities than M. smegmatis.
Table 1.
CarD mutants studied in this paper
| Mutation in CarD | M. smegmatis | M. tuberculosis | Activity Affected by Mutation |
|---|---|---|---|
| R25E | Viable | Unattainable | Interaction with RNAP |
| R47E | Viable | Viable | Interaction with RNAP |
| W85A | Viable | Unattainable | Unknown |
| K90A | Viable | Unattainable | Interaction with dsDNA |
| K90E | Viable | Unattainable | Interaction with dsDNA |
| K125A | Viable | Viable | None Determined |
| K125E | Viable | Unattainable | Interaction with dsDNA |
The possibility existed that changing the charge of the C-terminal basic patch would affect the stability or folding of CarD. In order to address this possibility, we constructed M. smegmatis strains expressing HA tagged CarDWT or CarD mutants and analyzed the amount of each mutant protein present in the cell by western blot (Fig. 2A) as a readout for protein stability. Using these same strains, we also performed co-immunoprecipitation experiments to determine if the CarD C-terminal point mutants retain the ability to associate with the RNAP β subunit (Fig. 2A) as a readout for proper folding. All of the C-terminal mutants were stably expressed in the bacteria and associated with RNAP β to the same degree as WT CarD except for CarDK90E, which was barely detectable in the cell lysate suggesting that stability of the protein is affected by this substitution (Fig. 2A). Therefore, we chose not to pursue this mutant further. Alanine substitution at the conserved tryptophan (CarDW85A) did not affect the amount of RNAP associated with CarD, demonstrating that not only does this residue not have a strong impact on binding dsDNA (Fig. 1B), it is also not involved in binding the RNAP (Fig. 2A) and indicates that this residue serves a unique function for CarD activity.
Figure 2. Each of CarD’s functional domains is required for optimal growth in mycobacteria.

A. Immunoprecipitation experiments with a monoclonal antibody specific for HA in the M. smegmatis strains expressing CarDWT-HA (lane 1), CarDR25E-HA (lane 2), CarDK90A-HA (lane 3), CarDK90E-HA (lane 4), CarDW85A-HA (lane 5), CarDK125A-HA (lane 6), or CarDK125E-HA (lane 7). Inputs (before immunoprecipitation) and eluates were analyzed by western blotting with antibodies specific for either RNAP β (Panel A and D) or CarD (Panel B, C, and E). Panel C is a longer exposure of the film from panel B in order to show the CarDK90E band.
B–C. The doubling time of each CarD strain was expressed as a ratio to the doubling time of the CarDWT expressing strain for the M. smegmatis strains expressing CarDWT, CarDR25E, CarDK90A, CarDW85A, CarDK125A, or CarDK125E (B) and M. tuberculosis strains expressing CarDWT, CarDK125A, or CarDR47E (C). Each graph shows the mean ± SEM of data from at least three replicates. Significance of the differences between mutant strains and WT were determined by calculating P values by Student’s t test. An asterisk indicates significance with a P value of <0.05, two asterisks indicate significance with a P value of <0.01, and three asterisks indicate significance with a P value of <0.005.
D. Plated dilutions of M. smegmatis strains expressing CarDWT, CarDR25E, CarDW85A, CarDK90A, CarDK125A, or CarDK125E on LB after 3 days of growth at 37°C.
E. Image of nondenaturing polyacrylamide gel from EMSAs with no protein, M. tuberculosis CarDWT, CarDR25E, or CarDR47E incubated with IRDye labeled M. smegmatis rrnAPL DNA. The reactions were separated on a nondenaturing polyacrylamide gel, which was then imaged using the Odyssey CLX imaging system (LI-COR).
To determine whether the CarD-DNA interaction and the conserved tryptophan in CarD are important for optimal growth, we measured the growth rate of the viable M. smegmatis (Fig. 2B) and M. tuberculosis (Fig. 2C) CarD mutants. All strains used in this manuscript contain only one carD allele. Based on at least 3 replicate experiments, M. smegmatis strains expressing the CarDW85A, CarDK90A, or CarDK125E mutant grew 1.23 (standard deviation, 0.084), 1.26 (standard deviation, 0.069), or 1.31 (standard deviation, 0.404) times slower than control strains expressing CarDWT, respectively (Fig. 2B). This degree of decrease in growth rate is similar to what was observed previously with the CarDR25E mutant with weakened affinity to RNAP β (Fig. 2B and (Weiss et al., 2012)). Therefore, even though these M. smegmatis strains are viable, efficient DNA binding and the conserved tryptophan are still required for optimal growth.
The K125A substitution in CarD, which did not affect DNA binding in EMSA experiments, slowed the growth of both M. smegmatis (Fig. 2B) and M. tuberculosis (Fig. 2C). Specifically, the M. smegmatis CarDK125A expressing strain grew 1.18 times slower than the control strain (standard deviation, 0.048) and the M. tuberculosis CarDK125A expressing strain grew 1.35 times (standard deviation, 0.026) slower than the control strain. This indicates that this mutation does affect CarD’s function, despite maintaining WT levels of DNA binding in the EMSA assay, supporting the importance of this lysine residue for CarD activity.
The growth defects were even more apparent on solid media where the colonies of the M. smegmatis CarDR25E, CarDW85A, CarDK90A, and CarDK125E mutant expressing strains were smaller than those of the WT strain, as illustrated in Fig. 2D at the 10−3 and 10−4 dilutions. In addition, we noticed that the colonies from the strains expressing the CarDR25E RID mutant and the CarDW85A mutant had defects in pigmentation, while this was not observed in the strains expressing CarD mutants that loss the ability to bind dsDNA. This illustrates an important distinction between the conserved tryptophan and the lysine residues involved in interacting with DNA in that mutating the tryptophan residue does not phenocopy the DNA binding mutants but rather appears more like a RID mutant without affecting the interaction with RNAP. Therefore, this supports that the tryptophan and lysine residues confer different functions for CarD.
Our discovery of CarD’s DNA binding activity also raised the question of whether the previously described mutations in the RID domain that weakened the interaction between CarD and the RNAP β subunit (Weiss et al., 2012) also affected the interaction with DNA. To examine this possibility, we investigated the DNA binding activity of the CarDR25E and CarDR47E mutants in EMSAs (Fig. 2E). These experiments showed that the CarDR25E and CarDR47E mutants retained the ability to bind and shift the rrnAPL DNA fragment, as evidenced by both the accumulation of a slower migrating complex containing the labeled DNA and the disappearance of the band corresponding to the unbound DNA fraction in these samples. The altered migration of the CarD-DNA complexes formed by these mutants as compared to CarDWT may be due to the altered net charge in the CarD mutants or changes in cooperativity of the sequence nonspecific binding of CarD. Regardless, the retained DNA binding by these mutants indicates that these substitutions in the CarD RID do not affect the ability of CarD to bind DNA in the absence of RNAP. Therefore, the RNAP and DNA binding activities of CarD are able to function independently of each other.
