

Abstract
Multiple myeloma is a hematological malignancy in which clonal plasma cells proliferate and accumulate within the bone marrow. The presence of osteolytic lesions due to increased osteoclast (OC) activity and suppressed osteoblast (OB) function is characteristic of the disease. The bone marrow mesenchymal stromal cells (MSCs) play a critical role in multiple myeloma pathophysiology, greatly promoting the growth, survival, drug resistance and migration of myeloma cells. Here, we specifically discuss on the relative contribution of MSCs to the pathophysiology of osteolytic lesions in light of the current knowledge of the biology of myeloma bone disease (MBD), together with the reported genomic, functional and gene expression differences between MSCs derived from myeloma patients (pMSCs) and their healthy counterparts (dMSCs). Being MSCs the progenitors of OBs, pMSCs primarily contribute to the pathogenesis of MBD because of their reduced osteogenic potential consequence of multiple OB inhibitory factors and direct interactions with myeloma cells in the bone marrow. Importantly, pMSCs also readily contribute to MBD by promoting OC formation and activity at various levels (i.e., increasing RANKL to OPG expression, augmenting secretion of activin A, uncoupling ephrinB2-EphB4 signaling, and through augmented production of Wnt5a), thus further contributing to OB/OC uncoupling in osteolytic lesions. In this review, we also look over main signaling pathways involved in the osteogenic differentiation of MSCs and/or OB activity, highlighting amenable therapeutic targets; in parallel, the reported activity of bone-anabolic agents (at preclinical or clinical stage) targeting those signaling pathways is commented.
Keywords: Mesenchymal stromal cells, Multiple myeloma, Osteolytic lesions, Myeloma bone disease, Bone-directed therapy, Bone-anabolic drugs
Core tip: In multiple myeloma, bone marrow mesenchymal stromal cells (MSCs) primarily contribute to associated osteolytic lesions because of their defective differentiation to mature osteoblasts. Importantly, these MSCs also contribute to myeloma bone disease by enhancing osteoclast formation and activity through various mechanisms (i.e., increasing the receptor activator of nuclear factor-κB ligand/osteoprotegerin ratio, augmenting activin A secretion, uncoupling ephrinB2-EphB4 signaling and because of heightened production of Wnt5a). In addition, we overview signaling pathways involved in the osteogenic differentiation of MSCs or osteoblast activity and comment on the reported activity of bone-anabolic agents (preclinical or clinical stage) to restore bone homeostasis in myeloma patients.
MESENCHYMAL STROMAL CELLS: IN VITRO AND IN VIVO PROPERTIES
Mesenchymal stromal cells
Bone marrow (BM)-derived mesenchymal stromal cells (MSCs) were initially described by Friedenstein et al[1] in the late 60-s as adherent cells of fibroblastic morphology with the ability to differentiate into osteogenic cells, although it was later demonstrated that these cells also have chondrogenic and adipogenic differentiation potential[2]. They were initially named as Colony Forming Unit-Fibroblasts[3], but soon they were referred to as MSCs, term that gained general acceptance[4]. Instead, the International Society for Cellular Therapy (ISCT) recommends the term “mesenchymal stromal cells” for MSCs[5] and published several years ago a number of minimal definition criteria for these cells[6], which are indicated in Table 1.
Table 1.
Minimal criteria for mesenchymal stromal cell definition (International Society for Cellular Therapy)
| Adherence to plastic surfaces in standard culture conditions | ||
| Positive (> 95% +) | Negative (< 2% +) | |
| Immunophenotype | CD105 | CD45 |
| CD73 | CD34 | |
| CD90 | CD14 or CD11b | |
| CD79a or CD19 | ||
| HLA-DR | ||
| In vitro differentiation to osteoblasts, adipocytes and chondroblasts (demonstrated by appropriate staining of cell cultures) | ||
MSC isolation, characterization and in vitro expansion
BM-derived MSCs may be isolated from mononuclear cells obtained after density-gradient centrifugation of BM aspirates and subsequent adherence to tissue culture plasticware. Since their proportion in a normal BM sample is really low (between 0.01% and 0.0001% of nucleated cells)[7], for most applications MSCs need to be in vitro expanded. The standard culture medium is based on Dulbecco’s Modified Eagle Medium or α-Minimum Essential Medium with 10% of fetal bovine serum, although the latter can be replaced by platelet lysate or a commercial concentrate of growth factors[8]. The expansion medium is replaced twice a week and thus non-adherent cells are removed. After two or three passages, the primary culture contains more than 95% of MSCs, and these cells are then used for most experiments[9].
According to the ISCT definition criteria[6] (Table 1), an immunophenotypic study is mandatory to evaluate the positivity for at least CD73, CD105 and CD90 and negativity for HLA-DR and hematopoietic antigens (CD45, CD34, CD19 or CD79α, CD14 or CD11b).
Differentiation and immunomodulatory properties of MSCs
Upon in vitro culture with the appropriate differentiation media, MSCs are able to differentiate into osteogenic, adipogenic and chondrogenic phenotypes[10]. This multi-lineage differentiation ability into mesodermal cell types is another definition criteria established by the ISCT[6] (Table 1), and is the basis for evaluating the therapeutic potential of MSCs in a number of clinical trials, especially for treating musculoskeletal diseases[11].
Being this property important, the range of diseases in which MSCs are of potential use has widely expanded when these cells demonstrated to display potent immunomodulatory and anti-inflammatory effects both in vitro and in vivo[12]. In this regard, MSCs are able to reduce the activation of both T cells[13] and B cells[14], and to increase the number of T-regulatory cells[15]. In addition, MSCs inhibit the maturation of dendritic cells and their capacity to process and present antigens[16]. MSCs also reduce neutrophil activation and proliferation of natural killer cells[17,18], thus regulating innate immune system responses. For these reasons, MSCs are currently being evaluated for the treatment of several immune-mediated diseases.
MULTIPLE MYELOMA AND THE BONE MARROW MICROENVIRONMENT. MYELOMA-ASSOCIATED BONE DISEASE
Multiple myeloma and the bone marrow microenvironment
Multiple myeloma (MM) is a hematological malignancy resulting from the clonal expansion of plasma cells in the BM. Diagnostic criteria of symptomatic myeloma include the presence of at least 10% BM myeloma cells and of monoclonal protein in serum and/or urine, together with myeloma-related end-organ or tissue damage (including hypercalcemia, renal dysfunction, anemia, immunodeficiency and bone destruction)[19]. In fact, almost 80% of myeloma patients develop osteolytic lesions, which are responsible for some of the most devastating characteristics of the disease. In most (if not all) cases, symptomatic myeloma is preceded by sequential asymptomatic stages of monoclonal gammopathy of undetermined significance (MGUS) and smoldering myeloma[20], with increasing BM plasmocytosis and monoclonal component as well as augmented risk of progression to active MM (around 1% per year for MGUS patients and 10%-20% for patients with smoldering myeloma)[21]. MM accounts for more than 1% of all cancers, with an incidence of 33400 new myeloma cases and 20300 deaths in the European Union 27 in 2012[22].
During last decade, substantial advances both in the knowledge of the biology of the disease and in the use of more effective molecular-targeted novel agents and combinatorial regimens[23,24] have led to improved responses and survival rates (median survival has increased from 3 to over 6 years[25]). It is expected that advances in remaining controversies of the pathophysiology of the disease[25] together with novel therapies currently in development and testing, may further improve myeloma survival in coming years, with future therapeutic aims rather focusing on increasing long-term remission rates and improving the quality of life of myeloma patients.
The biological behavior and clinical outcome in MM is partly dependent on the genomic and epigenetic abnormalities of myeloma cells[26]. Compelling evidence has accumulated, however, supporting a critical role of the BM microenvironment in the pathogenesis and progression of the disease[27-31]. MM cells establish complex interactions with other cellular components of the BM milieu [MSCs, osteoclasts (OCs), osteoblasts (OBs) and osteoprogenitor cells, endothelial cells, adipocytes, immune cells-dendritic cells, macrophages, T cells-], with components of the extracellular matrix (ECM) (e.g., laminin, collagen, proteoglycans, glycosaminoglycans), and also with secreted soluble factors (cytokines, chemokines and growth factors). These interactions have bidirectional consequences: on the one hand, interactions of MM cells mainly with MSCs and OCs lead to activation of multiple cellular signaling pathways on myeloma cells [phosphatidylinositol 3-kinase/AKT, Janus kinase/signal transducer and activator of transcription 3, Ras/Raf/mitogen-activated protein kinase kinase/extracellular signal-related kinase, nuclear factor-κB (NFκB)] which support their proliferation, survival, migration and even resistance to therapeutic agents (reviewed in[27,31,32]). On the other hand, myeloma cells perturb the BM homeostasis causing anemia, immunosuppression, and uncoupling of the bone remodeling process leading to the development of osteolytic bone lesions characteristic of the disease.
Myeloma bone disease: Mechanisms of OC activation and OB inhibition
Myeloma bone disease (MBD) is characterized by the presence of osteolytic lesions that result in skeletal-related events (SREs) including severe bone pain, osteopenia, diffuse osteoporosis, focal lytic lesions, pathological fractures and spinal cord compression[33,34]. Of importance, MBD not only severely affects the quality of life of MM patients, but is also linked to poor prognosis, shorter overall survival and progression-free survival[35,36]. This highlights the need of bone-targeted supportive treatments in addition to anti-myeloma therapy, which may reduce the risk of bone complications in MM patients. In addition, accumulating evidences of preclinical and clinical studies support the notion of an intimate relationship between tumor growth and the development of MBD, being one the necessity and consequence of the other[37]. Since many of the dysregulated factors involved in the pathophysiology of osteolytic lesions also promote MM growth and survival, it is expected that effective interventions on MBD would secondarily lead to myeloma inhibition[38].
Clinical observations, histomorphometric studies and measurements of serum/urine bone metabolism markers[39-41] showed that uncoupled bone remodeling in MM was associated to both increased bone resorption (with increased number and activity of OCs) and almost absent bone formation (impaired OB formation and function). Only recently, many of the cellular and molecular interplayers involved in the pathophysiology of MBD have been identified and excellently reviewed[34,37,38,42,43]. Next, we summarize the main factors and molecular mechanisms leading to the enhanced OC activation and suppressed OB function in MM.
Enhanced OC differentiation and resorptive activity
Enhanced OC formation from myeloid precursors and OC hyperactivation is primarily mediated through increased production of multiple “OC-activating factors (OAFs)” both by MM cells and other cells from the BM microenvironment {e.g., receptor activator of NF-κB ligand (RANKL), CCL3 [also known as macrophage inflammatory protein-1α (MIP-1α)], activin A, interleukin-3 (IL-3), IL-7, IL-1β, IL-6 and CCL20[44]; for reviews of cellular origin of OAFs, see[37,45]; also see Figure 1}.
Figure 1.
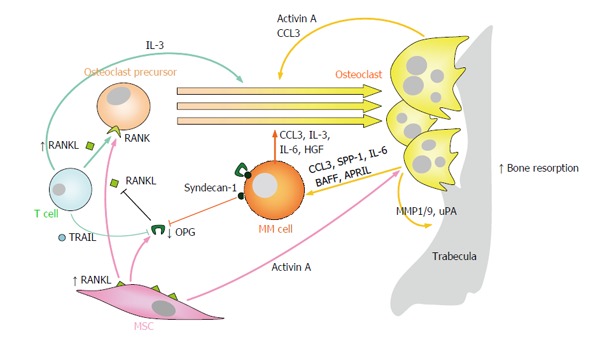
Enhanced osteoclast formation and resorption partially mediates the development of myeloma bone disease. Numerous “OC-activating factors” produced by multiple myeloma cells and other cells in the bone marrow microenvironment (including RANKL, CCL3, activin A, IL-3, HGF and IL-6) readily promote OC differentiation from OC precursors and/or stimulation of OC resorptive activity. In MM, the RANKL/OPG ratio is clearly favored towards RANKL, both because of increased expression of RANKL on MSCs and T lymphocytes, and because of reduced expression of OPG by MSCs and inactivation of OPG through binding to TRAIL or syndecan-1 on the surfaces of myeloma cells. On the other hand, OCs produce several factors (e.g., IL-6, CCL3, BAFF and APRIL) which promote the growth and survival of multiple myeloma cells. RANKL: Receptor activator of NFκB ligand; NFκB: Nuclear factor-κB; CCL3/MIP1α: Macrophage inflammatory protein 1-α; IL-3/6: Interleukin 3/6; HGF: Hepatocyte growth factor; OPG: Osteoprotegerin; RANK: Receptor activator of NFκB; TRAIL: Tumor necrosis factor-related apoptosis inducing ligand; BAFF: B-cell-activating factor; APRIL: A proliferation-inducing ligand; SPP-1: Osteopontin; MMP1/9: Matrix metalloprotease 1/9; uPA: Urokinase plasminogen activator; MSCs: Mesenchymal stromal cells; OC: Osteoclast; MM: Multiple myeloma.
RANKL: RANKL is a member of the tumor necrosis factor (TNF) superfamily expressed as a transmembrane protein by BM MSCs and OBs, and by T lymphocytes as a soluble form; whether MM cells are producers of RANKL is still a controversial issue[46,47]. Instead, the receptor of RANKL, RANK, is expressed on the surface of OCs and OC precursors. RANK-RANKL signaling has been shown to play an essential role in OC formation, activation and survival preventing OC apoptosis[48,49]. Osteoprotegerin (OPG) is a soluble glycoprotein secreted by MSCs and OBs which acts as a decoy receptor for RANKL, neutralizing its activity and thus inhibiting osteoclastogenenesis and bone resorption[50]. Myeloma cells induce MSCs and OBs in the BM to upregulate the expression of RANKL and to reduce the expression of OPG, leading to increased RANKL/OPG ratios. In addition, MM cells may also sequester OPG through its binding to syndecan-1 (CD138), which is subsequently internalized and degraded[51]. Furthermore, T lymphocytes in MM also overexpress TNF-related apoptosis inducing ligand (TRAIL), which binds and neutralizes OPG, further reducing its OC inhibitory activity[52]. Whereas in physiological conditions the RANKL/OPG ratio tightly regulates OC function for adequate bone remodeling, in MM it is clearly favored towards RANKL, promoting osteoclastogenesis and bone destruction[53,54]. Thus, the RANKL/OPG axis constitutes an important target for the treatment of MBD.
CCL3: The CCL3 (MIP-1α) chemokine is mainly produced by both myeloma cells and OCs, and functions as a major osteoclastogenic and OC survival factor, both directly and indirectly by enhancing the osteoclastogenic activity of RANKL and IL-6[55,56]. Interestingly, CCL3 has been found to have pleiotropic roles in MM, also inducing growth, survival and chemotaxis for malignant plasma cells[57] and, as will be commented later, inhibition of OB differentiation[58,59].
Activin A: Activin A is a transforming growth factor (TGF)β family member identified as a key component of MBD, having a dual role in stimulating OC formation and activity[34,60] and as an inhibitor of OB differentiation[61,62]. MSCs and OCs are the main sources of activin A and interacting myeloma cells further upregulate its expression in MSCs[61].
Other factors promoting OC formation and activity: IL-3 is majorly produced by activated T lymphocytes and by myeloma cells, and may stimulate OC formation and resorption directly or by further augmenting that of RANKL and CCL3[63,64]. Other chemokines such as IL-7, tumor necrosis factor α (TNFα) and IL-1β indirectly stimulate osteolytic processes, inducing RANKL expression on BM stromal cells (TNFα and IL-1β[65]) and by circulating T cells (IL-7)[66]. Several other OAFs are secreted by myeloma cells and/or stromal cells [e.g., hepatocyte growth factor (HGF)[67,68], IL-6[69], IL-8[70], and vascular endothelial growth factor (VEGF)[71]], or by dendritic cells, Th17 1 lymphocytes, osteocytes and megakaryocytes in the BM milieu [(e.g., IL-17, IL-11) reviewed in[37,72]], which further increase the gradient of osteoclastogenic factors in focal lesions and contribute to OC production and activity.
At the same time, OCs readily promote MM cell survival and growth by physical cell-cell contact and by the release of several soluble factors [including IL-6, CCL3, osteopontin, B-cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL)], and thus creating a vicious circle between bone lesions and tumour expansion (reviewed in[30,37]).
On the other hand, myeloma-OC interactions may directly contribute to bone matrix degradation via secreted metalloproteases 1/9 and urokinase-type plasminogen activator from OCs[73]. Besides, some myeloma cells may acquire resorbing capabilities and degrade bone[74,75], and dendritic cells in the BM may transdifferentiate to bone-resorbing OCs after myeloma interaction[76], further contributing to enhanced resorption.
Suppression of osteoblastogenesis and OB function
Myeloma-induced suppression of osteoblastogenesis and OB activity is exerted both by functional inhibition of existing OBs as well as by impaired differentiation of MSCs into mature OBs. This is in accord with the findings of a significant reduction in the number of active OBs in BM biopsies[77] and extremely low serum markers of osteoblastogenesis (such as osteocalcin and OPG) in patients with active osteolytic lesions as compared to myeloma patients not having bone lesions[45].
In recent years, many of the molecular mediators underlying suppression of OB differentiation and function in MM have been identified, involving both direct cellular interactions and soluble factors (Figure 2).
Figure 2.
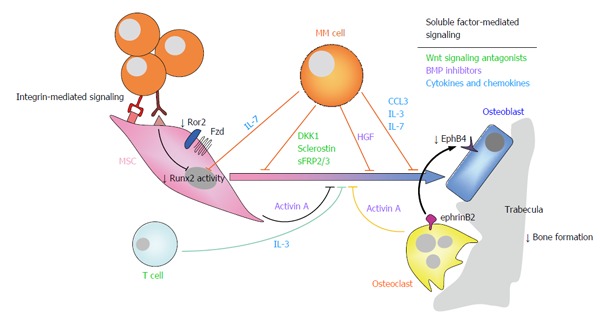
Suppression of osteoblastogenesis and osteoblast function in multiple myeloma is also involved in the pathophysiology of myeloma bone disease. Myeloma-induced OB suppression is partially mediated by direct cell to cell contact interactions with MSCs, leading to reduced activity of the Runx2/Cbfa1 transcription factor, and to inhibition of non-canonical Wnt5a signaling due to decreased expression of Ror2 in pre-OBs. In addition, soluble factors produced by myeloma cells and cells in the bone marrow microenvironment, such as Wnt signaling antagonists (e.g., DKK1, sclerostin, sFRP-2/3), BMP inhibitors (activin A, TGFβ, HGF), cytokines and chemokines (such as IL-7, TNFα, IL-3, CCL3) and apoptotic factors also contributed to inhibition of osteogenic differentiation and function. Finally, reduced ephrinB2-EphB4 signaling (from OCs to OBs) because of diminished EphB4 expression in MSCs, further contributes to impaired OB differentiation. Ror2: Receptor tyrosine kinase-like orphan receptor 2; DKK1: Dickkopf-1; sFRP-2/3: Secreted frizzled related protein-2/3; TGFβ: Transforming growth factor β; HGF: Hepatocyte growth factor; IL-7/3: Interleukin 7/3; ephrinB2: ephrin-B2 ligand; EphB4: Eph receptor B4; MSCs: Mesenchymal stromal cells; OB: Osteoblast; OC: Osteoclast; CCL3/MIP1α: Macrophage inflammatory protein 1-α; TNFα: Tumor necrosis factor α; BMP: Bone morphogenetic protein; MM: Multiple myeloma.
Soluble factors contributing to OB suppression include inhibitors of the two major signaling pathways governing osteoblastogenesis [i.e., Wnt and bone morphogenetic protein (BMP) signaling pathways], several cytokines and chemokines, as well as MM-induced apoptotic factors for OBs.
Wnt signaling antagonists (Dickkopf-1, sclerostin, secreted Frizzled-related proteins-2 and 3): MM cells secrete Dickkopf-1 (DKK1)[78] and sclerostin[79], both inhibiting Wnt canonical signaling and thus OB differentiation because of direct binding to the low-density lipoprotein receptor-related protein (LRP)5/6 co-receptor in osteoprogenitor cells[80]. Sclerostin is also produced by osteocytes, mediating osteocyte-OC communication necessary for bone homeostasis[81]. Interestingly, both DKK1 and sclerostin further increase the RANKL/OPG ratio on MSCs and osteoprogenitor cells by upregulating the expression of RANKL and reducing that of OPG, thus indirectly enhancing OC differentiation and activity[79,82]. A direct stimulatory effect of sclerostin on OC formation has also been reported[83]. Other Wnt antagonists produced by primary cells and MM cell lines are the secreted Frizzled-related proteins-2 and 3 (sFRP2 and sFRP3), which bind directly to secreted Wnt ligands and at least sFRP3 has been associated to the extent of MBD at diagnosis[84-86].
BMP inhibitors (activin A, TGFβ, hepatocyte growth factor): The BMP is another major molecular pathway involved in osteogenesis, in which members of the TGFβ superfamily of cytokines (BMPs, activin A, TGFβ) bind to heterodimeric receptors to activate Smad proteins, which may directly regulate the expression of osteoblastogenic genes as transcription factors (e.g., DLX5-distal-less 5) or indirectly via Runx2/Cbfa1[87]. Although some ligands (e.g., BMP2) directly stimulate osteogenesis through this pathway, others (such as activin A and TGFβ) have opposite effects. Activin A is produced by OCs and by MSCs after interaction with myeloma cells, and in addition to its commented pro-OC effect, it inhibits OB differentiation via Smad2-dependent DLX5 downregulation[61]. Similarly, TGFβ also downregulates DLX5[88], and inhibits OB differentiation. The HGF is produced by MM cells and is found at high concentrations in the BM of myeloma patients[89]. It has been shown to promote proliferation of human MSCs keeping cells in an undifferentiated state, and to inhibit BMP-induced Smad traslocation, thus inhibiting OB formation.
Other cytokines and chemokines: Other cytokines and chemokines may additionally contribute to suppression of OB activity (e.g., IL-7, TNFα, IL-3 and CCL3). IL-7 is produced by malignant plasma cells and partially mediates Runx2/Cbfa1 decreased activity in MSCs interacting with myeloma cells, and reduces the expression of OB markers[77,90]. The pro-inflammatory cytokine TNFα inhibits the expression of both Runx2/Cbfa1 and Osterix transcription factors[91,92] and increases the expression of sclerostin in OBs[93]. Interestingly, IL-7 and TNFα effects on osteoprogenitor cells in MM were found to be partly mediated by increased levels of the Gfi1 transcriptional repressor of Runx2/Cbfa[94]. IL-3 is majorly secreted by T lymphocytes (but also by myeloma cells) and besides its commented activity on OC formation and activation[63], it indirectly inhibits basal and BMP2-induced OB differentiation by stimulating CD45+ monocyte-macrophages[95,96]. In addition to the commented activity of CCL3 related to its pro-OC activity and in support of MM growth, CCL3 has been shown to inhibit OB differentiation and function through CCR1[58].
MM-induced apoptosis on OBs: MM-induced apoptosis on OBs and osteocytes may also account for OB suppression in MM. OBs from myeloma patients with extensive osteolytic lesions have been shown to overexpress the Fas Cell Surface Death Receptor, death receptors DR4/5 and receptors to TRAIL, and to promptly undergo apoptosis when co-cultured with myeloma cells[97,98]. Similarly, pre-osteocytes of patients with active bone disease in co-culture with myeloma cells showed increased apoptosis and upregulated expression of IL-11, thus increasing their pro-osteoclastogenic properties[99].
In addition to soluble factors, direct contact interactions of myeloma and pre-OBs further contribute to OB suppression in MBD leading to reduced activity of Runt-related transcription factor 2/core-binding factor Runt domain α subunit 1 (Runx2/Cbfa1), which is a critical transcriptional regulator of OB differentiation[77]. Blocking of very late antigen 4 (VLA4)-vascular cell adhesion molecule 1 (VCAM1) interactions with a neutralizing anti-VLA4 antibody reduced the inhibitory effect on Runx2/Cbfa1 activity, thus making these adhesion molecules partially responsible for the inhibition of OB differentiation and function[77]. Further, long-term inhibition of Runx2/Cbfa1 and Osterix in pre-OBs seems to be mediated by overexpression of the transcriptional repressor 4EBP1[100].
Only recently, Giuliani et al[101] identified another mechanism of myeloma-induced OB impairment through contact interactions. Although canonical Wnt signaling is known to play a critical role in osteoblastogenesis[87], the non-canonical Wnt5a ligand has been shown to mediate the osteogenic differentiation of BM human MSCs through activation of co-receptor receptor tyrosine kinase-like orphan receptor 2 (Ror2)[102,103]. Myeloma cells were found to inhibit the expression of Ror2 when in co-culture interaction with pre-OB cells, therefore inhibiting non-canonical Wnt5a signaling and the osteogenic differentiation of MSCs[101].
Finally, suppressed osteoblastogenesis in MM is further mediated by dysregulation of cell surface molecules involved in OB-OC communication (i.e., ephrinB2-EphB4). Bidirectional signaling between the cell-surface molecules ephrin ligands and Eph receptors controls numerous processes including OB-OC communication[104]. Specifically, MSCs and OBs express both ephrinB2 and EphB4, whereas OC precursors mainly express ephrinB2. The ephrinB2-EphB4 signaling (from OCs to OBs) stimulates OB differentiation and leads to new bone formation; on the other hand, EphB4-ephrinB2 signaling (from OBs to OCs) blocks OC differentiation[105]. MSCs from myeloma patients have reduced expression of both ephrinB2 and EphB4 due to interacting myeloma cells, which results in reduced osteogenic differentiation as compared to dMSCs, and in stimulation of osteoclastogenesis[105].
A secondary consequence of suppression of OB differentiation is that it renders an excess of MSCs/immature OBs in the BM which would enhance OC activation due to higher expression of RANKL, activin A and reduced secretion of OPG, as compared to mature OBs[106,107]. These MSCs/immature OBs pool would also further support myeloma progression and survival by providing higher levels of cytokines and growth factors than mature OBs.
COMPARISON BETWEEN MSCs FROM MM PATIENTS AND HEALTHY DONORS. CONTRIBUTION OF pMSCs TO MYELOMA BONE DISEASE
A number of studies have compared MSCs derived from the BM of newly diagnosed myeloma patients (pMSCs) and those from healthy donors (dMSCs) (reviewed in[108]), in an attempt to gain insight into their role in the pathophysiology of MM and MBD. Although MSCs from both origins similar adipogenic and chondrogenic potential, pMSCs functionally and genetically differ from their healthy counterparts. A summary of main similarities and differences found between BM-derived pMSCs and dMSCs is shown in Table 2.
Table 2.