The CarD-DNA interaction is dispensable for resistance to killing by oxidative stress
We have previously shown that both depletion of CarD and mutations that weaken the interaction between CarD and the RNAP dramatically compromise survival upon exposure to oxidative stress (Fig. 3 and (Stallings et al., 2009; Weiss et al., 2012)), a stress encountered by M. tuberculosis during infection (Cooper et al., 2000). To test whether the interaction between CarD and DNA is necessary for survival during oxidative stress, log-phase cultures of M. smegmatis strains expressing different carD alleles were treated with 25 mM H2O2 for 1 hour before dilutions were plated to count the surviving colony forming units (CFU) (Fig. 3). Strains expressing the CarDK90A or CarDK125E mutants, which lose the ability to bind DNA in EMSA experiments, were as resistant to oxidative stress as CarDWT expressing strains, indicating that the interaction between CarD and DNA is not required for the response to reactive oxygen species. These results further support that the CarD-RNAP and CarD-DNA interactions are independent, as mutations that weaken the interaction with RNAP but not with DNA cause sensitivity to reactive oxygen species. Interestingly, the W85A mutation in CarD sensitizes M. smegmatis to oxidative stress to a similar degree as mutations that weaken the interaction with RNAP (Fig. 3), but without affecting association with the RNAP (Fig. 2A). Therefore, the W85A substitution in CarD diverges from the CarD DNA binding mutants in the C-terminal basic patch in terms of the effect on pigmentation and withstanding exposure to reactive oxygen species and we conclude that this conserved tryptophan is performing another function in CarD distinct from the RNAP and dsDNA binding activity of this protein. These data demonstrate that CarD encodes three activities that are distinct and operate independently of one another: 1) interaction with the RNAP, 2) interaction with dsDNA, and 3) an activity conferred by the conserve tryptophan at position 85. The role of CarD in combating oxidative stress requires a robust interaction with the RNAP β subunit and the conserved tryptophan residue, but not efficient binding to DNA.
Figure 3.

The CarD-DNA interaction is dispensable for resistance to killing by oxidative stress. Log-phase M. smegmatis strains expressing CarDWT, CarDR25E, CarDR47E, CarDW85A, CarDK90A, CarDK125A, or CarDK125E in LB were treated for 1 hr with 25 mM H2O2. After treatment, dilutions were plated on LB and the surviving CFUs were counted. Survival of each replicate is graphed as a ratio of CFU in treated cultures to that in untreated cultures along with the mean ± SEM of the ratios for each set of replicates. Each sample is represented by a black circle.
A mutation in the DNA binding domain of CarD affects the pathogenesis of M. tuberculosis in murine tissues
We have shown that CarD is required for acute and chronic M. tuberculosis infection in mice (Stallings et al., 2009) and that weakening the interaction between CarD and the RNAP results in a decrease in bacterial burden in both the lungs and the spleen during chronic infection (Weiss et al., 2012). To investigate the effect of mutations in the DNA binding domain of CarD on pathogenesis, we infected C57BL/6 mice with M. tuberculosis strains expressing either CarDWT or the CarDK125A mutant. Although the K125A substitution did not affect DNA binding in vitro, since it is our only viable M. tuberculosis CarD C-terminal mutant, we used it to study the effect of mutations in the basic patch in vivo. The CarDK125A mutant displayed WT kinetics and levels of virulence during early acute infection in the lungs, but peaked at a bacterial burden half a log lower than the CarDWT expressing strain (Fig. 4A). The lower bacterial burden in animals infected with the M. tuberculosis CarDK125A mutant continued into the chronic phase of infection. The virulence defect of the CarDK125A strain during the chronic phase of infection was also apparent in the spleen where the titers of the CarDK125A expressing strain were more than a half log lower than the CarDWT expressing strain by 5 weeks post infection (Fig. 4B). The K125A substitution itself did not affect CarD binding to the DNA template tested in vitro (Fig. 1B). However, the loss of DNA binding by the CarDK125E mutant indicates that this lysine residue is involved in the association with DNA (Fig. 1B) and these data show that this lysine residue is also necessary for maintaining WT titers during chronic infection. Since both the CarD-RNAP and CarD-DNA interactions are required for maintaining high titers during chronic infection of mice (Fig. 4 and (Weiss et al., 2012)), this excludes the source of attenuation being sensitivity to oxidative stress, since interfering with the ability of CarD to interact with DNA does not sensitize mycobacteria to reactive oxygen species (Fig. 3). Accordingly, we have previously shown that a CarD mutant with decreased affinity for the RNAP retains a virulence defect regardless of whether the mouse is able to mount a functional phagocytic oxidative burst (Weiss et al., 2012).
Figure 4.

A mutation in the DNA binding domain of CarD affects the pathogenesis of M. tuberculosis in murine tissues. C57BL/6 mice were infected by the aerosol route with the M. tuberculosis strains expressing either CarDWT (black circles) or CarDK125A (grey squares). Shown are bacterial titers in the lungs (A) and spleens (B) of the infected mice. Each time point is the mean ± SEM of data from 6 mice per strain, combined from two experiments. The significance of the differences between the CarDK125A and the CarDWT strain in both panels were determined as described for Figure 2.
Each of CarD’s functional domains is important for resistance to clinically relevant antibiotics
We have previously shown that depleting CarD sensitizes M. smegmatis to killing by ciprofloxacin (Stallings et al., 2009) and weakening the CarD-RNAP interaction increases the sensitivity of M. smegmatis to rifampicin and streptomycin (Weiss et al., 2012). To determine if the interaction between CarD and DNA is also necessary for antibiotic tolerance, we performed zone of inhibition assays and determined the sensitivity of M. smegmatis and M. tuberculosis strains expressing WT or mutant alleles of carD to clinically relevant antibiotics. We specifically tested sensitivity to ciprofloxacin (inhibition of DNA replication), rifampicin (inhibition of transcription), and streptomycin (inhibition of translation) since perturbations in CarD function have been associated with sensitivity to these drugs (Stallings et al., 2009; Weiss et al., 2012). Each antibiotic targets a different essential cellular process, thereby testing the role of CarD’s DNA binding activity in response to antibiotics with effects on diverse cellular processes. We found that mutations in the basic patch important for interacting with DNA (K90A and K125E) caused increased sensitivity to ciprofloxacin, rifampicin, and streptomycin in M. smegmatis in zone of inhibition assays (Fig. 5A–C), indicating that the association of CarD with DNA is important in tolerating treatment with these antibiotics. Both the K90A and the K125E mutations abolish the interaction between CarD and the rrnAPL DNA fragment in EMSAs (Fig. 1B), but expression of CarDK125E causes strains to be more sensitive to rifampicin than expression of CarDK90A. This could be due to the more dramatic change in charge in the C-terminus of CarD in the K125E mutant. Expression of the CarDK125A mutant, which retains binding to the DNA template tested in vitro (Fig. 1B), also increases sensitivity of M. smegmatis to the antibiotics tested. The effect of the K125A mutation in CarD on antibiotic sensitivity, growth rate, and M. tuberculosis virulence indicates that this lysine residue is important for an activity that is affected by the alanine substitution. Using similar assays, we observed that the conserved tryptophan residue at position 85 in CarD is also important for tolerance to rifampicin, ciprofloxacin, and streptomycin in M. smegmatis and the W85A mutation results in sensitivity to these antibiotics that is comparable to mutations that either weaken the interaction with DNA (Fig. 5A–C).
Figure 5. Each of CarD’s functional domains is important for resistance to clinically relevant antibiotics.

A–C. Survival of M. smegmatis strains during the disk zone of inhibition assays. Five hundred microliters of log-phase M. smegmatis strains expressing CarDWT, CarDR25E, CarDR47E, CarDW85A, CarDK90A, CarDK125A, or CarDK125E were plated on LB agar, and a disk spotted with 5μl of 1 mg ml−1 ciprofloxacin (A), 100 mg ml−1 rifampicin (B), or 200 mg ml−1 streptomycin (C) was placed in the middle of the bacterial lawn. A representative experiment is shown above the x-axis. The radius of the zone of inhibition for each replicate and the mean ± SEM for each set of replicates is graphed, with each sample represented by a black circle.
D. Survival of M. smegmatis strains during transient ciprofloxacin treatment. Log-phase cultures of the M. smegmatis strains expressing CarDWT, CarDR25E, CarDR47E, CarDW85A, CarDK90A, CarDK125A, or CarDK125E growing in LB were treated for 2 hr with 10 μg ml−1 ciprofloxacin before the dilutions were plated to determine the surviving CFU. Graphed is the ratio of CFUs in treated cultures to that in untreated cultures for each replicate and the mean ± SEM for each set of replicates, with each sample represented by a black circle.