Main similarities and differences between bone marrow mesenchymal stromal cells from myeloma patients and mesenchymal stromal cells from healthy donors
| Study | Assay | Description |
| Similarities | ||
| Adipogenic differentiation | Oil O Red staining | Both pMSCs and dMSCs showed accumulation of lipid-rich vacuoles[109,194] |
| Chondrogenic differentiation | Toluidine Blue staining | Chondrogenic differentiation potential was similar between pMSCs and dMSCs[109] |
| Differences | ||
| Chromosomic alterations | CGH arrays, FISH | pMSCs did not carry the genomic abnormalities detectable in their correspondent myeloma cells[111,194,195]. Only pMSCs showed several non-recurrent chromosomal gains and losses (> 1 Mb size) and “hot-spot” regions with discrete (< 1 Mb) genomic alterations[195] |
| Gene expression profiling | Gene expression microarray | Among 145 differentially expressed genes between pMSCs and dMSCs, 46% accounted for tumor-microenvironment cross-talk. Functional assignment revealed their implication in tumor-support (e.g., GDF15), angiogenesis (e.g., ANGPTL4, PAI1, SCG2), and contribution to bone disease (e.g., NPR3, WISP1, EDG2)[111]. Even a distinct transcriptional pattern was found associated to the occurrence of bone lesions in pMSCs[113] |
| Immunophenotype | Flow cytometry | Although few significant differences in cell surface marker expression were found between dMSCs and pMSCs, the latter expressed reduced VCAM1 and fibronectin[196], and higher ICAM1[197] compared to dMSCs |
| Bone formation markers | qPCR, WB | Expression of bone formation markers (i.e., osteocalcin and osteopontin), master transcription factors of osteogenic differentiation (i.e., Runx2/Cbfa1 and Osterix) and TAZ (a Runx2/Cbfa1 transcriptional co-activator) was lower in pMSCs than in dMSCs[109] |
| Expression and secretion of growth factors/cytokines/ chemokines | RT-PCR, ELISA | Compared to dMSCs, pMSCs showed increased expression of IL-1β[111], IL-3[112], IL-6[111,112,194,198], IL-10[199], BAFF[199], GDF15[111,198], TNFα[112], TGFβ1[112,198], DKK1[111,121,198], RANKL[112], AREG[111], and decreased expression of TGFβ2, TGFβ3 and FasL[112] |
| Senescence profile | β-gal staining, propidium iodide DNA staining, qPCR | pMSCs showed an early senescence state compared to dMSCs, as assessed by increased expression of senescence-associated β-galactosidase, increased cell size and accumulation of cells in S phase[198] |
| Immunoability | Co-cultures of MSCs and lymphocytes or PBMCs | pMSCs exhibited reduced efficiency to suppress T-cell proliferation compared to that of dMSCs[112,194,198] |
| Angiogenic potential | qPCR, ELISA, tube formation assay | Angiogenic factors (bFGF, HGF and VEGF) were elevated in the CM of pMSCs compared to dMSCs. Besides, CM from pMSCs significantly promoted proliferation, chemotaxis and capillary formation of HUVECs compared to dMSCs[200] |
| Controversial points | ||
| Proliferation rate | Cell density, CFU-F | Whereas some studies did not find differences in CFU-F number and cell density between dMSCs and pMSCs[111], others found a deficient proliferative potential in pMSCs which could be partly explained by the reduced expression of receptors for several growth factors[121] |
| ALP expression and activity | BCIP-NBT staining and pNPP hydrolysis | ALP expression/activity did not differ between MSCs from both origins[111], whereas other authors found it was significantly reduced in pMSCs compared to dMSCs, with lowest levels in pMSCs from patients with bone lesions[110] |
| Matrix mineralization | Alizarin Red and Von Kossa staining | Some groups have reported a significative reduction of matrix mineralization by pMSCs relative to dMSCs[110,111,198], although others have not observed those differences[121,194] |
| Hematopoietic stem cell support | Long-term co-cultures | Some authors reported that the ability to support the growth of hematopoietic stem cells did not differ between dMSCs and pMSCs[111,194], whilst others found that pMSCs better supported CD34+ progenitor expansion[198] |
pMSCs: Mesenchymal stromal cells from myeloma patients; dMSCs: Mesenchymal stromal cells from healthy donors; CGH: Comparative genomic hybridization; FISH: Fluorescence in situ hybridization; qPCR: Quantitative PCR; WB: Western blot; RT-PCR: Reverse transcription-PCR; PBMC: Peripheral blood mononuclear cells; CM: Conditioned media; HUVECs: Human umbilical vein endothelial cells; CFU-F: Colony forming unit-fibroblast assay; BCIP-NBT: Bromo-chloro-indolyl-phosphate and nitro blue tetrazolium staining; pNPP: p-nitrophenyl phosphate; ALP: Alkaline phosphatase; IL-3: Interleukin-3; TNFα: Tumor necrosis factor α; TGFβ1: Transforming growth factor β1; DKK1: Dickkopf-1; RANKL: Receptor activator of NFκB ligand; NFκB: Nuclear factor-κB.
Relative to the contribution of MSCs to the pathogenesis of MBD, and despite some opposite results within groups, several features of pMSCs readily reflect their reduced osteogenic potential as compared to their healthy counterparts (e.g., reduced expression of bone formation markers and critical transcription factors in OB differentiation-Runx2/Cbfa1, Osterix and TAZ)[109]; reduced expression and activity of early OB marker alkaline phosphatase (ALP)[110]; reduced matrix mineralization under osteogenic conditions[110,111]; increased expression of OB inhibitory factors (DKK1, IL-3, IL-1β, TGFβ)[111,112], and a discernible gene expression signature for pMSCs with or without osteolytic lesions[113]. These characteristics on pMSCs are likely the consequence of myeloma cell interactions and exposure to multiple soluble OB inhibitory factors and microenvironment conditions (e.g., hypoxia)[114] as occurring in the BM milieu of myeloma patients. Most of these studies have been conducted in MSCs after in vitro expansion and, in the case of pMSCs, after long-term absence of interaction with myeloma cells; thus, the presented differences between dMSCs and pMSCs may have been retained in vitro likely by epigenetic mechanisms.
Importantly, pMSCs not only contribute to MBD because of their reduced osteogenic potential, but also because they ultimately lead to the differentiation and/or activation of OCs at various levels (Figure 3): (1) increased RANKL/OPG ratio: interacting myeloma cells upregulate the expression of RANKL in MSCs, whereas the expression of OPG is reduced, thus favouring osteoclastogenesis and OC activation through RANKL-RANK signaling; (2) augmented secretion of activin A: interaction with myeloma cells leads to increased secretion of activin A in MSCs via adhesion-mediated c-Jun N-terminal kinase (JNK) activation[61]. Besides inhibiting OB differentiation, increased activin A levels would stimulate OC formation and activity[60]; (3) diminished expression of eprhinB2 and EphB4 in pMSCs: myeloma cells reduce the expression of both ephrinB2 and EphB4 in pMSCs[105] thereby dysregulating the ephrinB2/EphB4 signaling between OCs and OBs. Besides an impaired OB differentiation (due to reduced ephrinB2-EphB4 signaling from OCs to OBs), diminished EphB4-ephrinB2 signaling (from OBs to OCs) would no longer prevent OC formation, allowing increased osteoclastogenesis; and (4) increased Wnt5a production by MSCs: Wnt5a has been identified as a myeloma growth factor being overexpressed by myeloma plasma cells and pMSCs (as compared to their healthy counterparts)[115]. Interestingly, we have found the upregulated expression of Wnt5a in MSCs after interaction with myeloma cells (our unpublished results), which would further contribute to its enhanced production in the BM. Recently, a link between Wnt5a and increased osteoclastogenesis has been found by identification of signaling between Wnt5a (secreted by OB-lineage cells) and the membrane Ror2 receptor (expressed on OC precursors), leading to upregulated RANK expression in the latter and increased sensitivity to RANKL[116]. Accordingly, myeloma interacting-MSCs would heighten the production of Wnt5a which in turn would increase OC formation and activity.
Figure 3.
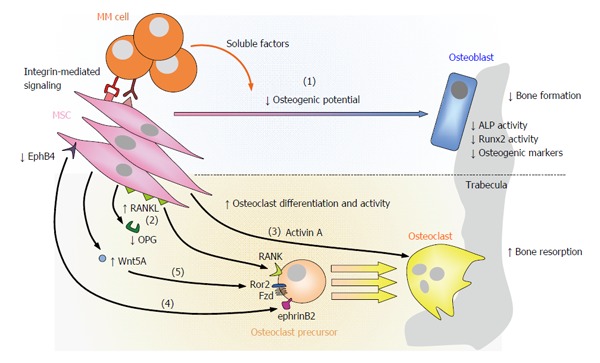
Contribution of mesenchymal stromal cells to myeloma bone disease. In MM, MSCs contribute to the development of osteolytic lesions not only because of their reduced osteogenic potential [(1)], but also because they promote OC differentiation and hyperactivation at various levels: pMSCs upregulate the expression of RANKL and reduce that of OPG [(2)]; pMSCs augment the secretion of activin A [(3)]; diminished EphB4-ephrinB2 signaling from pMSCs/OBs to OCs allows osteoclastogenesis [(4)]; increased Wnt5a secretion by pMSCs interacting with myeloma cells enhances RANK expression in OC precursors through Ror2, ultimately increasing their sensibility to RANKL [(5)]. RANKL: Receptor activator of NFκB ligand; NFκB: Nuclear factor-κB; OPG: Osteoprotegerin; RANK: Receptor activator of NFκB; EphrinB2: Ephrin-B2 ligand; EphB4: Eph receptor B4; Ror2: Receptor tyrosine kinase-like orphan receptor 2; MSCs: Mesenchymal stromal cells; MM: Multiple myeloma; OC: Osteoclast; OB: Osteoblast; ALP: Alkaline phosphatase; MM: Multiple myeloma.
Thus, both a reduced osteogenic capacity and a hyperstimulation of OCs[68] at various levels constitute the two major contributions of MSCs to the development of MBD. Other contributions of MSCs, such as modification of ECM components (in relation with retention of OC-activating and OB-inhibiting factors or growth factors) have not been addressed here, but would likely participate in the pathophysiology of the disease.
PATHOPHYSIOLOGY OF IMPAIRED OSTEOGENIC DIFFERENTIATION OF pMSCs: THERAPEUTIC OPPORTUNITIES BASED ON MSC TARGETING
In this section, we will review the major signaling pathways involved in OB differentiation and OB function [e.g., Wnt, Notch, BMP and CCL3 signaling, ephrin-Eph axis, unfolded protein response (UPR)]; at the same time, we also discuss potential therapies targeting members of these pathways in order to restore OB differentiation and activity in patients with MBD.
Bisphosphonates (BPs) are the current mainstay for the treatment of bone complications in MM patients, generally administered as supportive therapy in addition to anti-myeloma agents. BPs are pyrophosphate analogs with great affinity for mineralized matrix surfaces, causing inhibition of OC function and OC apoptosis[117]. Second generation nitrogen-containing BPs now in use, such as pamidronate and zoledronic acid, have been shown to be superior to previous BPs and to more effectively reduce the incidence of SREs[33]. However, adverse side-effects after long BP treatment (i.e., osteonecrosis of the jaw, kidney failure and accumulation of bone microfractures[34,118]) and eventual progress of bone disease under treatment[33], has prompted preclinical and clinical studies for the use of alternate bone anabolic agents which may achieve a more efficacious and improved management of MBD (Table 3).
Table 3.
Therapeutic targets, bone anabolic drugs and preclinical/clinical studies in the context of myeloma bone disease or other bone diseases
| Drug | Mechanism of action | Signaling pathway | Cell target | Preclinical studies | Phase of clinical trials |
| BHQ880 | Neutralizing anti-DKK1 antibody | Wnt | MSC, MMPC | [122,123,125] | II[126,127] |
| Romosozumab (AMG785) | Neutralizing anti-sclerostin antibody | MSC | [79,83] | II (postmenopausal osteoporosis)[129] | |
| LiCl | GSK3β inhibitor | MSC, MMPC | [131] | NA | |
| DAPT | γ-secretase inhibitor | Notch | MSC, MMPC | [110,142] | NA |
| GSI15 | OC, MMPC | [139] | |||
| Bortezomib and second generation PIs | Proteasome inhibitor | UPR | MSC, OC, MMPC | [179,180,201] | Bortezomib and carfilzomib: Approved Oprozomib: I/II Ixazomib: III |
| RAP-011 (mouse) Sotatercept/ACE-011 (human) | Decoy receptor neutralizing activin A | BMP | MSC, OC, MMPC | [61,62] | II[153,154] |
| SB431542 | TGFβ inhibitor | MSC | [150] | NA | |
| Ki26894 | MSC | ||||
| MLN3897 | CCR1 antagonists | CCL3 | MSC, OC, MMPC | [58,160] | NA |
| CCX721 (mouse) CCX354-C (human) | OC, MMPC | [157] | II (rheumatoid arthritis)[161] |
MSC: Mesenchymal stromal cell; MMPC: Multiple myeloma plasma cell; OC: Osteoclast; NA: Not applicable; DKK1: Dickkopf-1; GSK3β: Glycogen synthase kinase 3β; DAPT: N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester; TGFβ: Transforming growth factor β; UPR: Unfolded protein response; BMP: Bone morphogenetic protein; CCR1: Chemokine (C-C Motif) receptor 1; CCL3/MIP1α: Macrophage inflammatory protein 1-α.
Wnt signaling
Role in MBD: Wnts are a family of 19 secreted glycoproteins that trigger several pathways involved in cell face determination, proliferation, migration and polarity, both in embryogenesis and regeneration of adult tissues. Specifically, Wnt signaling in MSCs is critical for OB differentiation and hence, for bone metabolism[119]. Literature categorizes this pathway in canonical or non-canonical, depending on the requirement of β-catenin or not, respectively. In the Wnt canonical pathway and in the absence of Wnt stimulation, cytoskeletal β-catenin is phosphorylated by a multi-protein destruction complex and undergoes ubiquitin-mediated degradation in the proteasome. Upon binding of canonical Wnt ligands to a Frizzled (Fzd) receptor and a LRP co-receptor, the destruction complex is inhibited allowing β-catenin to translocate into the nucleus where it interacts with T-cell factor/lymphoid enhancer factors to activate transcription of target genes involved in osteoblastogenesis. On the other hand, the two better characterized Wnt non-canonical pathways are the planar cell polarity and the Wnt/Ca2+ pathways, mainly implicated in cell polarity and cell migration mediated by cytoskeletal-actin rearrangements[119].
Several secreted factors may negatively regulate canonical and non-canonical Wnt signaling: DKK1-4 and sclerostin directly bind to the LRP5/6 co-receptor limiting its availability to Wnt ligands; on the other hand, sFRP1-5 or Wnt inhibitory factor 1 (Wif1), directly bind to Wnt ligands, preventing their functional association with Fzd receptors. Since Wnt signaling plays such a critical role in the osteogenic differentiation of MSCs, alterations in this pathway may lead to skeletal disorders as observed in MBD. In fact, newly diagnosed MM patients showed elevated DKK1[78], sclerostin[120] and sFRP3[84] levels compared to that of healthy donors both in BM and peripheral blood plasma, correlating with the presence of bone lesions.
Although malignant plasma cells are the main source of these Wnt antagonists in the BM[78,79,84-86], pMSCs secreted higher DKK1 levels than their healthy counterparts[111,121]. Similarly, sclerostin was found to be produced by OBs derived from pMSCs co-cultured with a MM cell line, further contributing to suppression of OB differentiation and function in MBD[83]. Even though non-canonical Wnt signaling is not usually associated to osteogenic functions, Wnt5a ligand seems to be at least partially responsible for the osteogenic differentiation of MSCs in the BM[102,103]. As we commented before, this pathway was inhibited in MSCs from myeloma patients, due to downregulation of Ror2 co-receptor expression[101].