For all panels, the significance of differences between mutant strains and WT were determined as described in Figure 2.
Previous experiments that examined the antibiotic sensitivity of M. smegmatis strains expressing CarD mutants with weakened affinity to the RNAP employed transient treatment assays in liquid cultures (Stallings et al., 2009; Weiss et al., 2012). Since we were now using a zone of inhibition assay where the bacteria are continuously exposed to the antibiotics from a disk on solid agar, we also confirmed that the mutations that weaken the interaction between CarD and the RNAP (R25E and R47E) also increase the sensitivity of M. smegmatis to ciprofloxacin (Fig. 5A), rifampicin (Fig. 5B), and streptomycin (Fig. 5C) in these assays. These data demonstrate that the role of CarD during tolerance to these antibiotics requires all three of CarD’s proposed activities. This is distinct from the response to oxidative stress, where CarD’s ability to bind DNA is dispensable.
We had previously reported that depletion of CarD but not mutations in the RID domain increased sensitivity to ciprofloxacin in transient liquid kill assays in which exponential cultures of M. smegmatis were treated for 2 hours with antibiotic and then survival was monitored by plating the surviving CFUs (Stallings et al., 2009; Weiss et al., 2012). Despite the increased sensitivity of all of the CarD mutants to ciprofloxacin in the zone of inhibition assays, we found no significant differences in sensitivity after 2 hours of antibiotic treatment in liquid culture (Fig. 5D), which is similar to previous reports. The difference between these results may lie in the experimental design where bacteria are only transiently exposed to the antibiotic in liquid cultures as opposed to continuous exposure in the zone of inhibition assays. These data suggest that, unlike cells depleted for CarD, strains encoding mutant alleles of carD are able to mount enough of a response to survive transient exposure to ciprofloxacin, but this response is less effective than in bacteria expressing CarDWT, thus resulting in sensitivity to prolonged exposure.
Interfering with the DNA binding activity of CarD affects rRNA levels and transcription from rRNA promoters in mycobacteria
CarD is a global transcriptional regulator that has been most well-studied for its role in regulating rRNA (Stallings et al., 2009; Weiss et al., 2012; Srivastava et al., 2013). We predicted that the CarD mutants with weakened affinity to DNA display slower growth kinetics and increased sensitivity to antibiotics due to improper transcriptional regulation by CarD. We performed quantitative real-time PCR (qRT-PCR) to measure the levels of 16S rRNA in exponentially growing cultures of M. smegmatis and M. tuberculosis expressing WT or mutant alleles of carD. In M. smegmatis, mutations in CarD that affected the interaction with RNAP, the association with DNA, or the conserved tryptophan each led to decreased levels of rRNA in the bacteria, indicating that all of CarD’s activities are involved in regulating rRNA transcription (Fig. 6A). The strain expressing CarDK125E had a more severe defect in 16S rRNA transcript levels than the CarDK90A expressing strain, which mirrors the trend in survival defects during exposure to rifampicin (Fig. 5B). The importance of the lysine residue at position 125 was further confirmed when the CarDK125A mutant also had decreased levels of rRNA in M. smegmatis and M. tuberculosis as compared to CarDWT (Fig. 6A–B).
Figure 6. Each of CarD’s functional domains are important for transcriptional regulation.

A–B. 16S rRNA levels in log phase cultures of M. smegmatis strains expressing CarDWT, CarDR25E, CarDR47E, CarDW85A, CarDK90A, CarDK125A, or CarDK125E grown in LB (A) and M. tuberculosis strains expressing CarDWT or CarDK125A grown in 7H9 (B) as determined by qRT-PCR and expressed as a ratio to the levels in CarDWT expressing strains. Each graph shows the mean ± SEM of data from at least 3 replicates and the significance of the differences between mutant strains and WT.
C. M. smegmatis strains expressing CarDWT, CarDR25E, CarDR47E, CarDW85A, CarDK90A, CarDK125A, or CarDK125E were transformed with the promoter-lacZ fusion constructs diagramed above each graph. The ratio of β-galactosidase activity that resulted from lacZ expression from the M. smegmatis rrnAP1, rrnAP2, or rrnAP3 promoter in each fusion in the CarD mutant versus CarDWT expressing strains was calculated and graphed as the mean of at least 3 replicates ± SEM. Shown above each bar is the significance of the difference between the mutant strain and WT from the same promoter. The bracket designates the significance of the difference between the activation of the rrnAP1 and rrnAP2 promoters in the CarDK125E expressing strain.
For all panels, the significance of differences were calculated as described in Figure 2.
We have previously shown that CarD can directly activate transcription from rRNA promoters and that this activity is dependent on both the interaction with RNAP and the presence of the conserved tryptophan (W85) (Srivastava et al., 2013). Since rRNA is also down-regulated in strains expressing mutations in the CarD DNA binding domain (Fig. 6A–B), we examined whether the interaction with DNA is necessary for CarD’s ability to activate transcription from rRNA promoters. In M. tuberculosis there is only one rRNA operon, rrnA, and transcription is driven by two promoters, P1 and P3 (Gonzalez-y-merchand et al., 1996; Gonzalez-y-merchand et al., 1997). Transcription of the homologous rrnA operon in M. smegmatis is derived from similar P1 and P3 promoters as well as an additional P2 promoter (Gonzalez-y-merchand et al., 1996; Gonzalez-y-merchand et al., 1997). Individual promoters from the M. smegmatis rrnA operon were cloned upstream of the lacZ gene, transformed into M. smegmatis strains expressing WT or mutant alleles of carD, and β-galactosidase activity resulting from the expression of lacZ was measured to determine promoter activity in each M. smegmatis strain (Fig. 6C). Interfering with the ability of CarD to bind DNA decreased transcription from M. smegmatis rrnA promoters P1 and P2 (Fig. 6C). These are the same two promoters affected by weakening CarD’s interaction with the RNAP as well as mutating the conserved tryptophan residue. β-galactosidase activity measured from the rrnAP3 promoter was highly variable between replicates and any differences were not statistically significant, therefore we could not make conclusions regarding the role of CarD’s activities at this promoter in the lacZ fusion experiments. The mutations in the CarD DNA binding domain (K90A, K125A, and K125E) had more dramatic effects on transcription from the rrnAP2 promoter as compared to the rrnAP1 promoter, resulting in a greater decrease in β-galactosidase activity from that seen in CarDWT strains at the rrnAP2 promoters (Fig. 6C). This difference suggests that the strength of interaction between CarD and DNA is more important for initiation of transcription from the rrnAP2 promoter than from the rrnAP1 promoter. Conversely, the R25E and the W85A mutations in CarD lead to a similar fold decrease at both promoters (0.47 and 0.41 fold lower transcription in CarDR25E expressing strains and 0.31 and 0.35 fold lower in CarDW85A expressing strains for rrnAP1 and rrnAP2, respectively; Fig. 6C), indicating that the promoters require the same levels of the CarD-RNAP interaction and the activity conferred by the conserved tryptophan. The CarDK125E mutant also had a more dramatic defect in activating both the rrnAP1 and rrnAP2 promoter as compared to the CarDK90A mutant, which agrees with the observation that the levels of 16S rRNA were lower in the CarDK125E expressing strain as compared to the CarDK90A expressing strain (Fig. 6A and C). These data demonstrate that the interaction with DNA is critical for the activation of rRNA promoters by CarD. Unlike the other functions encoded by CarD, its interaction with DNA is the first activity ascribed to CarD that may exhibit some promoter specificity in terms of the importance of its activity.