Therapeutic approaches: Given that Wnt inhibition (by DKK1, sclerostin and sFRPs) has been involved in the development of osteolytic lesions, modulation of Wnt signaling by different approaches constitutes a potential clinical strategy in MBD.
BHQ880 is a humanized monoclonal antibody against DKK1, which has been shown to reverse the hampering effects of this Wnt inhibitor on OB formation. Treatment with anti-DKK1 or BHQ880 therapy prevented OB suppression and reduced the development of osteolytic lesions in in vivo studies with mouse models of murine/human MM[122-125]. Furthermore, BHQ880 showed an anti-myeloma effect, overcoming the growth advantage conferred by MSCs to MM cells in co-culture through downregulation of cell adhesion and IL-6 production by MSCs[123], which was also corroborated in in vivo models[122,123,125]. A phase I/II study of BHQ880 in relapsed or refractory MM patients with or without BPs besides standard chemotherapy (NCT00741377), assessed the bone anabolic efficacy of this DKK1 inhibitor through an increase in bone mineral density and regulation of bone metabolism markers[126]. Ongoing phase II studies of BHQ880 in untreated patients with high risk smoldering myeloma (NCT01302886), have reported increased vertebral strength but no anti-MM activity[127].
Inhibition of sclerostin by monoclonal antibodies has been explored in different bone disorders, leading to increased bone formation, bone mass and bone strength in preclinical models in mouse, rats and monkeys (reviewed in)[128]. The development of romosozumab (AMG785), a humanized monoclonal antibody to sclerostin, has allowed its translation to clinical trials. In phase I studies, romosozumab was administered to healthy men and postmenopausal women resulting in a dose-related increase in bone formation markers, a decrease in bone resorption markers, and significatively increased bone mineral density at the lumbar spine and total hip[129]. A phase II trial is currently ongoing to compare the efficacy of romosozumab with alendronate and teriparatide in the treatment of postmenopausal women with low bone mineral density (NCT00896532). In the MM setting, in vitro assays with neutralizing anti-sclerostin antibodies restored OB function as assessed by increased expression of bone formation markers and transcription factors Fra-1, Fra-2 and JunD, modulation of the unbalanced OPG-RANKL ratio and accumulation of β-catenin[79,83].
Wnt3a administration was also shown to enhance Wnt signaling on OB progenitors, and promoted bone formation and attenuated MM growth in a myeloma SCID-hu mouse model[130]. Inhibition of glycogen synthase kinase 3β (GSK3β), a serine-threonine kinase involved in the phosphorylation of β-catenin for proteasome degradation has also been explored. GSK3β inhibitors such as lithium chloride[131] ameliorated the development of MBD and inhibited tumor growth in a disseminated 5TGM1 mouse model of MM, despite some concerns about the possibility that this strategy may stimulate myeloma growth[132].
Notch signaling
Role in MBD: Evolutionarily conserved Notch signaling plays an important role during embryonic and postnatal life by regulating cell fate determination, proliferation, differentiation and apoptosis in a spatio-temporal manner[133]. Notch is a family of four (Notch1-4) transmembrane receptors activated by single-pass membrane ligands (Jagged1-2 and Delta like-1/3/4). Upon Notch-ligand interactions, the γ-secretase complex cleaves the Notch intracellular domain, which then translocates to the nucleus to regulate the transcription of target genes, including Hairy enhancer of split (Hes) and Hes-related to YRPW motif[133]. Notch signaling plays a key role in skeletal development and remodeling maintaining MSCs in an undifferentiated stage by suppressing OB differentiation (directly repressing Runx2/Cbfa1 activity[134] or inhibiting Wnt/β-catenin pathway[135]). However, once Notch signaling is activated in MSCs, it stimulates early osteoblastic proliferation[134]) leading to the maintenance of an immature OB pool. Considering the well-established role of Notch in osteogenic differentiation, dysregulation of this pathway is associated with human diseases affecting the skeleton. In this sense, alterations in Notch signaling have been reported in pMSCs[110], which maintain high gene expression levels of some Notch signaling molecules (e.g., Notch1 receptor and the transcription factors Hes1 and Hes5) as compared to dMSCs, which suggests an inhibitory role of these molecules in OB differentiation.
On the other hand, it has also been reported that activation of Notch signaling may regulate osteoclastogenesis depending on the ligands and receptor isoforms involved. Notch1 and Notch3 are able to suppress OC differentiation and activity via ligand-mediated receptor activation[136], whereas Notch2 is upregulated during RANKL-induced osteoclastogenesis and enhances OC formation through increased NFATc1 expression[137].
Therapeutic approaches: To date, Notch signaling blockade has focused on inhibition of the γ-secretase complex, the intramembrane-cleaving protease with a growing list of protein substrates, including Notch receptors and the amyloid precursor protein involved in Alzheimer’s disease[138]. Treatment with N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT), a γ-secretase inhibitor (GSI), restored the osteogenic ability of MSCs both in vitro (by increasing the gene expression of bone formation-related markers, ALP activity and matrix mineralization) and in vivo (as assessed by increased OB cell number at the endocortical surface in naive mice)[110]. However, GSI treatment failed to stimulate OB formation in a MM model, probably due to the lack of activity over MM cells[110]. Looking at the OC compartment, treatment with the Notch inhibitor GSI15 blocked MM cell-induced activation of OCs, reinforcing evidences for the use of GSIs as a therapeutic option in MBD[139]. Other preclinical studies performed with GSIs (GSI-XII, MRK003, DAPT) in the myeloma setting prevented MM cell migration, proliferation, clonogenic ability, resistance to apoptosis, angiogenesis as well as tumor growth in vitro and in a SCID-human model of MM[140-144]. Moreover, it has been found that combined treatment of GSIs and established anti-MM drugs (such as bortezomib[145], melphalan, doxorubicin[144]), or other agents such as ABT-737[143] or Akt1/2 inhibitors[141], results in a synergistic cytotoxic effect on myeloma cells. In this sense, combining Notch inhibitors with anti-MM drugs holds promise as a valuable therapeutic approach for the treatment of both MM and MBD.
BMP signaling (activin A and TGFβ)
Role in MBD: Activin A is a TGFβ superfamily member mainly secreted by BM-derived MSCs from myeloma patients and OCs[61]. MM cell lines and primary MM plasma cells secrete very low or undetectable levels of activin A, but co-culture with MSCs induces the secretion of activin A in the latter via JNK pathway activation[61]. Activin A binds to the serine/threonine kinase activin A receptor, type IIA (ActRIIA), which recruits and phosphorylates the receptor type IB (ActRIB), leading to phosphorylation of cytoplasmic Smad2/3 proteins. This complex associates with Smad4, which translocates into the nucleus and controls gene expression[146]. Activin A has several roles in the development of osteolytic lesions: it enhances OC formation and activity (inducing nuclear translocation of NFκB and RANK expression in OC precursors)[60], inhibits OB differentiation (via Smad2-mediated DLX5 downregulation)[61] and alters the extracellular matrix maturation phase[147]. Accordingly, high levels of circulating activin A correlate with extensive bone disease and inferior survival[148].
TGFβ is abundantly deposited in the bone matrix and the enhanced bone resorption in MM bone lesions causes a marked increase in the release and activation of this factor[149,150]. Although TGFβ enhances the recruitment and proliferation of OBs progenitors, it potently suppresses later phases of OB differentiation, maturation and matrix mineralization[149,150].
Therapeutic approaches: Sotatercept (ACE-011) or RAP-011 are chimeric proteins derived from the fusion of the extracellular domain of ActRIIA and the Fc domain of human IgG1 or murine IgG2a, respectively. These proteins sequester ligands of ActRIIA (activin A among others), interfering with Smad signaling and restoring the uncoupled bone remodeling.
Treatment of MSCs with RAP-011 increased OB differentiation, even in the presence of MM cells, by rescuing DLX5 expression[61]. The bone anabolic effect of RAP-011 could be translated to the in vivo setting on a SCID-hu model of MBD. RAP-011 treatment prevented bone destruction and reduced MM tumor burden[61], providing the basis for clinical testing in myeloma patients suffering from severe bone disease. Similar results were obtained in healthy and ovariectomized mice[151], in murine models of osteolytic disease induced by MM cells and breast cancer cells[62], and in non-human primates[152].
The human counterpart of RAP-011, sotatercept, has been evaluated in phase II studies in MM patients with osteolytic lesions receiving a regimen of melphalan, prednisone and thalidomide (NCT00747123); after sotatercept treatment, patients showed an increase in bone formation markers (bone-specific ALP), improvement in osteolytic lesions, reduction of bone pain and myeloma burden[153]. Other studies of sotatercept in combination with lenalidomide and dexamethasone in patients with relapsed and/or refractory myeloma are currently recruiting participants (NCT01562405). In accordance with these studies, a phase I trial of sotatercept in postmenopausal women has evidenced a bone anabolic and anti-resorptive effect, as observed by sustained increase in bone formation markers (bone-specific ALP) and decrease in bone resorption markers (CTX and TRACP-5b)[154].
Relative to TGFβ, pharmacological inhibition of the TGFβ type I receptor kinase (TβRI), SB431542 and Ki26894, potently enhanced OB differentiation in vitro, releasing MSCs from their differentiation arrest and facilitating the formation of terminally differentiated OBs[150]. In vivo administration of these agents showed anabolic and anti-catabolic effects on bone, in parallel with suppression of MM cell growth[150,155]. Therefore, TGFβ appears to be an important therapeutic target in MBD.
CCL3 signaling
Role in MBD: CCL3 (MIP1α) is a chemokine mainly secreted by OCs and MM cells, which binds to G-protein-coupled receptors CCR1 and CCR5. Both chemokine receptors are expressed in MM cells, MSCs/OBs and OCs[58,156], being CCR1 the major receptor on OC precursors and mature OCs[157]. The CCL3 pathway is not only involved in the survival, growth and migration of MM cells[156], but CCL3 also readily contributes to the imbalance between bone formation and bone resoption by enhancing OC formation[158] and hampering OB function[58]. BM plasma CCL3 levels were found to be elevated in MM patients, correlating directly with the extent of MBD and inversely with survival[159] and osteocalcin expression[58].
Therapeutic approaches: Preclinical in vitro and in vivo studies have been performed either targeting CCL3 (antisense construct to human CCL3[56] and neutralizing antibody against CCL3[57]), or the CCR1 receptor (small-molecule CCR1 antagonists MLN3897[160] and CCX721[157]). These treatments reduced myeloma tumor burden and prevented osteolysis, thus providing a strong rationale for the clinical evaluation of these compounds in the treatment of MBD.
Therapeutic strategies towards the inhibition of the CCL3 pathway have mainly focused on their effect on the OC compartment[56,57,157,160], although there is also preclinical evidence of the anabolic effect of the CCR1 inhibitor MLN3897 in osteogenic differentiation[58]. In the latter study, in vitro CCR1 inhibition suppressed CCL3-induced ERK activation and restored both Osterix and osteocalcin expression in OBs differentiated from a human stromal cell line; in the SCID-hu murine model of MM, treatment with MLN3897 reduced tumor burden, decreased OC number and increased both the trabecular bone area and the percentage of osteocalcin-positive area in the trabeculae[58]. These studies set the stage for development of clinical trials to assess the effects of CCR1 inhibitors in MM. CCX354, the human structural analog of CCX721, is currently in phase II studies for rheumatoid arthritis, exhibiting clinical activity with a good safety and tolerability profile[161].
Eph/ephrin signaling
Role in MBD: Another example of a bidirectional signaling pathway capable of regulating both osteoblastic and osteoclastic lineages is the one mediated by Eph receptors and ephrins (Eph receptor-interacting ligands). There are two classes of ephrins: the B class (ephrin B1 to B3), which are ligands for EphB tyrosine kinase receptors (B1 to B6), and the A class (ephrin A1 to A5), which are ligands for glycophosphatidylinositol-anchored EphA receptors (A1 to A10)[162]. Eph-ephrin complexes signal bidirectionally to orchestrate several cellular processes including immune regulation, neuronal development and cancer metastasis. The Eph/ephrin system is expressed by BM microenvironment cells (including OBs and OCs), and growing evidence point out the pivotal role of this pathway in the control of normal and pathological bone remodeling[163]. Specifically, the ephrinB2/EphB4 axis has been involved in bone homeostasis: reverse signaling through ephrinB2 ligand (expressed by OCs and MSCs/OBs) limits OC activity, whereas forward signaling through EphB4 receptor (expressed by MSCs and OBs) enhances OB differentiation[104]. Dysregulation of Eph/ephrin function may also contribute to other bone pathological conditions such as osteoarthritis, rheumatoid arthritis or osteosarcoma[163].
In the MBD context, Pennisi et al[105] have found reduced levels of ephrinB2 and EphB4 in MSCs from MM patients as compared to their healthy counterparts, and also in OBs/OCs of myelomatous bones compared to non-myelomatous bones. In co-culture experiments, MM cell lines markedly downregulated EphB4 receptor and ephrinB2 ligand in human MSCs, thus confirming a MM cell-induced imbalance of ephrinB2/EphB4 signaling in the MSC-OB lineage[105].
In addition to the EphB4/ephrinB2 axis, OB-OC, OB-OB and OC-OC interactions through other ephrins and Eph receptors do in fact occur and participate in bone homeostasis. For example, it has been reported that OC-derived ephrinA2/EphA2 interaction enhanced OC differentiation via reverse signaling, whereas ephrinA2 inhibited osteoblastogenesis through OB-derived EphA2 receptor via forward signaling, contributing to the transition phase of bone remodeling from bone formation to bone resorption[164]. Future studies about the expression/function of A class ephrins/Eph in MSCs from MM patients may thus unravel new governing mechanisms of impaired OB differentiation in MBD.