CarD’s interaction with DNA is critical for the stabilization of RNAP-promoter complexes
In vitro transcription experiments have shown that CarD can stabilize a competitor resistant RNAP-promoter complex and the interaction between CarD and the RNAP is required for this activity (Srivastava et al., 2013). This ability of CarD to stabilize RNAP-promoter complexes could be a source of higher levels of promoter activity in vivo. Since CarD mutants with weakened affinity for RNAP are unable to stabilize these complexes in vitro (Srivastava et al., 2013), this could explain the lower levels of 16S rRNA and rrnA promoter activity in strains expressing these mutants (Fig. 6). Mycobacterial strains expressing CarD mutants with weakened affinity to DNA also have lower levels of rRNA (Fig. 6A–B) and rrnA promoter activity (Fig. 6C), suggesting that the interaction between CarD and DNA may also be necessary for stabilizing RNAP-promoter complexes during transcription initiation. To investigate this possibility, we performed single round in vitro transcription assays. Specifically, mycobacterial RNAP σA holoenzyme was incubated in the presence or absence of CarD with circular supercoiled DNA plasmids containing rrnA promoters driving transcription of a test transcript to allow for formation of transcription initiation complexes. dsDNA competitor was added to compete away free RNAP and prevent formation of new RNAP-promoter complexes. NTPs were added at successive time points after the addition of competitor and the amount of transcript produced was measured. As transcripts can only be initiated from transcription competent complexes formed prior to the addition of competitor, by monitoring the amount of transcription that occurs at different times following the addition of competitor, the half-life of the RNAP-promoter complexes can be calculated. Addition of CarD to single round in vitro transcription assays increased the half-life of the RNAP-promoter complexes formed at the M. tuberculosis rrnAP3 promoter by 14.7 fold (Fig. 7A–C).
Figure 7. CarD’s interaction with DNA is important for stabilization of RNAP-promoter complexes.

A. Schematic of the M. tuberculosis and M. smegmatis rRNA promoter constructs used during in vitro transcription assays. The length of the rrnAP3-derived transcripts initiated at the promoter and ending at the terminator (T) are shown.
B. Autoradiographs of 32P-labeled transcripts on denaturing polyacrylamide gels from a representative complex stability assay using M. bovis RNAP-σA holoenzyme and the rrnAP3 construct. The amount of transcript formed at the indicated timepoints following the addition of competitor is shown when no factor is added versus when CarDWT is added. The intensity of each band was quantified and the ratio to the intensity at time 0 was calculated and annotated under the gel.
C. The half-life (t1/2) of RNAP-promoter complexes formed at the rrnAP3 promoter in the M. tuberculosis rrnAP3 and the M. tuberculosis rrnAP13 constructs in the presence (hatched bars) and absence (solid grey bars) of CarDWT was calculated in GraphPad Prism based on the amount of transcripts formed during complex stability assays. The graph shows the mean of at least 3 replicates ± SEM and the significance of the differences in the comparisons designated by the brackets are shown.
D–E. Ratio of the t1/2 of RNAP-promoter complexes containing CarDWT, CarDR25E, CarDW85A, CarDK90A, or CarDK125E to the t1/2 of RNAP-promoter complexes in the absence of CarD as determined by single round complex stability in vitro transcription assays at the rrnAP3 promoter on the M. tuberculosis rrnAP13 construct (D) or the M. smegmatis rrnAP123 construct (E). The significance of differences as compared to the reactions containing no factor are shown.
For all panels, the significance of differences were determined as described for Figure 2.
In these experiments, the M. tuberculosis rrnAP3 promoter is present in isolation of its natural genomic context. In the genome, the beginning of the −35 region of rrnAP3 is located 48 bp downstream from the end of the −10 region of rrnAP1. It is possible that the in vitro transcription assays with only one promoter have neglected effects of the adjacent rrnAP1 promoter on the stability of initiation complexes formed at rrnAP3, which may alter how the initiation complexes respond to CarD activity. To study the stability of RNAP-promoter complexes formed at the M. tuberculosis rrnAP3 promoter in its native context, we generated DNA templates containing nucleotides 1,469,982–1,470,234 of the M. tuberculosis genome, which harbored both of the rrnA promoters and their intervening sequences. We designated this DNA fragment M. tuberculosis rrnAP13. rrnAP1 and rrnAP3 each produce a transcript of a unique length that can be quantified independently (Fig. 7A). However, the rrnAP1 promoter is relatively weak (Gonzalez-y-merchand et al., 1996; Gonzalez-y-merchand et al., 1997) and transcription from the rrnAP1 promoter is not reliably detected in our assays. Single round in vitro transcription assays showed that in the absence of CarD, the RNAP-promoter complexes formed at M. tuberculosis rrnAP3 were 3 fold more stable when rrnAP1 was located upstream (Fig. 7C). Therefore, the upstream rrnAP1 promoter has a polar effect on transcription initiation complexes that form at rrnAP3. The addition of CarDWT to the reactions containing the rrnAP13 construct further stabilized the half-life of the RNAP-promoter complexes formed at rrnAP3 (Fig. 7C). Interestingly, the addition of CarDWT resulted in a similar final RNAP-promoter complex half- life regardless of the context of rrnAP3. This data indicates that CarD has a maximum effect on the stability of the RNAP-promoter complexes at rrnAP3 that is independent of the stabilizing effect of the upstream rrnAP1 promoter (Fig. 7C).
Using our point mutant collection, we investigated the contribution of each of CarD’s activities to its ability to stabilize RNAP-promoter complexes at the M. tuberculosis rrnAP3 promoter in the context of the rrnAP13 construct (Fig. 7D). Addition of CarDWT increased the half-life of the RNAP-promoter complex at the M. tuberculosis rrnAP3 promoter 5.3 fold relative to the half-life of the complex in the absence of CarD (Fig. 7D). The residues involved in the interaction between CarD and the RNAP as well as the W85 residue in CarD were essential for the ability of CarD to stabilize RNAP-promoter complexes at M. tuberculosis rrnAP3 in the rrnAP13 construct, and mutations in these residues abolish this activity at this promoter (Fig. 7D). CarD mutants with decreased affinity for DNA, CarDK90A and CarDK125E, were also attenuated in their ability to stabilize the RNAP-promoters complexes at rrnAP3 (Fig. 7D). These data demonstrate that CarD’s DNA binding activity is required for CarD’s ability to stabilize RNAP-rrnAP3 complexes, which could explain the lower levels of rRNA in strains expressing CarD proteins with mutations in the DNA binding domain (Fig. 6A).
Similar experiments were also performed to assay the effect of CarD on the stability RNAP-promoter complexes formed at M. smegmatis rrnAP3 in the context of the rrnAP123 construct, which harbors nucleotides 5,029,667–5,029,905 of the M. smegmatis genome thus encoding all 3 rrnA promoters (Fig. 7A). Unlike CarDWT, the CarDR25E, CarDK90A, and CarDK125E mutants were unable to increase the half-life of RNAP-promoter complexes at M. smegmatis rrnAP3, demonstrating that the CarD-RNAP and CarD-DNA interactions are required for CarD’s activity at M. smegmatis rrnAP3 (Fig. 7E). The CarDW85A mutant had only a partial defect in the ability to stabilize complexes at this promoter as compared to CarDWT.