Therapeutic approaches: The dual role of EphB4/ephrinB2 signaling in the OB/OC compartment is especially attractive as a therapeutic approach in MBD, since its activation is able to promote both OB differentiation and function and attenuate OC formation and bone resorption. Pennisi et al[105] performed experiments with two chimeric proteins (ephrinB2-Fc and EphB4-Fc) in an attempt to induce forward and reverse signaling in MSCs and OC progenitors respectively, and to observe their effects on OB/OC differentiation. Treatment of MSCs with ephrinB2-Fc induced forward signaling (as assessed by phosphorylation of the EphB4 receptor), and increased osteocalcin expression and matrix mineralization of OBs under osteogenic conditions[105]. On the other hand, EphB4-Fc treatment had an inhibitory effect in OC progenitors (as checked by phosphorylation of ephrinB2 and downregulated expression of NFATc1 and reduced numbers of TRAP+ OCs), but no effect in MSCs. In the same line of reasoning, both ephrinB2-Fc and EphB4-Fc treatments in the SCID-hu model of MM, increased bone formation and OB number, but only EphB4-Fc reduced the number of OCs[105] (since no expression of EphB4 was found in the OC lineage). These results supported the notion that activation of either forward or reverse EphB4/ephrinB2 signaling affects bone remodeling, resulting in increased bone formation. Moreover, the anti-myeloma effect of ephrinB2-Fc and EphB4-Fc treatments was evaluated in myelomatous bones, as assessed by the area of myeloma infiltration and the human Ig monoclonal component; however, only EphB4-Fc-treated SCID-hu mice showed a reduction in tumor burden. Since no effect was found for EphB4-Fc on MM cells in vitro, the anti-myeloma activity of this molecule was probably due to its modulatory effects on the BM environment (inhibition of osteoclastogenesis and neovascularization and stimulation of OB activity)[105]. In this sense, upregulation of the endogenous expression of EphB4 in pMSCs or osteoprogenitor cells of myeloma patients (e.g., by Wnt3a administration-since EphB4 receptor is a Wnt signaling target-or directly by EphB4-Fc treatment) could restore coupling of bone homeostasis and simultaneously reduce MM tumor burden in MM patients with bone affection.
Unfolded protein response pathway
Role in MBD: The endoplasmic reticulum (ER) is a membranous compartment present in eukaryotic cells which controls the synthesis, folding and trafficking of proteins to be secreted, as well as calcium storage and synthesis of membranes[165]. Increased load of unfolded or misfolded proteins within the ER triggers a sophisticated mechanism known as the UPR, in an attempt to refold those proteins and to allow cellular adaptation to the imbalance in the protein folding homeostasis, referred as ER stress. Briefly, when unfolded proteins accumulate in the lumen of the ER, three coordinated pathways are activated by the transmembrane ER stress-sensor proteins, namely: PKR-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1). The activation of these ER sensor proteins leads to the induction of a battery of transcription factors [orchestrated by ATF4, ATF6 and X box-binding proteins (XBP1s)] to promote the transcription of ER chaperone proteins and folding enzymes to increase the protein folding capacity of the ER, as well as proteins controlling the ER-associated degradation machinery, a mechanism by which misfolded proteins are retro-translocated into the cytosol for degradation by the proteasome. Alternatively, prolonged or severe exposure to ER stress may result in the cell undergoing apoptosis[165-167].
Although ER stress often arises in pathological situations, specialized secretory cells such as hepatocytes, insulin-producing β cells of the pancreas, plasma cells and connective tissue cells (fibroblasts, chondrocytes and OBs) are particularly sensitive to ER stress induction in their normal development and function[167]. Therefore, ER stress is essential during osteoblastogenesis through the three arms of the UPR: IRE1-XBP1s (promoting Osterix transcription)[168], PERK-ATF4 (increasing osteocalcin and bone sialoprotein expression)[169] and ATF6 (enhancing osteocalcin expression)[170].
On the other hand, a recent study showed that MSCs from MM patients displayed elevated mRNA and protein levels of endogenous XBP1s (an active transcription factor involved in the clearance of unfolded/misfolded proteins) compared with dMSCs, suggesting that the IRE1-XBP1s pathway is activated in pMSCs[171]. Experiments with overexpression of XBP1s in MSCs led to an increase in IL-6 and RANKL secretion and VCAM1 expression, which translated into an enhanced in vitro ability of MSCs to support MM cell growth and OC formation[171]. Future studies exploring the expression and role of the other components of the UPR in MSCs would be of particular value for disrupting the protective effects of the MM microenvironment on tumor cell growth and bone destruction.
Therapeutic approaches: Plasma cells seem to be exquisitely sensitive to their core protein handling machinery due to the large amounts of immunoglobulins that these cells produce and secrete. The ubiquitine-proteasome pathway, linked to the UPR response to discard misfolded proteins, has become a potential drug target for the treatment of several tumors including MM[172]. Bortezomib was the first-in-class proteasome inhibitor (PI) introduced in the clinical practice with a significant benefit in terms of anti-myeloma response rate and overall survival in both front-line and relapsed/refractory settings[23]. Moreover, bortezomib not only reduces myeloma tumor burden, but directly restrains the progression of MBD, clinically evidenced by changes in bone turnover markers and radiologic data favouring bone healing[173,174]. The beneficial impact of bortezomib on bone metabolism is not merely secondary to its anti-myeloma activity, but rather this agent directly targets the OC and MSCs/OB populations, both hampering osteoclastogenesis and OC resorption and promoting osteoblastogenesis and OB function[175-177].
A next-generation of PIs, including peptide boronic acid analogs (delanzomib and ixazomib), peptide epoxyketones (carfilzomib and oprozomib) and a β-lactone compound (marizomib) are have been developed to address the shortcomings of bortezomib treatment with the aim of retaining or improving bortezomib efficacy[178]. Our group has investigated the potential bone anabolic and anti-resorptive effects of three of these second-generation PIs (i.e., carfilzomib, its orally bioavailable analog oprozomib and ixazomib) in preclinical models of MM[179,180]. In vitro studies evidenced that the three PIs were able to promote osteoblastogenesis and OB function (as assessed by augmented expression of bone formation markers, increased ALP activity and enhanced bone matrix mineralization), and to inhibit OC formation and resorption (through disruption of RANKL-induced NFκB signaling together with reduced expression of integrin αVβ3 and F-actin ring disruption)[179,180]. These effects were subsequently corroborated in vivo, since the three PIs provided a marked benefit in associated bone disease, sustained by bone anabolic and anti-resorptive activities[179,180].
Moreover, the UPR was identified as a crucial pathway affected by PI-treatment of MSCs and osteoprogenitors resulting in enhanced osteoblastogenesis. Treatment of a BM-derived mesenchymal stromal cell line with PIs led to increased protein levels of the ER stress sensor IRE1α. IRE1α knockdown by siRNAs significantly diminished PI-enhanced mineralized bone formation, thus underscoring the crucial role of IRE1α in the promotion of OB activity by these agents[180]. In the same line, Nakamura et al[181] recently reported a critical role for other ER stress mediator, ATF4, in bortezomib-mediated osteoblastogenesis, and suggested the optimization of a dose regimen for PI-treatment in order to obtain a maximal bone anabolic response (lower doses) avoiding the induction of pro-apoptotic pathways in the MSC-OB lineage (higher doses). It is thought that the adaptative threshold for myeloma plasma cells and OBs is quite different, since UPR induced by PIs (at the same range of doses) results in a cytotoxic effect in MM cells[172] whereas promotes OB differentiation on mesenchymal precursors[179].
Other therapeutical approaches on OB differentiation and function
Inhibition of tyrosine kinases: Several studies showed that the tyrosine kinase inhibitor imatinib mesylate directly promoted OB differentiation and stimulated osteogenic gene expression and mineralization, majorly by inhibiting PDGFR function on osteoprogenitors[182,183]. This partially explained the increased trabecular bone volume and bone mineral density of long-term imatinib treated patients[182]. As expected, subsequent studies with dasatinib, a second generation tyrosine kinase inhibitor with more potency and broader target profile, also evidenced enhanced OB differentiation from mesenchymal precursors and promotion of OB activity both in vitro and in vivo[184-187]. Preclinical anti-myeloma and anti-angiogenic efficacy of dasatinib was also reported, but attained at higher concentrations than those required for the bone anabolic effect of this drug, and which were cytotoxic for mesenchymal osteoprogenitors and OBs[52,187]. Therefore, the latter suggests that if dasatinib is to be used for the treatement of MBD, it should be administered in combination with another anti-myeloma agent.
MSC cytotherapy for MBD: MSCs have been considered as excellent candidates for cytotherapy studies due to their immunoprivileged nature, their ability to migrate to damaged and tumor tissues, together with their capacity to differentiate to several mesenchymal lineages[188]. Some concerns have been raised, however, for the use of MSCs in the treatment of MBD since interacting BM MSCs have been shown to support the proliferation, survival, migration and chemotherapeutic resistance of MM cells[27,30,31,189]. When genetically-modified human MSCs overexpressing OPG were administered to a model of medullary myeloma with associated bone disease, they reduced OC activation and restored bone volume[190]. Moreover, human placenta or BM derived MSCs were intrabone or systemically administered in the severe combined immunodeficiency (SCID)-rab model, and found to promote bone formation, prevent MM-induced bone disease and tumor growth[191,192].
Specific delivery of RNAi-based anabolic therapy: The use of siRNA-based bone anabolic therapies in the clinic has been hampered by lack of specific targeting to bone-formation surfaces. The (AspSerSer)6 has been found to be a targeting moiety for bone formation sites in vivo, due to its great affinity to lowly crystallized hydroxyapatite and amorphous calcium phosphonate. Systemic administration of (AspSerSer)6-labeled liposomes containing osteogeneic siRNAs has been shown to be an effective therapeutic approach in a model of osteoporosis[193] and its use may also be explored in MM to promote OB function.
CONCLUSION
In conclusion, MSCs from myeloma patients are important contributors to the development of osteolytic lesions because of their reduced osteogenic potential and because they also promote OC differentiation and/or activity at various levels (increased RANKL/OPG ratio, augmented activin A secretion, uncoupled ephrinB2/EphB4 axis and because of increased Wnt5a production). We have reviewed current therapeutic approaches targeting components of signaling pathways involved in the osteogenic differentiation and maintenance of OB activity. It is likely that due to the multifactorial character of MBD, combinations of both anti-resorptive and bone-anabolic agents may be required for an effective restoration of bone homeostasis and for an additional anti-myeloma benefit.