Discussion
In this manuscript, we have dissected three independent activities encoded by CarD: binding the RNAP, binding dsDNA, and the yet to be elucidated function of the conserved tryptophan residue within the C-terminal basic patch. We have generated a panel of mutants that individually affect one of these activities and have used them to probe the functional importance of each activity by both in vitro and in vivo assays. Biochemically, we have demonstrated that all three of these activities are required for enhancing the stability of RNAP-promoter complexes formed during transcription initiation (Fig. 7D–E). Mutating any single activity of CarD also compromises CarD’s ability to activate transcription in vivo, resulting in decreased transcription from rRNA promoters in transcriptional fusion assays (Fig. 6C) and lower rRNA levels (Fig. 6A–B). Furthermore, we have shown that all three of CarD’s activities are required for optimal growth rate and resistance to diverse stresses (Figs. 2 and 5). Together, the results from these studies allow us to propose a model for CarD’s activity at transcription complexes (Fig. 8). In this model, CarD is first directed to promoters via its interaction with RNAP β, which is based on ChIP-seq data where CarD is never localized to DNA in the absence of RNAP β (Srivastava et al., 2013). Once at promoters, the basic patch in the C-terminus of CarD contacts the DNA. The association of RNAP-bound CarD with the promoter DNA stabilizes the RNAP-promoter complex. Within this stabilized CarD-RNAP-promoter complex, the function of the conserved tryptophan in CarD is unknown, but is predicted to either promote formation of open complexes, in a way analogous to the tryptophan residues in σ factors (Panaghie et al., 2000), or stabilize the open complex by intercalating into the distorted backbone of the DNA at the upstream edge of the open complex (Srivastava et al., 2013). Together, these three activities of CarD promote a gene expression profile that increases viability and resistance to stress.
Figure 8.
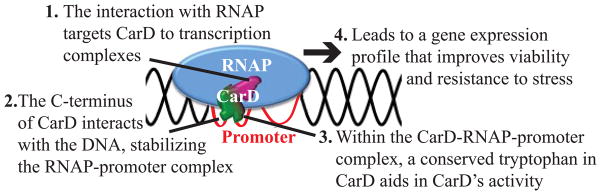
Model of CarD activity at promoters. CarD is targeted to the initiation complex via its interaction with RNAP-β (1). The C-terminus of CarD then interacts with the DNA (2). The combined association of CarD with the RNAP and CarD with the promoter DNA stabilizes the RNAP-promoter complex. Within the CarD-RNAP-promoter complex, the conserved tryptophan in CarD performs a function to facilitate CarD activity (3). The efficient functioning of all three of CarD’s activities improves viability and resistance to stress and results in changes in gene expression (4).
Much of the characterization of CarD’s role in mycobacteria has been focused on its importance in regulating rRNA transcription. The inability of CarD mutants to activate transcription of rRNA promoters in transcriptional fusion experiments (Fig. 6C) as well as the lower levels of 16S rRNA in CarD mutants (Fig. 6A–B) indicates that CarD functions as a transcriptional activator of these promoters (Srivastava et al., 2013). This is in contrast to initial data from CarD depletion experiments that suggested that CarD may repress rRNA transcription since depletion of CarD caused increased levels of rRNA (Stallings et al., 2009). However, CarD depletion is lethal to the bacteria and it is possible that changes in rRNA content during CarD depletion may be due to pleiotropic effects resulting from cell death. Therefore, we favor the data from experiments with CarD point mutants where the bacteria maintain viability. In all bacteria, the rates of ribosomal protein transcription, ribosome biogenesis, and cell growth correlate to the levels of rRNA production (Masayasu et al., 1984; Bremer and Dennis, 1996; Gourse et al., 1996). One aspect of M. tuberculosis biology that contributes to its success as a pathogen is its ability to persist within the host by entering a state of dormancy (Chao and Rubin, 2010; Gengenbacher and Kaufmann, 2012), which most likely requires stringent regulation of ribosome biogenesis and translation. However, the regulation of rRNA transcription in mycobacteria and its relationship to pathogenesis are under-investigated. Investigations into the mechanism by which CarD regulates rRNA transcription will shed light onto these important topics.
While current data supports a role for CarD as an activator of rRNA transcription, investigations into CarD activity at non-ribosomal promoters has yet to be performed. As CarD is localized to promoters throughout the genome (Srivastava et al., 2013), it remains possible that CarD may not always function as a transcriptional activator. For instance, if an RNAP holoenzyme has a high affinity for a promoter in the absence of CarD, further increasing the stability of this complex could repress transcription by inhibiting promoter escape by RNAP. The ability of a transcription factor to activate or repress transcription in this manner has been demonstrated for the phage ϕ 29 protein p4 which represses strong promoters but activates weak promoters by binding upstream of the promoter and interacting with the C-terminal domain of the RNAP α subunit (Monsalve et al., 1997).
CarD homologs are essential in M. tuberculosis, M. smegmatis (Stallings et al., 2009) and Myxococcus xanthus (García-Moreno et al., 2010) and knockouts were not attainable in Borrelia burgdorferi (Yang et al., 2008). However, the reasons for CarD essentiality in these bacteria and how CarD confers antibiotic and stress tolerance in mycobacteria remain unknown. It is possible that CarD is essential due to its role in regulating rRNA transcription, however more studies are necessary to determine if this is the case. Since CarD is associated with all RNAP-σA transcription initiation complexes within the M. smegmatis genome (Srivastava et al., 2013), it is likely that CarD is a general component of the transcription initiation machinery rather than designed for regulation of specific transcripts. Organisms lacking CarD, such as E. coli, must have an alternative means to fulfill CarD’s role in transcription initiation. This may be accomplished if the bacteria lacking CarD encode a different accessory protein that renders CarD unnecessary or if there are inherent differences in the transcription complexes of these bacteria that allow them to function efficiently in the absence of CarD. Likewise, our data has shown that all three functional modules of CarD are essential in M. tuberculosis but not in M. smegmatis. This implies that there is a more stringent requirement for CarD’s function in M. tuberculosis as compared to M. smegmatis. As an obligate pathogen, M. tuberculosis has evolved for its very specialized niche, whereas M. smegmatis and other environmental mycobacteria must be more versatile and generally contain larger genomes with greater redundancy in stress response pathways. Therefore, it is possible that M. smegmatis expresses other factors that allow for loss of the functions conferred by the conserved tryptophan and the DNA binding activity in CarD. Further comparison of the physiology of these two mycobacterial species could provide insight into the reasons behind the essentiality of CarD.
Of the CarD mutants with amino acid substitutions in the C-terminus, we were only able to obtain a CarDK125A expressing strain in M. tuberculosis. The CarDK125A mutant retains WT levels of interaction with DNA in EMSA assays (Fig. 1B) and WT levels of association with the RNAP in co-immunoprecipitation assays (Fig. 2A) but results in decreased growth (Fig. 2B–C) and rRNA levels (Fig. 6A–B) and increased sensitivity to antibiotic treatment (Fig. 5A–C). Therefore, the K125A mutation does have functional consequences for CarD. These consequences could be the result of this mutation affecting the binding to sequences other than what was used in the EMSA or a defect in DNA binding that is undetectable by the EMSA. It also remains possible that the mutation affects an unknown function of CarD distinct from its interaction with either RNAP or DNA.
One of the most surprising findings from our studies was that while the majority of CarD mutant phenotypes are similar regardless of which of CarD’s activities were targeted, increased sensitivity to H2O2 and loss of pigmentation are specific for mutations at the conserved tryptophan or that weaken the interaction with RNAP (Fig. 3). This suggests that either dsDNA binding is dispensable for regulation of transcripts required for tolerating H2O2 treatment or CarD has a cellular function independent of transcriptional regulation that does not require an interaction with DNA but is necessary to survive oxidative stress. Because the mutation of the conserved tryptophan results in sensitivity to reactive oxygen species without affecting the interaction with RNAP (Fig. 2A), it is unlikely that the CarD-mediated resistance is due to CarD physically shielding the RNAP from oxidative damage. The differential requirement for the association with the RNAP but not the DNA to withstand oxidative stress could be linked to the observation that the CarD/DNA interaction, but not the CarD/RNAP interaction, exhibits some promoter specificity in terms of the importance of its activity. However, the mechanism by which H2O2 is killing mycobacteria is currently unknown and the reason for increased sensitivity in some CarD mutants remains elusive.
While this manuscript was in preparation, another article was published that made triple mutants of basic residues in the C-terminus of CarD and showed a loss of the ability of CarD to bind and shift DNA in EMSAs (Gulten and Sacchettini, 2013). Importantly, this paper did not discuss whether these mutations affected the folding of CarD nor were there any further correlations made to CarD activity beyond DNA binding in vitro. We have shown that a single mutation within the C-terminal basic patch, K90E, affects protein stability (Fig. 2A), demonstrating that mutations at these residues can have deleterious effects on the protein beyond affecting DNA binding. In contrast, our studies focus on single point mutants that specifically affect DNA binding without affecting protein stability or CarD’s interaction with the RNAP. This same recently published report also suggested that CarD could be acting like a histone-like protein (Gulten and Sacchettini, 2013), however our ChIP-seq data (Srivastava et al., 2013) strongly argues against this and rather supports our model that CarD is directed to the DNA via its interaction with RNAP and does not bind and coat the DNA in the absence of RNAP initiation complexes.
Experimental Procedures
Media and bacterial strains
(i) M. tuberculosis
All M. tuberculosis strains were derived from the Erdman strain and were grown at 37°C in 7H9 (broth) or 7H10 (agar) (Difco) media supplemented with 60 μl L−1 oleic acid, 5 g L−1 bovine serum albumin (BSA), 2 g L−1 dextrose, 0.003 g L−1 catalase (OADC), 0.5% glycerol, and 0.05% Tween 80 (broth). Gene switching was used to construct strains of mycobacteria expressing different carD alleles and to test for their viability (Pashley and Parish, 2003). Specifically, the M. tuberculosis ΔcarD attB::tet-carD strain (described previously in (Stallings et al., 2009)) was transformed with pMSG430smcarD, pMSG430smcarDW85A, pMSG430smcarDK90A, pMSG430smcarDK90E, pMSG430smcarDK125A, or pMSG430smcarDK125E (expresses M. smegmatis CarDWT, CarDW85A, CarDK90A, CarDK90E, CarDK125A, or CarDK125E, respectively, from a constitutive Pmyc1-tetO promoter, kanamycin resistant) to replace the pDB19-Rv3583c construct (expresses M. tuberculosis CarD from a constitutive Pmyc1-tetO promoter, zeocin resistant) at the attB site of M. tuberculosis ΔcarD attB::tet-carD. The transformants were selected on kanamycin. The carD gene from each transformant was sequenced to confirm the presence of the correct sequence. The M. tuberculosis ΔcarD attB::tet-carD strains transformed with pMSG430smcarD and pMSG430smcarDK125A were named csm41 and csm45, respectively. Previously described M. tuberculosis ΔcarD attB::tet-carD strains transformed with pDB19-Rv3583cWT and pDB19-Rv3583cR47E, named mgm3080 and mgm3081 respectively, were also used in this paper (Weiss et al., 2012). The M. tuberculosis and M. smegmatis CarDs have only 3 conservative amino acid differences and complement each other both in vivo and in vitro.
(ii) M. smegmatis
All M. smegmatis strains were derived from mc2155 and were grown at 37°C in LB supplemented with 0.5% dextrose, 0.5% glycerol, and 0.05% Tween 80 (broth). The M. smegmatis strains expressing either hemagglutinin (HA)-tagged or untagged CarDWT, CarDR25E, CarDR47E, CarDW85A, CarDK90A, CarDK90E, CarDK125A, or CarDK125E were engineered as described for the analogous M. tuberculosis strains using pMSG430 expression plasmids and the M. smegmatis ΔcarD attB::tet-carD strain (described previously in (Stallings et al., 2009)). The M. smegmatis ΔcarD attB::tet-carD strains expressing M. tuberculosis CarDWT, M. tuberculosis CarDR25E, M. tuberculosis CarDR47E, M. smegmatis CarDW85A, M. smegmatis CarDK90A, M. smegmatis CarDK90E, M. smegmatis CarDK125A, or M. smegmatis CarDK125E from a constitutive Pmyc1-tetO promoter at the attB site of M. smegmatis ΔcarD attB::tet-carD were named mgm3043, mgm3044, mgm3045, csm34, csm32, csm33, csm35, and csm36, respectively. The strains expressing these same alleles but as C-terminal HA-tagged versions of CarD were named mgm3090, mgm3091, mgm3092, csm50, csm48, csm49, csm51, and csm52, respectively.
All M. tuberculosis and M. smegmatis strains used in this manuscript contain only one carD allele.
Antibiotics and chemicals
In mycobacterial cultures, 20 μg ml−1 kanamycin, 12.5 μg ml−1 zeocin, 20 μg ml−1 streptomycin, 50 μg ml−1 hygromycin, and 50 ng ml−1 of anhydrotetracycline (ATc) were used. H2O2 (Fisher Scientific) was used at 25mM.
Native Gel Electrophoresis Mobility Shift Assays (EMSAs)
A DNA fragment containing the M. smegmatis rrnA promoter and leader sequences, called rrnAPL and corresponding to M. smegmatis mc2155 nucleotides 5029577 – 5029909, was used for EMSAs as previously described (Srivastava et al., 2013). The DNA (250 ng) was amplified with IRDye labeled primers. 1pmol of labeled DNA, 1μg of LightShift Poly(di/dc) competitor DNA (Thermo Scientific), 10 μg BSA, and 200 pmol of CarD proteins were mixed with binding buffer (20 mM Tris, pH 8, 150 mM NaCl) in a total volume of 20 μL and incubated for 20 min at room temperature. Samples were then electrophoresed on 4–20% nondenaturing TBE polyacrylamide gels (Invitrogen) and imaged using an Odyssey CLX imaging system (LI-COR).
Western blotting and immunoprecipitation
For immunoprecipitation, 50 ml cultures were washed and lysed in 500 μl of NP-40 buffer (10 mM sodium phosphate, pH 8.0, 150 mM NaCl, 1% Nonidet® P-40, and Roche Complete protease inhibitor cocktail) by bead beating (FastPrep; MP Bio). Twenty-five microliters of lysate was used for the input sample and the rest was treated with DNase I (New England BioLabs), added to monoclonal anti-HA agarose (Sigma), and rotated overnight at 4°C. The matrix was washed 3 times with NP-40 buffer, and immunoprecipitated protein complexes were eluted with 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 500 μg ml−1 HA peptide (Roche), and protease inhibitors. For the western blot analyses, CarD-HA and RNAP β were detected using mouse monoclonal antibodies specific for CarD (clone 10F05; Memorial Sloan-Kettering Cancer Center Monoclonal Antibody Core Facility) and RNAP β (clone 8RB13; Neoclone, Madison, WI), respectively.
Animal infections
Before infection, exponentially replicating M. tuberculosis csm41 (CarDWT) and csm45 (CarDK125A) strains were washed in PBS plus 0.05% Tween-80 and sonicated to disperse clumps. Eight- to nine-week-old female C57BL/6 mice (Jackson Laboratory) were exposed to 8 × 107 CFU of the appropriate strain in an inhalation exposure system (Glas-Col) that delivers ~100 bacteria per animal. The bacterial burden was determined by plating serial dilutions of lung and spleen homogenates onto 7H10 agar plates. The plates were incubated at 37°C in 5% CO2 for 3 weeks prior to counting colonies. All procedures involving animals were conducted according to the National Institutes of Health (NIH) guidelines for the housing and care of laboratory animals, and they were performed in accordance with institutional regulations after protocol review and approval by the Institutional Animal Care and Use Committee of the Washington University in St. Louis School of Medicine (protocol 20130156, Analysis of Mycobacterial Pathogenesis). Washington University is registered as a research facility with the United States Department of Agriculture and is fully accredited by the American Association of Accreditation of Laboratory Animal Care. The Animal Welfare Assurance documentation is on file with the Office for Protection from Research Risks of the NIH. All animals used in these experiments were subjected to no or minimal discomfort. All mice were euthanized by CO2 asphyxiation, which is approved by the American Veterinary Medical Association Panel on Euthanasia.