Footnotes
Supported by Grants from the Spanish Ministry of Economía y Competitividad-Instituto de Salud Carlos III, No. PI12/02591; European Funds for Regional Development; the Spanish Health Thematic Networks of Cooperative Research in Cancer, No. RTICC RD12/0036/0058; Cellular Therapy, No. TerCel RD12/0019/0001, group 8; the Network of Centers for Regenerative Medicine and Cellular Therapy from Castilla y León; and the Spanish Society of Hematology and Hemotherapy (to Garcia-Gomez A)
P- Reviewers: Marfe G, Ribatti D, Stuppia L S- Editor: Song XX L- Editor: A E- Editor: Liu SQ
References
- 1.Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6:230–247. [PubMed] [Google Scholar]
- 2.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 3.Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970;3:393–403. doi: 10.1111/j.1365-2184.1970.tb00347.x. [DOI] [PubMed] [Google Scholar]
- 4.Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991;9:641–650. doi: 10.1002/jor.1100090504. [DOI] [PubMed] [Google Scholar]
- 5.Horwitz EM, Le Blanc K, Dominici M, Mueller I, Slaper-Cortenbach I, Marini FC, Deans RJ, Krause DS, Keating A. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy. 2005;7:393–395. doi: 10.1080/14653240500319234. [DOI] [PubMed] [Google Scholar]
- 6.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop Dj, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 7.Dazzi F, Ramasamy R, Glennie S, Jones SP, Roberts I. The role of mesenchymal stem cells in haemopoiesis. Blood Rev. 2006;20:161–171. doi: 10.1016/j.blre.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Pérez-Simon JA, López-Villar O, Andreu EJ, Rifón J, Muntion S, Campelo MD, Sánchez-Guijo FM, Martinez C, Valcarcel D, Cañizo CD. Mesenchymal stem cells expanded in vitro with human serum for the treatment of acute and chronic graft-versus-host disease: results of a phase I/II clinical trial. Haematologica. 2011;96:1072–1076. doi: 10.3324/haematol.2010.038356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrancio S, López-Holgado N, Sánchez-Guijo FM, Villarón E, Barbado V, Tabera S, Díez-Campelo M, Blanco J, San Miguel JF, Del Cañizo MC. Optimization of mesenchymal stem cell expansion procedures by cell separation and culture conditions modification. Exp Hematol. 2008;36:1014–1021. doi: 10.1016/j.exphem.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Vater C, Kasten P, Stiehler M. Culture media for the differentiation of mesenchymal stromal cells. Acta Biomater. 2011;7:463–477. doi: 10.1016/j.actbio.2010.07.037. [DOI] [PubMed] [Google Scholar]
- 11.Chanda D, Kumar S, Ponnazhagan S. Therapeutic potential of adult bone marrow-derived mesenchymal stem cells in diseases of the skeleton. J Cell Biochem. 2010;111:249–257. doi: 10.1002/jcb.22701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi M, Liu ZW, Wang FS. Immunomodulatory properties and therapeutic application of mesenchymal stem cells. Clin Exp Immunol. 2011;164:1–8. doi: 10.1111/j.1365-2249.2011.04327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, Grisanti S, Gianni AM. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–3843. doi: 10.1182/blood.v99.10.3838. [DOI] [PubMed] [Google Scholar]
- 14.Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, Risso M, Gualandi F, Mancardi GL, Pistoia V, et al. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006;107:367–372. doi: 10.1182/blood-2005-07-2657. [DOI] [PubMed] [Google Scholar]
- 15.Di Ianni M, Del Papa B, De Ioanni M, Moretti L, Bonifacio E, Cecchini D, Sportoletti P, Falzetti F, Tabilio A. Mesenchymal cells recruit and regulate T regulatory cells. Exp Hematol. 2008;36:309–318. doi: 10.1016/j.exphem.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, Galun E, Rachmilewitz J. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105:2214–2219. doi: 10.1182/blood-2004-07-2921. [DOI] [PubMed] [Google Scholar]
- 17.Raffaghello L, Bianchi G, Bertolotto M, Montecucco F, Busca A, Dallegri F, Ottonello L, Pistoia V. Human mesenchymal stem cells inhibit neutrophil apoptosis: a model for neutrophil preservation in the bone marrow niche. Stem Cells. 2008;26:151–162. doi: 10.1634/stemcells.2007-0416. [DOI] [PubMed] [Google Scholar]
- 18.Spaggiari GM, Capobianco A, Abdelrazik H, Becchetti F, Mingari MC, Moretta L. Mesenchymal stem cells inhibit natural killer-cell proliferation, cytotoxicity, and cytokine production: role of indoleamine 2,3-dioxygenase and prostaglandin E2. Blood. 2008;111:1327–1333. doi: 10.1182/blood-2007-02-074997. [DOI] [PubMed] [Google Scholar]
- 19.Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351:1860–1873. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 20.Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113:5418–5422. doi: 10.1182/blood-2008-12-195008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kyle RA, Durie BG, Rajkumar SV, Landgren O, Blade J, Merlini G, Kröger N, Einsele H, Vesole DH, Dimopoulos M, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24:1121–1127. doi: 10.1038/leu.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49:1374–1403. doi: 10.1016/j.ejca.2012.12.027. [DOI] [PubMed] [Google Scholar]
- 23.Ocio EM, Mateos MV, San-Miguel JF. Novel agents derived from the currently approved treatments for MM: novel proteasome inhibitors and novel IMIDs. Expert Opin Investig Drugs. 2012;21:1075–1087. doi: 10.1517/13543784.2012.691164. [DOI] [PubMed] [Google Scholar]
- 24.Lonial S, Kaufman JL. The era of combination therapy in myeloma. J Clin Oncol. 2012;30:2434–2436. doi: 10.1200/JCO.2011.40.6967. [DOI] [PubMed] [Google Scholar]
- 25.Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol. 2012;9:135–143. doi: 10.1038/nrclinonc.2012.15. [DOI] [PubMed] [Google Scholar]
- 26.Munshi NC, Avet-Loiseau H. Genomics in multiple myeloma. Clin Cancer Res. 2011;17:1234–1242. doi: 10.1158/1078-0432.CCR-10-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007;7:585–598. doi: 10.1038/nrc2189. [DOI] [PubMed] [Google Scholar]
- 28.Mitsiades CS, Mitsiades NS, Munshi NC, Richardson PG, Anderson KC. The role of the bone microenvironment in the pathophysiology and therapeutic management of multiple myeloma: interplay of growth factors, their receptors and stromal interactions. Eur J Cancer. 2006;42:1564–1573. doi: 10.1016/j.ejca.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 29.Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia. 2009;23:10–24. doi: 10.1038/leu.2008.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basak GW, Srivastava AS, Malhotra R, Carrier E. Multiple myeloma bone marrow niche. Curr Pharm Biotechnol. 2009;10:345–346. doi: 10.2174/138920109787847493. [DOI] [PubMed] [Google Scholar]
- 31.Yasui H, Hideshima T, Richardson PG, Anderson KC. Novel therapeutic strategies targeting growth factor signalling cascades in multiple myeloma. Br J Haematol. 2006;132:385–397. doi: 10.1111/j.1365-2141.2005.05860.x. [DOI] [PubMed] [Google Scholar]
- 32.Podar K, Richardson PG, Hideshima T, Chauhan D, Anderson KC. The malignant clone and the bone-marrow environment. Best Pract Res Clin Haematol. 2007;20:597–612. doi: 10.1016/j.beha.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 33.Longo V, Brunetti O, D’Oronzo S, Dammacco F, Silvestris F. Therapeutic approaches to myeloma bone disease: an evolving story. Cancer Treat Rev. 2012;38:787–797. doi: 10.1016/j.ctrv.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Raje N, Roodman GD. Advances in the biology and treatment of bone disease in multiple myeloma. Clin Cancer Res. 2011;17:1278–1286. doi: 10.1158/1078-0432.CCR-10-1804. [DOI] [PubMed] [Google Scholar]
- 35.Saad F, Lipton A, Cook R, Chen YM, Smith M, Coleman R. Pathologic fractures correlate with reduced survival in patients with malignant bone disease. Cancer. 2007;110:1860–1867. doi: 10.1002/cncr.22991. [DOI] [PubMed] [Google Scholar]
- 36.Sonmez M, Akagun T, Topbas M, Cobanoglu U, Sonmez B, Yilmaz M, Ovali E, Omay SB. Effect of pathologic fractures on survival in multiple myeloma patients: a case control study. J Exp Clin Cancer Res. 2008;27:11. doi: 10.1186/1756-9966-27-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yaccoby S. Advances in the understanding of myeloma bone disease and tumour growth. Br J Haematol. 2010;149:311–321. doi: 10.1111/j.1365-2141.2010.08141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu P, Morgan GJ. Targeting bone as a therapy for myeloma. Cancer Microenviron. 2011;4:299–311. doi: 10.1007/s12307-011-0079-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edwards CM, Zhuang J, Mundy GR. The pathogenesis of the bone disease of multiple myeloma. Bone. 2008;42:1007–1013. doi: 10.1016/j.bone.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valentin-Opran A, Charhon SA, Meunier PJ, Edouard CM, Arlot ME. Quantitative histology of myeloma-induced bone changes. Br J Haematol. 1982;52:601–610. doi: 10.1111/j.1365-2141.1982.tb03936.x. [DOI] [PubMed] [Google Scholar]
- 41.Terpos E, Dimopoulos MA, Sezer O, Roodman D, Abildgaard N, Vescio R, Tosi P, Garcia-Sanz R, Davies F, Chanan-Khan A, et al. The use of biochemical markers of bone remodeling in multiple myeloma: a report of the International Myeloma Working Group. Leukemia. 2010;24:1700–1712. doi: 10.1038/leu.2010.173. [DOI] [PubMed] [Google Scholar]
- 42.Giuliani N, Rizzoli V, Roodman GD. Multiple myeloma bone disease: Pathophysiology of osteoblast inhibition. Blood. 2006;108:3992–3996. doi: 10.1182/blood-2006-05-026112. [DOI] [PubMed] [Google Scholar]
- 43.Vallet S, Raje N. Bone anabolic agents for the treatment of multiple myeloma. Cancer Microenviron. 2011;4:339–349. doi: 10.1007/s12307-011-0090-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giuliani N, Lisignoli G, Colla S, Lazzaretti M, Storti P, Mancini C, Bonomini S, Manferdini C, Codeluppi K, Facchini A, et al. CC-chemokine ligand 20/macrophage inflammatory protein-3α and CC-chemokine receptor 6 are overexpressed in myeloma microenvironment related to osteolytic bone lesions. Cancer Res. 2008;68:6840–6850. doi: 10.1158/0008-5472.CAN-08-0402. [DOI] [PubMed] [Google Scholar]
- 45.Silvestris F, Lombardi L, De Matteo M, Bruno A, Dammacco F. Myeloma bone disease: pathogenetic mechanisms and clinical assessment. Leuk Res. 2007;31:129–138. doi: 10.1016/j.leukres.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 46.Giuliani N, Colla S, Morandi F, Barille-Nion S, Rizzoli V. Lack of receptor activator of nuclear factor-kB ligand (RANKL) expression and functional production by human multiple myeloma cells. Haematologica. 2005;90:275–278. [PubMed] [Google Scholar]
- 47.Heider U, Langelotz C, Jakob C, Zavrski I, Fleissner C, Eucker J, Possinger K, Hofbauer LC, Sezer O. Expression of receptor activator of nuclear factor kappaB ligand on bone marrow plasma cells correlates with osteolytic bone disease in patients with multiple myeloma. Clin Cancer Res. 2003;9:1436–1440. [PubMed] [Google Scholar]
- 48.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 49.Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, Tan HL, Elliott G, Kelley MJ, Sarosi I, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci USA. 1999;96:3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Standal T, Seidel C, Hjertner Ø, Plesner T, Sanderson RD, Waage A, Borset M, Sundan A. Osteoprotegerin is bound, internalized, and degraded by multiple myeloma cells. Blood. 2002;100:3002–3007. doi: 10.1182/blood-2002-04-1190. [DOI] [PubMed] [Google Scholar]
- 52.Coluccia AM, Cirulli T, Neri P, Mangieri D, Colanardi MC, Gnoni A, Di Renzo N, Dammacco F, Tassone P, Ribatti D, et al. Validation of PDGFRbeta and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood. 2008;112:1346–1356. doi: 10.1182/blood-2007-10-116590. [DOI] [PubMed] [Google Scholar]
- 53.Pearse RN, Sordillo EM, Yaccoby S, Wong BR, Liau DF, Colman N, Michaeli J, Epstein J, Choi Y. Multiple myeloma disrupts the TRANCE/ osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc Natl Acad Sci USA. 2001;98:11581–11586. doi: 10.1073/pnas.201394498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giuliani N, Bataille R, Mancini C, Lazzaretti M, Barillé S. Myeloma cells induce imbalance in the osteoprotegerin/osteoprotegerin ligand system in the human bone marrow environment. Blood. 2001;98:3527–3533. doi: 10.1182/blood.v98.13.3527. [DOI] [PubMed] [Google Scholar]
- 55.Han JH, Choi SJ, Kurihara N, Koide M, Oba Y, Roodman GD. Macrophage inflammatory protein-1alpha is an osteoclastogenic factor in myeloma that is independent of receptor activator of nuclear factor kappaB ligand. Blood. 2001;97:3349–3353. doi: 10.1182/blood.v97.11.3349. [DOI] [PubMed] [Google Scholar]
- 56.Choi SJ, Oba Y, Gazitt Y, Alsina M, Cruz J, Anderson J, Roodman GD. Antisense inhibition of macrophage inflammatory protein 1-alpha blocks bone destruction in a model of myeloma bone disease. J Clin Invest. 2001;108:1833–1841. doi: 10.1172/JCI13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oyajobi BO, Franchin G, Williams PJ, Pulkrabek D, Gupta A, Munoz S, Grubbs B, Zhao M, Chen D, Sherry B, et al. Dual effects of macrophage inflammatory protein-1alpha on osteolysis and tumor burden in the murine 5TGM1 model of myeloma bone disease. Blood. 2003;102:311–319. doi: 10.1182/blood-2002-12-3905. [DOI] [PubMed] [Google Scholar]
- 58.Vallet S, Pozzi S, Patel K, Vaghela N, Fulciniti MT, Veiby P, Hideshima T, Santo L, Cirstea D, Scadden DT, et al. A novel role for CCL3 (MIP-1α) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leukemia. 2011;25:1174–1181. doi: 10.1038/leu.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vallet S, Anderson KC. CCR1 as a target for multiple myeloma. Expert Opin Ther Targets. 2011;15:1037–1047. doi: 10.1517/14728222.2011.586634. [DOI] [PubMed] [Google Scholar]
- 60.Sugatani T, Alvarez UM, Hruska KA. Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. J Cell Biochem. 2003;90:59–67. doi: 10.1002/jcb.10613. [DOI] [PubMed] [Google Scholar]
- 61.Vallet S, Mukherjee S, Vaghela N, Hideshima T, Fulciniti M, Pozzi S, Santo L, Cirstea D, Patel K, Sohani AR, et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc Natl Acad Sci USA. 2010;107:5124–5129. doi: 10.1073/pnas.0911929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chantry AD, Heath D, Mulivor AW, Pearsall S, Baud’huin M, Coulton L, Evans H, Abdul N, Werner ED, Bouxsein ML, et al. Inhibiting activin-A signaling stimulates bone formation and prevents cancer-induced bone destruction in vivo. J Bone Miner Res. 2010;25:2633–2646. doi: 10.1002/jbmr.142. [DOI] [PubMed] [Google Scholar]
- 63.Lee JW, Chung HY, Ehrlich LA, Jelinek DF, Callander NS, Roodman GD, Choi SJ. IL-3 expression by myeloma cells increases both osteoclast formation and growth of myeloma cells. Blood. 2004;103:2308–2315. doi: 10.1182/blood-2003-06-1992. [DOI] [PubMed] [Google Scholar]
- 64.Giuliani N, Rizzoli V. Myeloma cells and bone marrow osteoblast interactions: role in the development of osteolytic lesions in multiple myeloma. Leuk Lymphoma. 2007;48:2323–2329. doi: 10.1080/10428190701648281. [DOI] [PubMed] [Google Scholar]
- 65.Hofbauer LC, Lacey DL, Dunstan CR, Spelsberg TC, Riggs BL, Khosla S. Interleukin-1beta and tumor necrosis factor-alpha, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone. 1999;25:255–259. doi: 10.1016/s8756-3282(99)00162-3. [DOI] [PubMed] [Google Scholar]
- 66.Giuliani N, Colla S, Sala R, Moroni M, Lazzaretti M, La Monica S, Bonomini S, Hojden M, Sammarelli G, Barillè S, et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: a potential role in multiple myeloma bone disease. Blood. 2002;100:4615–4621. doi: 10.1182/blood-2002-04-1121. [DOI] [PubMed] [Google Scholar]
- 67.Hjertner O, Torgersen ML, Seidel C, Hjorth-Hansen H, Waage A, Børset M, Sundan A. Hepatocyte growth factor (HGF) induces interleukin-11 secretion from osteoblasts: a possible role for HGF in myeloma-associated osteolytic bone disease. Blood. 1999;94:3883–3888. [PubMed] [Google Scholar]
- 68.Sanderson RD, Epstein J. Myeloma bone disease. J Bone Miner Res. 2009;24:1783–1788. doi: 10.1359/jbmr.090901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gunn WG, Conley A, Deininger L, Olson SD, Prockop DJ, Gregory CA. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: a potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells. 2006;24:986–991. doi: 10.1634/stemcells.2005-0220. [DOI] [PubMed] [Google Scholar]
- 70.Bendre MS, Montague DC, Peery T, Akel NS, Gaddy D, Suva LJ. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone. 2003;33:28–37. doi: 10.1016/s8756-3282(03)00086-3. [DOI] [PubMed] [Google Scholar]
- 71.Tanaka Y, Abe M, Hiasa M, Oda A, Amou H, Nakano A, Takeuchi K, Kitazoe K, Kido S, Inoue D, et al. Myeloma cell-osteoclast interaction enhances angiogenesis together with bone resorption: a role for vascular endothelial cell growth factor and osteopontin. Clin Cancer Res. 2007;13:816–823. doi: 10.1158/1078-0432.CCR-06-2258. [DOI] [PubMed] [Google Scholar]
- 72.Oranger A, Carbone C, Izzo M, Grano M. Cellular mechanisms of multiple myeloma bone disease. Clin Dev Immunol. 2013;2013:289458. doi: 10.1155/2013/289458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hecht M, von Metzler I, Sack K, Kaiser M, Sezer O. Interactions of myeloma cells with osteoclasts promote tumour expansion and bone degradation through activation of a complex signalling network and upregulation of cathepsin K, matrix metalloproteinases (MMPs) and urokinase plasminogen activator (uPA) Exp Cell Res. 2008;314:1082–1093. doi: 10.1016/j.yexcr.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 74.Calvani N, Cafforio P, Silvestris F, Dammacco F. Functional osteoclast-like transformation of cultured human myeloma cell lines. Br J Haematol. 2005;130:926–938. doi: 10.1111/j.1365-2141.2005.05710.x. [DOI] [PubMed] [Google Scholar]
- 75.Tucci M, De Palma R, Lombardi L, Rodolico G, Berrino L, Dammacco F, Silvestris F. beta(3) Integrin subunit mediates the bone-resorbing function exerted by cultured myeloma plasma cells. Cancer Res. 2009;69:6738–6746. doi: 10.1158/0008-5472.CAN-09-0949. [DOI] [PubMed] [Google Scholar]
- 76.Kukreja A, Radfar S, Sun BH, Insogna K, Dhodapkar MV. Dominant role of CD47-thrombospondin-1 interactions in myeloma-induced fusion of human dendritic cells: implications for bone disease. Blood. 2009;114:3413–3421. doi: 10.1182/blood-2009-03-211920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Giuliani N, Colla S, Morandi F, Lazzaretti M, Sala R, Bonomini S, Grano M, Colucci S, Svaldi M, Rizzoli V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood. 2005;106:2472–2483. doi: 10.1182/blood-2004-12-4986. [DOI] [PubMed] [Google Scholar]
- 78.Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 79.Colucci S, Brunetti G, Oranger A, Mori G, Sardone F, Specchia G, Rinaldi E, Curci P, Liso V, Passeri G, et al. Myeloma cells suppress osteoblasts through sclerostin secretion. Blood Cancer J. 2011;1:e27. doi: 10.1038/bcj.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qiang YW, Barlogie B, Rudikoff S, Shaughnessy JD. Dkk1-induced inhibition of Wnt signaling in osteoblast differentiation is an underlying mechanism of bone loss in multiple myeloma. Bone. 2008;42:669–680. doi: 10.1016/j.bone.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 81.Sims NA, Gooi JH. Bone remodeling: Multiple cellular interactions required for coupling of bone formation and resorption. Semin Cell Dev Biol. 2008;19:444–451. doi: 10.1016/j.semcdb.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 82.Qiang YW, Chen Y, Stephens O, Brown N, Chen B, Epstein J, Barlogie B, Shaughnessy JD. Myeloma-derived Dickkopf-1 disrupts Wnt-regulated osteoprotegerin and RANKL production by osteoblasts: a potential mechanism underlying osteolytic bone lesions in multiple myeloma. Blood. 2008;112:196–207. doi: 10.1182/blood-2008-01-132134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eda H, Santo L, Cirstea D, Yee AJ, Mahindra A, Scullen T, Nemani N, Mishima Y, Vallet S, Raje N. Increased Sclerostin Secretion in Multiple Myeloma Plays a Central Role in Osteolytic Bone Disease. Blood (ASH Annual Meeting Abstracts) 2012;120:3989. [Google Scholar]
- 84.Giuliani N, Morandi F, Tagliaferri S, Lazzaretti M, Donofrio G, Bonomini S, Sala R, Mangoni M, Rizzoli V. Production of Wnt inhibitors by myeloma cells: potential effects on canonical Wnt pathway in the bone microenvironment. Cancer Res. 2007;67:7665–7674. doi: 10.1158/0008-5472.CAN-06-4666. [DOI] [PubMed] [Google Scholar]
- 85.Oshima T, Abe M, Asano J, Hara T, Kitazoe K, Sekimoto E, Tanaka Y, Shibata H, Hashimoto T, Ozaki S, et al. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP-2. Blood. 2005;106:3160–3165. doi: 10.1182/blood-2004-12-4940. [DOI] [PubMed] [Google Scholar]
- 86.Kristensen IB, Haaber J, Lyng MB, Knudsen LM, Rasmussen T, Ditzel HJ, Abildgaard N. Myeloma plasma cell expression of osteoblast regulatory genes: overexpression of SFRP3 correlates with clinical bone involvement at diagnosis. Leuk Lymphoma. 2013;54:425–427. doi: 10.3109/10428194.2012.708027. [DOI] [PubMed] [Google Scholar]
- 87.Lin GL, Hankenson KD. Integration of BMP, Wnt, and notch signaling pathways in osteoblast differentiation. J Cell Biochem. 2011;112:3491–3501. doi: 10.1002/jcb.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee MH, Kim YJ, Kim HJ, Park HD, Kang AR, Kyung HM, Sung JH, Wozney JM, Kim HJ, Ryoo HM. BMP-2-induced Runx2 expression is mediated by Dlx5, and TGF-beta 1 opposes the BMP-2-induced osteoblast differentiation by suppression of Dlx5 expression. J Biol Chem. 2003;278:34387–34394. doi: 10.1074/jbc.M211386200. [DOI] [PubMed] [Google Scholar]
- 89.Standal T, Abildgaard N, Fagerli UM, Stordal B, Hjertner O, Borset M, Sundan A. HGF inhibits BMP-induced osteoblastogenesis: possible implications for the bone disease of multiple myeloma. Blood. 2007;109:3024–3030. doi: 10.1182/blood-2006-07-034884. [DOI] [PubMed] [Google Scholar]
- 90.Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R. Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest. 2002;110:1643–1650. doi: 10.1172/JCI15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu X, Gilbert L, He X, Rubin J, Nanes MS. Transcriptional regulation of the osterix (Osx, Sp7) promoter by tumor necrosis factor identifies disparate effects of mitogen-activated protein kinase and NF kappa B pathways. J Biol Chem. 2006;281:6297–6306. doi: 10.1074/jbc.M507804200. [DOI] [PubMed] [Google Scholar]
- 92.Jourdan M, Tarte K, Legouffe E, Brochier J, Rossi JF, Klein B. Tumor necrosis factor is a survival and proliferation factor for human myeloma cells. Eur Cytokine Netw. 1999;10:65–70. [PMC free article] [PubMed] [Google Scholar]
- 93.Vincent C, Findlay DM, Welldon KJ, Wijenayaka AR, Zheng TS, Haynes DR, Fazzalari NL, Evdokiou A, Atkins GJ. Pro-inflammatory cytokines TNF-related weak inducer of apoptosis (TWEAK) and TNFalpha induce the mitogen-activated protein kinase (MAPK)-dependent expression of sclerostin in human osteoblasts. J Bone Miner Res. 2009;24:1434–1449. doi: 10.1359/jbmr.090305. [DOI] [PubMed] [Google Scholar]
- 94.D’Souza S, del Prete D, Jin S, Sun Q, Huston AJ, Kostov FE, Sammut B, Hong CS, Anderson JL, Patrene KD, et al. Gfi1 expressed in bone marrow stromal cells is a novel osteoblast suppressor in patients with multiple myeloma bone disease. Blood. 2011;118:6871–6880. doi: 10.1182/blood-2011-04-346775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Giuliani N, Morandi F, Tagliaferri S, Colla S, Bonomini S, Sammarelli G, Rizzoli V. Interleukin-3 (IL-3) is overexpressed by T lymphocytes in multiple myeloma patients. Blood. 2006;107:841–842. doi: 10.1182/blood-2005-07-2719. [DOI] [PubMed] [Google Scholar]
- 96.Ehrlich LA, Chung HY, Ghobrial I, Choi SJ, Morandi F, Colla S, Rizzoli V, Roodman GD, Giuliani N. IL-3 is a potential inhibitor of osteoblast differentiation in multiple myeloma. Blood. 2005;106:1407–1414. doi: 10.1182/blood-2005-03-1080. [DOI] [PubMed] [Google Scholar]
- 97.Silvestris F, Cafforio P, Calvani N, Dammacco F. Impaired osteoblastogenesis in myeloma bone disease: role of upregulated apoptosis by cytokines and malignant plasma cells. Br J Haematol. 2004;126:475–486. doi: 10.1111/j.1365-2141.2004.05084.x. [DOI] [PubMed] [Google Scholar]
- 98.Silvestris F, Cafforio P, Tucci M, Grinello D, Dammacco F. Upregulation of osteoblast apoptosis by malignant plasma cells: a role in myeloma bone disease. Br J Haematol. 2003;122:39–52. doi: 10.1046/j.1365-2141.2003.04374.x. [DOI] [PubMed] [Google Scholar]
- 99.Giuliani N, Ferretti M, Bolzoni M, Storti P, Lazzaretti M, Dalla Palma B, Bonomini S, Martella E, Agnelli L, Neri A, et al. Increased osteocyte death in multiple myeloma patients: role in myeloma-induced osteoclast formation. Leukemia. 2012;26:1391–1401. doi: 10.1038/leu.2011.381. [DOI] [PubMed] [Google Scholar]
- 100.Silvestris F, Cafforio P, De Matteo M, Calvani N, Frassanito MA, Dammacco F. Negative regulation of the osteoblast function in multiple myeloma through the repressor gene E4BP4 activated by malignant plasma cells. Clin Cancer Res. 2008;14:6081–6091. doi: 10.1158/1078-0432.CCR-08-0219. [DOI] [PubMed] [Google Scholar]
- 101.Bolzoni M, Donofrio G, Storti P, Guasco D, Toscani D, Lazzaretti M, Bonomini S, Agnelli L, Capocefalo A, Dalla Palma B, et al. Myeloma cells inhibit non-canonical wnt co-receptor ror2 expression in human bone marrow osteoprogenitor cells: effect of wnt5a/ror2 pathway activation on the osteogenic differentiation impairment induced by myeloma cells. Leukemia. 2013;27:451–463. doi: 10.1038/leu.2012.190. [DOI] [PubMed] [Google Scholar]
- 102.Baksh D, Boland GM, Tuan RS. Cross-talk between Wnt signaling pathways in human mesenchymal stem cells leads to functional antagonism during osteogenic differentiation. J Cell Biochem. 2007;101:1109–1124. doi: 10.1002/jcb.21097. [DOI] [PubMed] [Google Scholar]
- 103.Baksh D, Tuan RS. Canonical and non-canonical Wnts differentially affect the development potential of primary isolate of human bone marrow mesenchymal stem cells. J Cell Physiol. 2007;212:817–826. doi: 10.1002/jcp.21080. [DOI] [PubMed] [Google Scholar]
- 104.Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4:111–121. doi: 10.1016/j.cmet.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 105.Pennisi A, Ling W, Li X, Khan S, Shaughnessy JD, Barlogie B, Yaccoby S. The ephrinB2/EphB4 axis is dysregulated in osteoprogenitors from myeloma patients and its activation affects myeloma bone disease and tumor growth. Blood. 2009;114:1803–1812. doi: 10.1182/blood-2009-01-201954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Atkins GJ, Kostakis P, Pan B, Farrugia A, Gronthos S, Evdokiou A, Harrison K, Findlay DM, Zannettino AC. RANKL expression is related to the differentiation state of human osteoblasts. J Bone Miner Res. 2003;18:1088–1098. doi: 10.1359/jbmr.2003.18.6.1088. [DOI] [PubMed] [Google Scholar]
- 107.Eijken M, Swagemakers S, Koedam M, Steenbergen C, Derkx P, Uitterlinden AG, van der Spek PJ, Visser JA, de Jong FH, Pols HA, et al. The activin A-follistatin system: potent regulator of human extracellular matrix mineralization. FASEB J. 2007;21:2949–2960. doi: 10.1096/fj.07-8080com. [DOI] [PubMed] [Google Scholar]
- 108.Reagan MR, Ghobrial IM. Multiple myeloma mesenchymal stem cells: characterization, origin, and tumor-promoting effects. Clin Cancer Res. 2012;18:342–349. doi: 10.1158/1078-0432.CCR-11-2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li B, Shi M, Li J, Zhang H, Chen B, Chen L, Gao W, Giuliani N, Zhao RC. Elevated tumor necrosis factor-alpha suppresses TAZ expression and impairs osteogenic potential of Flk-1+ mesenchymal stem cells in patients with multiple myeloma. Stem Cells Dev. 2007;16:921–930. doi: 10.1089/scd.2007.0074. [DOI] [PubMed] [Google Scholar]