Survival assays
Zone of inhibition assays: 500 μl of a log-phase M. smegmatis culture was plated on LB agar supplemented with 0.5% dextrose and 0.5% glycerol. A single 6-mm disk (Sigma) was placed in the middle of the freshly plated bacterial lawn, and 5 μl of 1 mg ml−1 ciprofloxacin, 100 mg ml−1 rifampicin, or 200 mg ml−1 streptomycin was spotted on the disk. The plates were then incubated for 2 days at 37 °C before the radius of the zone of inhibition was measured. For transient treatment in liquid culture assays, log-phase M. smegmatis cultures in LB broth supplemented with 0.5% dextrose, 0.5% glycerol, and 0.05% Tween 80 were treated with 10 μg ml−1 ciprofloxacin for 2 hrs or 25 mM H2O2 for 1 hr before plating dilutions made directly from the treated cultures.
qRT-PCR
RNA was prepared from 5–10 mL of log-phase M. smegmatis mgm3043 (CarDWT), mgm3044 (CarDR25E), mgm3045 (CarDR47E), csm32 (carDK90A), csm34 (CarDW85A), csm35 (CarDK125A), and csm36 (CarDK125E) or M. tuberculosis csm41 (CarDWT) and csm45 (CarDK125A) and 16S rRNA levels were measured and normalized to sigA transcript levels as previously described (Stallings et al., 2009).
β-galactosidase Assays
M. smegmatis mgm3043, mgm3044, mgm3045, csm32, csm34, csm35, and csm36 were transformed with pHMG147-AP1-lacZ, pHMG147-AP2-lacZ, or pHMG147-AP3-lacZ (HygR episomal plasmid that expresses lacZ from the indicated promoters) to perform β-galactosidase assays in strains expressing CarD mutants. β-galactosidase activity was measured from log-phase cultures by pelleting the cells and washing them in Z buffer (60 mM Na2HPO4, 60 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, pH 7) before resuspending them in 200 μL of Z buffer and bead beating four times (FastPrep; MP Bio) to lyse the cells. Total protein content in each lysate was measured by BCA assay (Pearce). A 50 mM final concentration of BME was added to the remaining lysate preparation and β-galactosidase activity was measured by adding a final concentration of 2 mg mL−1 ortho-nitrophenyl-β-galactoside (ONPG) solution in Z buffer. Change in OD λ420 absorbance was measured over time using the Epoch Microplate Spectrophotometer (BioTek). β-galactosidase activity was calculated using the equation with V max being the rate of change in OD λ420 absorbance (measured as the slope).
In Vitro Transcription Assays
The rrnAP123 (nucleotides 5,029,667–5,029,905 of the M. smegmatis mc2155 genome), rrnAP13 (nucleotides 1,469,982–1,470,234 of the M. tuberculosis Erdman strain), and rrnAP3 (nucleotides 1,470,113–1,470,157 of the M. tuberculosis Erdman strain) promoter fragments used in the in vitro transcription assays were cloned into the EcoRI and HindIII restriction enzyme recognition sites in the pRLG770 plasmid, which has been used previously for similar assays (Gaal et al., 2001). The DNA segment from pRLG770 (Ross et al., 1990) containing the promoter, test transcript template, and terminator was then cloned into the NcoI and PstI sites in the pGem-T (Promega) plasmid. The plasmids were prepared from E. coli by midiprep (Qiagen) and phenol/chloroform extraction. CarD proteins used in in vitro transcription assays were diluted into 1 × dialysis buffer (20 mM Tris pH8.0, 150 mM NaCl, and 1 mM BME). Recombinant M. bovis core RNAP was purified from E. coli using a system kindly supplied by Dr. Robert Landick (Huff et al., 2010; Czyz et al., 2014). Recombinant M. bovis σA was also purified from E. coli and added to the core RNAP to reconstitute the RNAP holoenzyme. For complex stability assays (Srivastava et al., 2013), 200 nM (for rrnAP3 or rrnAP13 assays) or 400 nM (rrnAP123 assays) M. smegmatis CarDWT, M. tuberculosis CarDR25E, M. smegmatis CarDW85A, M. smegmatis CarDK90A, or M. smegmatis CarDK125E was preincubated with 20 nM (for rrnAP3 or rrnAP13 assays) or 40 nM (rrnAP123 assays) Mycobacterium bovis core RNAP in 1x storage buffer (10 mM Tris·HCl, pH 8.0, 3 mM MgCl2, 50 mM NaCl, 1 mM BME, and 50% (vol/vol) glycerol) for 10 min on ice, followed by the addition of 40 nM (for rrnAP3 or rrnAP13 assays) or 80 nM (rrnAP123 assays) M. bovis σA (in 1x storage buffer). After 10 min, 50 ng of a supercoiled plasmid DNA containing a test promoter was added and the reaction was brought to 12.5μL by dilution such that the final solutions contained 1 × transcription buffer (10 mM Tris-Cl, pH 8.0, 10 mM MgCl2, 50 μg mL−1 BSA, 40 mM NaCl, and 1 mM DTT). 400 nM competitor DNA (double-stranded FullCon promoter DNA fragment (Gaal et al., 2001)) was added and at the designated times after the addition of competitor, transcription was initiated by the addition of NTPs (100 μM GTP, CTP, and ATP; 10 μM UTP; and 0.1 μL [α-32P]UTP for rrnAP3 and rrnAP13 or 100 μM CTP and ATP, 500 μM GTP, 10 μM UTP, and 0.1 μL [α-32P]UTP for rrnAP123). A higher concentration of the initiating nucleotide GTP is required to achieve enough transcript from the M. smegmatis rrnAP3 promoter to be reliably detectable in these assays. After 15 min, the reactions were stopped with 2 × formamide buffer (98% (vol/vol) formamide, 5 mM EDTA) and run on a 6% urea PAGE gel.
Acknowledgments
The authors thank Dr. Bob Landick for assistance with protocols and reagents related to the isolation of M. bovis RNAP-σA holoenzyme and Drs. Seth Darst and Elizabeth Campbell for structural studies that help identify residues involved in DNA binding by CarD. C.L.S. is supported by a Biomedical Research Grant from the American Lung Association and C.L.S. and E.A.G are supported by Grant GM107544 from the National Institutes of Health. A.L.G. is supported by the NIGMS Cell and Molecular Biology Training Grant GM007067 and the Stephen I. Morse Graduate Fellowship.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- Bremer H, Dennis PP. Escherichia Coli and Salmonella Typhimurium: Cellular and Molecular Biology. ASM Press; 1996. Modulation of Chemical Composition and Other Parameters of the Cell by Growth Rate; pp. 1553–69. [Google Scholar]
- Chao MC, Rubin EJ. Letting sleeping dos lie: does dormancy play a role in tuberculosis? Annu Rev Microbiol. 2010;64:293–311. doi: 10.1146/annurev.micro.112408.134043. [DOI] [PubMed] [Google Scholar]
- Cooper AM, Segal BH, Frank AA, Holland SM, Orme IM, Orme IANM. Transient Loss of Resistance to Pulmonary Tuberculosis in p47 phox −/− Mice Transient Loss of Resistance to Pulmonary Tuberculosis. Infect Immun. 2000;68:1231–1234. doi: 10.1128/iai.68.3.1231-1234.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czyz A, Mooney RA, Iaconi A. Mycobacterial RNA Polymerase Requires a U-Tract at Intrinsic Terminators and Is Aided by NusG at Suboptimal Terminators. MBio. 2014;5:1–10. doi: 10.1128/mBio.00931-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaal T, Ross W, Estrem ST, Nguyen LH, Burgess RR, Gourse RL. Promoter recognition and discrimination by EsigmaS RNA polymerase. Mol Microbiol. 2001;42:939–54. doi: 10.1046/j.1365-2958.2001.02703.x. [DOI] [PubMed] [Google Scholar]
- Gangwar SP, Meena SR, Saxena AK. Structure of the carboxy-terminal domain of Mycobacterium tuberculosis CarD protein: an essential rRNA transcriptional regulator. Acta Crystallogr Sect F, Struct Biol Commun. 2014;70:160–5. doi: 10.1107/S2053230X13034407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Moreno D, Abellón-Ruiz J, García-Heras F, Murillo FJ, Padmanabhan S, Elías-Arnanz M. CdnL, a member of the large CarD-like family of bacterial proteins, is vital for Myxococcus xanthus and differs functionally from the global transcriptional regulator CarD. Nucleic Acids Res. 2010;38:4586–98. doi: 10.1093/nar/gkq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gengenbacher M, Kaufmann SHE. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol Rev. 2012;36:514–32. doi: 10.1111/j.1574-6976.2012.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-y-merchand JA, Colston MJ, Cox RA. The rRNA operons of Mycobacterium smegmatis and Mycobacterium tuberculosis: comparison of promoter elements and of neighbouring upstream genes. Microbiology. 1996;142:667–674. doi: 10.1099/13500872-142-3-667. [DOI] [PubMed] [Google Scholar]
- Gonzalez-y-merchand JA, Garcia MJ, Gonzalez-Rico S, Colston MJ, Cox RA. Strategies used by pathogenic and nonpathogenic mycobacteria to synthesize rRNA. J Bacteriol. 1997;179:6949. doi: 10.1128/jb.179.22.6949-6958.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourse RL, Gaal T, Bartlett MS, Appleman Ja, Ross W. rRNA transcription and growth rate-dependent regulation of ribosome synthesis in Escherichia coli. Annu Rev Microbiol. 1996;50:645–77. doi: 10.1146/annurev.micro.50.1.645. [DOI] [PubMed] [Google Scholar]
- Gulten G, Sacchettini JC. Structure of the Mtb CarD/RNAP β-Lobes Complex Reveals the Molecular Basis of Interaction and Presents a Distinct DNA-Binding Domain for Mtb CarD. Structure. 2013;21:1859–69. doi: 10.1016/j.str.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J, Czyz A, Landick R, Niederweis M. Taking phage integration to the next level as a genetic tool for mycobacteria. Gene. 2010;468:8–19. doi: 10.1016/j.gene.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RC, Michael F, Simon MI. Host Protein Requirements for In Vitro Site-Specific DNA Inversion. Cell. 1986;46:531–539. doi: 10.1016/0092-8674(86)90878-0. [DOI] [PubMed] [Google Scholar]
- Johnson RC, Simon Hin-mediated site-specific recombination requires two 26 bp recombination sites and a 60 bp recombination enhancer. Cell. 1985;41:781–791. doi: 10.1016/s0092-8674(85)80059-3. [DOI] [PubMed] [Google Scholar]
- Kahmann R, Rudt F, Koch C, Mertens G. G inversion in bacteriophage Mu DNA is stimulated by a site within the invertase gene and a host factor. Cell. 1985;41:771–80. doi: 10.1016/s0092-8674(85)80058-1. [DOI] [PubMed] [Google Scholar]
- Kang PILJ, Craig EA. Identification and characterization of a new Identification and Characterization of a New Escherichia coli Gene That Is a Dosage-Dependent Suppressor of a dnaK Deletion Mutation. J Bacteriol. 1990;172:2055. doi: 10.1128/jb.172.4.2055-2064.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, Dutta D, Thakur KG. Crystal structure of Mycobacterium tuberculosis CarD, an essential RNA polymerase binding protein, reveals a quasidomain-swapped dimeric structural architecture. Proteins. 2014;82:879–84. doi: 10.1002/prot.24419. [DOI] [PubMed] [Google Scholar]
- Koch C, Kahmann R. Purification and properties of the Escherichia coli host factor required for inversion of the G segment in bacteriophage Mu. J Biol Chem. 1986;261:15673–8. [PubMed] [Google Scholar]
- Masayasu N, Gourse R, Baughman G. Regulation of the Synthesis of Ribosomes and Ribosomal Components. Annu Rev Biochem. 1984;53:75–117. doi: 10.1146/annurev.bi.53.070184.000451. [DOI] [PubMed] [Google Scholar]
- Monsalve M, Calles B, Mencía M, Salas M, Rojo F. Transcription activation or repression by phage psi 29 protein p4 depends on the strength of the RNA polymerase-promoter interactions. Mol Cell. 1997;1:99–107. doi: 10.1016/s1097-2765(00)80011-8. [DOI] [PubMed] [Google Scholar]
- Newlands JT, Josaitis Ca, Ross W, Gourse RL. Both fis-dependent and factor-independent upstream activation of the rrnB P1 promoter are face of the helix dependent. Nucleic Acids Res. 1992;20:719–26. doi: 10.1093/nar/20.4.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaghie G, Aiyar SE, Bobb KL, Hayward RS, de Haseth PL. Aromatic amino acids in region 2.3 of Escherichia coli sigma 70 participate collectively in the formation of an RNA polymerase-promoter open complex. J Mol Biol. 2000;299:1217–30. doi: 10.1006/jmbi.2000.3808. [DOI] [PubMed] [Google Scholar]
- Pashley Ca, Parish T. Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol Lett. 2003;229:211–215. doi: 10.1016/S0378-1097(03)00823-1. [DOI] [PubMed] [Google Scholar]
- Rao L, Ross Wi, Appleman Ja, Gaal T, Leirmo S, Schlax P, et al. Factor Independent Activation of rrnB P1 An “extended” promoter with an Upstream Element that Dramatically increases Promoter Strength. J Mol Biol. 1994;235:1421–1435. doi: 10.1006/jmbi.1994.1098. [DOI] [PubMed] [Google Scholar]
- Ross W, Thompson JF, Newlands JT, Gourse RL. E. coli Fis protein activates ribosomal RNA transcription in vitro and in vivo. EMBO J. 1990;9:3733–42. doi: 10.1002/j.1460-2075.1990.tb07586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava DB, Leon K, Osmundson J, Garner AL, Weiss La, Westblade LF, et al. Structure and function of CarD, an essential mycobacterial transcription factor. Proc Natl Acad Sci U S A. 2013;110:12619–24. doi: 10.1073/pnas.1308270110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings CL, Stephanou NC, Chu L, Hochschild A, Nickels BE, Glickman MS. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell. 2009;138:146–59. doi: 10.1016/j.cell.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers AA. Promoter Sequence for Stringent Control of Bacterial Ribonucleic Acid Synthesis Promoter. J Bacteriol. 1980;141 doi: 10.1128/jb.141.2.973-976.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss La, Harrison PG, Nickels BE, Glickman MS, Campbell Ea, Darst Sa, Stallings CL. Interaction of CarD with RNA polymerase mediates Mycobacterium tuberculosis viability, rifampin resistance, and pathogenesis. J Bacteriol. 2012;194:5621–31. doi: 10.1128/JB.00879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO. Tuberculosis WHO Global Tuberculosis Report 2013 2013 [Google Scholar]
- Yang XF, Goldberg MS, He M, Xu H, Blevins JS, Norgard MV. Differential expression of a putative CarD-like transcriptional regulator, LtpA, in Borrelia burgdorferi. Infect Immun. 2008;76:4439–44. doi: 10.1128/IAI.00740-08. [DOI] [PMC free article] [PubMed] [Google Scholar]