- 110.Xu S, Evans H, Buckle C, De Veirman K, Hu J, Xu D, Menu E, De Becker A, Vande Broek I, Leleu X, et al. Impaired osteogenic differentiation of mesenchymal stem cells derived from multiple myeloma patients is associated with a blockade in the deactivation of the Notch signaling pathway. Leukemia. 2012;26:2546–2549. doi: 10.1038/leu.2012.126. [DOI] [PubMed] [Google Scholar]
- 111.Corre J, Mahtouk K, Attal M, Gadelorge M, Huynh A, Fleury-Cappellesso S, Danho C, Laharrague P, Klein B, Rème T, et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia. 2007;21:1079–1088. doi: 10.1038/sj.leu.2404621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li B, Fu J, Chen P, Zhuang W. Impairment in immunomodulatory function of mesenchymal stem cells from multiple myeloma patients. Arch Med Res. 2010;41:623–633. doi: 10.1016/j.arcmed.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 113.Todoerti K, Lisignoli G, Storti P, Agnelli L, Novara F, Manferdini C, Codeluppi K, Colla S, Crugnola M, Abeltino M, et al. Distinct transcriptional profiles characterize bone microenvironment mesenchymal cells rather than osteoblasts in relationship with multiple myeloma bone disease. Exp Hematol. 2010;38:141–153. doi: 10.1016/j.exphem.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 114.Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest. 2012;122:3456–3463. doi: 10.1172/JCI61188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mahtouk K, Moreaux J, Hose D, Rème T, Meissner T, Jourdan M, Rossi JF, Pals ST, Goldschmidt H, Klein B. Growth factors in multiple myeloma: a comprehensive analysis of their expression in tumor cells and bone marrow environment using Affymetrix microarrays. BMC Cancer. 2010;10:198. doi: 10.1186/1471-2407-10-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Maeda K, Kobayashi Y, Udagawa N, Uehara S, Ishihara A, Mizoguchi T, Kikuchi Y, Takada I, Kato S, Kani S, et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 2012;18:405–412. doi: 10.1038/nm.2653. [DOI] [PubMed] [Google Scholar]
- 117.Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83:1032–1045. doi: 10.4065/83.9.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Terpos E, Sezer O, Croucher PI, García-Sanz R, Boccadoro M, San Miguel J, Ashcroft J, Bladé J, Cavo M, Delforge M, et al. The use of bisphosphonates in multiple myeloma: recommendations of an expert panel on behalf of the European Myeloma Network. Ann Oncol. 2009;20:1303–1317. doi: 10.1093/annonc/mdn796. [DOI] [PubMed] [Google Scholar]
- 119.Monroe DG, McGee-Lawrence ME, Oursler MJ, Westendorf JJ. Update on Wnt signaling in bone cell biology and bone disease. Gene. 2012;492:1–18. doi: 10.1016/j.gene.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Terpos E, Christoulas D, Katodritou E, Bratengeier C, Gkotzamanidou M, Michalis E, Delimpasi S, Pouli A, Meletis J, Kastritis E, et al. Elevated circulating sclerostin correlates with advanced disease features and abnormal bone remodeling in symptomatic myeloma: reduction post-bortezomib monotherapy. Int J Cancer. 2012;131:1466–1471. doi: 10.1002/ijc.27342. [DOI] [PubMed] [Google Scholar]
- 121.Garderet L, Mazurier C, Chapel A, Ernou I, Boutin L, Holy X, Gorin NC, Lopez M, Doucet C, Lataillade JJ. Mesenchymal stem cell abnormalities in patients with multiple myeloma. Leuk Lymphoma. 2007;48:2032–2041. doi: 10.1080/10428190701593644. [DOI] [PubMed] [Google Scholar]
- 122.Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood. 2007;109:2106–2111. doi: 10.1182/blood-2006-09-047712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fulciniti M, Tassone P, Hideshima T, Vallet S, Nanjappa P, Ettenberg SA, Shen Z, Patel N, Tai YT, Chauhan D, et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood. 2009;114:371–379. doi: 10.1182/blood-2008-11-191577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Heath DJ, Chantry AD, Buckle CH, Coulton L, Shaughnessy JD, Evans HR, Snowden JA, Stover DR, Vanderkerken K, Croucher PI. Inhibiting Dickkopf-1 (Dkk1) removes suppression of bone formation and prevents the development of osteolytic bone disease in multiple myeloma. J Bone Miner Res. 2009;24:425–436. doi: 10.1359/jbmr.081104. [DOI] [PubMed] [Google Scholar]
- 125.Pozzi S, Fulciniti M, Yan H, Vallet S, Eda H, Patel K, Santo L, Cirstea D, Hideshima T, Schirtzinge L, et al. In vivo and in vitro effects of a novel anti-Dkk1 neutralizing antibody in multiple myeloma. Bone. 2013;53:487–496. doi: 10.1016/j.bone.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Padmanabhan S, Beck JT, Kelly KR, Munshi NC, Dzik-Jurasz A, Gangolli E, Ettenberg S, Miner K, Bilic S, Whyte W, et al. A Phase I/II Study of BHQ880, a Novel Osteoblat Activating, Anti-DKK1 Human Monoclonal Antibody, in Relapsed and Refractory Multiple Myeloma (MM) Patients Treated with Zoledronic Acid (Zol) and Anti-Myeloma Therapy (MM Tx) Blood (ASH Annual Meeting Abstracts) 2009;114:750. [Google Scholar]
- 127.Munshi NC, Abonour R, Beck JT, Bensinger W, Facon T, Stockerl-Goldstein K, Baz R, Siegel DS, Neben K, Lonial S, et al. Early Evidence of Anabolic Bone Activity of BHQ880, a Fully Human Anti-DKK1 Neutralizing Antibody: Results of a Phase 2 Study in Previously Untreated Patients with Smoldering Multiple Myeloma At Risk for Progression. Blood (ASH Annual Meeting Abstracts) 2012;120:331. [Google Scholar]
- 128.Ke HZ, Richards WG, Li X, Ominsky MS. Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr Rev. 2012;33:747–783. doi: 10.1210/er.2011-1060. [DOI] [PubMed] [Google Scholar]
- 129.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 130.Qiang YW, Shaughnessy JD, Yaccoby S. Wnt3a signaling within bone inhibits multiple myeloma bone disease and tumor growth. Blood. 2008;112:374–382. doi: 10.1182/blood-2007-10-120253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Edwards CM, Edwards JR, Lwin ST, Esparza J, Oyajobi BO, McCluskey B, Munoz S, Grubbs B, Mundy GR. Increasing Wnt signaling in the bone marrow microenvironment inhibits the development of myeloma bone disease and reduces tumor burden in bone in vivo. Blood. 2008;111:2833–2842. doi: 10.1182/blood-2007-03-077685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Derksen PW, Tjin E, Meijer HP, Klok MD, MacGillavry HD, van Oers MH, Lokhorst HM, Bloem AC, Clevers H, Nusse R, et al. Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc Natl Acad Sci USA. 2004;101:6122–6127. doi: 10.1073/pnas.0305855101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Engin F, Lee B. NOTCHing the bone: insights into multi-functionality. Bone. 2010;46:274–280. doi: 10.1016/j.bone.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Engin F, Yao Z, Yang T, Zhou G, Bertin T, Jiang MM, Chen Y, Wang L, Zheng H, Sutton RE, et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat Med. 2008;14:299–305. doi: 10.1038/nm1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zanotti S, Smerdel-Ramoya A, Stadmeyer L, Durant D, Radtke F, Canalis E. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology. 2008;149:3890–3899. doi: 10.1210/en.2008-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F, Ross FP, Teitelbaum SL. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2008;283:6509–6518. doi: 10.1074/jbc.M707000200. [DOI] [PubMed] [Google Scholar]
- 137.Fukushima H, Nakao A, Okamoto F, Shin M, Kajiya H, Sakano S, Bigas A, Jimi E, Okabe K. The association of Notch2 and NF-kappaB accelerates RANKL-induced osteoclastogenesis. Mol Cell Biol. 2008;28:6402–6412. doi: 10.1128/MCB.00299-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Colombo M, Mirandola L, Platonova N, Apicella L, Basile A, Figueroa AJ, Cobos E, Chiriva-Internati M, Chiaramonte R. Notch-directed microenvironment reprogramming in myeloma: a single path to multiple outcomes. Leukemia. 2013;27:1009–1018. doi: 10.1038/leu.2013.6. [DOI] [PubMed] [Google Scholar]
- 139.Schwarzer R, Kaiser M, Acikgoez O, Heider U, Mathas S, Preissner R, Sezer O, Doerken B, Jundt F. Notch inhibition blocks multiple myeloma cell-induced osteoclast activation. Leukemia. 2008;22:2273–2277. doi: 10.1038/leu.2008.138. [DOI] [PubMed] [Google Scholar]
- 140.Mirandola L, Apicella L, Colombo M, Yu Y, Berta DG, Platonova N, Lazzari E, Lancellotti M, Bulfamante G, Cobos E, et al. Anti-Notch treatment prevents multiple myeloma cells localization to the bone marrow via the chemokine system CXCR4/SDF-1. Leukemia. 2013;27:1558–1566. doi: 10.1038/leu.2013.27. [DOI] [PubMed] [Google Scholar]
- 141.Ramakrishnan V, Ansell S, Haug J, Grote D, Kimlinger T, Stenson M, Timm M, Wellik L, Halling T, Rajkumar SV, et al. MRK003, a γ-secretase inhibitor exhibits promising in vitro pre-clinical activity in multiple myeloma and non-Hodgkin’s lymphoma. Leukemia. 2012;26:340–348. doi: 10.1038/leu.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Xu D, Hu J, Xu S, De Bruyne E, Menu E, Van Camp B, Vanderkerken K, Van Valckenborgh E. Dll1/Notch activation accelerates multiple myeloma disease development by promoting CD138+ MM-cell proliferation. Leukemia. 2012;26:1402–1405. doi: 10.1038/leu.2011.332. [DOI] [PubMed] [Google Scholar]
- 143.Li M, Chen F, Clifton N, Sullivan DM, Dalton WS, Gabrilovich DI, Nefedova Y. Combined inhibition of Notch signaling and Bcl-2/Bcl-xL results in synergistic antimyeloma effect. Mol Cancer Ther. 2010;9:3200–3209. doi: 10.1158/1535-7163.MCT-10-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nefedova Y, Sullivan DM, Bolick SC, Dalton WS, Gabrilovich DI. Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood. 2008;111:2220–2229. doi: 10.1182/blood-2007-07-102632. [DOI] [PubMed] [Google Scholar]
- 145.Chen F, Pisklakova A, Li M, Baz R, Sullivan DM, Nefedova Y. Gamma-secretase inhibitor enhances the cytotoxic effect of bortezomib in multiple myeloma. Cell Oncol (Dordr) 2011;34:545–551. doi: 10.1007/s13402-011-0060-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lotinun S, Pearsall RS, Horne WC, Baron R. Activin receptor signaling: a potential therapeutic target for osteoporosis. Curr Mol Pharmacol. 2012;5:195–204. doi: 10.2174/1874467211205020195. [DOI] [PubMed] [Google Scholar]
- 147.Alves RD, Eijken M, Bezstarosti K, Demmers JA, van Leeuwen JP. Activin A suppresses osteoblast mineralization capacity by altering extracellular matrix (ECM) composition and impairing matrix vesicle (MV) production. Mol Cell Proteomics. 2013;12:2890–2900. doi: 10.1074/mcp.M112.024927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Terpos E, Kastritis E, Christoulas D, Gkotzamanidou M, Eleutherakis-Papaiakovou E, Kanellias N, Papatheodorou A, Dimopoulos MA. Circulating activin-A is elevated in patients with advanced multiple myeloma and correlates with extensive bone involvement and inferior survival; no alterations post-lenalidomide and dexamethasone therapy. Ann Oncol. 2012;23:2681–2686. doi: 10.1093/annonc/mds068. [DOI] [PubMed] [Google Scholar]
- 149.Matsumoto T, Abe M. TGF-β-related mechanisms of bone destruction in multiple myeloma. Bone. 2011;48:129–134. doi: 10.1016/j.bone.2010.05.036. [DOI] [PubMed] [Google Scholar]
- 150.Takeuchi K, Abe M, Hiasa M, Oda A, Amou H, Kido S, Harada T, Tanaka O, Miki H, Nakamura S, et al. Tgf-Beta inhibition restores terminal osteoblast differentiation to suppress myeloma growth. PLoS One. 2010;5:e9870. doi: 10.1371/journal.pone.0009870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pearsall RS, Canalis E, Cornwall-Brady M, Underwood KW, Haigis B, Ucran J, Kumar R, Pobre E, Grinberg A, Werner ED, et al. A soluble activin type IIA receptor induces bone formation and improves skeletal integrity. Proc Natl Acad Sci USA. 2008;105:7082–7087. doi: 10.1073/pnas.0711263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lotinun S, Pearsall RS, Davies MV, Marvell TH, Monnell TE, Ucran J, Fajardo RJ, Kumar R, Underwood KW, Seehra J, et al. A soluble activin receptor Type IIA fusion protein (ACE-011) increases bone mass via a dual anabolic-antiresorptive effect in Cynomolgus monkeys. Bone. 2010;46:1082–1088. doi: 10.1016/j.bone.2010.01.370. [DOI] [PubMed] [Google Scholar]
- 153.Abdulkadyrov KM, Salogub GN, Khuazheva NK, Woolf R, Haltom E, Borgstein NG, Knight R, Renshaw G, Yang Y, Sherman ML. ACE-011, a Soluble Activin Receptor Type Iia IgG-Fc Fusion Protein, Increases Hemoglobin (Hb) and Improves Bone Lesions in Multiple Myeloma Patients Receiving Myelosuppressive Chemotherapy: Preliminary Analysis. Blood (ASH Annual Meeting Abstracts) 2009;114:749. [Google Scholar]
- 154.Ruckle J, Jacobs M, Kramer W, Pearsall AE, Kumar R, Underwood KW, Seehra J, Yang Y, Condon CH, Sherman ML. Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J Bone Miner Res. 2009;24:744–752. doi: 10.1359/jbmr.081208. [DOI] [PubMed] [Google Scholar]
- 155.Mohammad KS, Chen CG, Balooch G, Stebbins E, McKenna CR, Davis H, Niewolna M, Peng XH, Nguyen DH, Ionova-Martin SS, et al. Pharmacologic inhibition of the TGF-beta type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS One. 2009;4:e5275. doi: 10.1371/journal.pone.0005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lentzsch S, Gries M, Janz M, Bargou R, Dörken B, Mapara MY. Macrophage inflammatory protein 1-alpha (MIP-1 alpha ) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood. 2003;101:3568–3573. doi: 10.1182/blood-2002-08-2383. [DOI] [PubMed] [Google Scholar]
- 157.Dairaghi DJ, Oyajobi BO, Gupta A, McCluskey B, Miao S, Powers JP, Seitz LC, Wang Y, Zeng Y, Zhang P, et al. CCR1 blockade reduces tumor burden and osteolysis in vivo in a mouse model of myeloma bone disease. Blood. 2012;120:1449–1457. doi: 10.1182/blood-2011-10-384784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tsubaki M, Kato C, Isono A, Kaneko J, Isozaki M, Satou T, Itoh T, Kidera Y, Tanimori Y, Yanae M, et al. Macrophage inflammatory protein-1α induces osteoclast formation by activation of the MEK/ERK/c-Fos pathway and inhibition of the p38MAPK/IRF-3/IFN-β pathway. J Cell Biochem. 2010;111:1661–1672. doi: 10.1002/jcb.22907. [DOI] [PubMed] [Google Scholar]
- 159.Terpos E, Politou M, Szydlo R, Goldman JM, Apperley JF, Rahemtulla A. Serum levels of macrophage inflammatory protein-1 alpha (MIP-1alpha) correlate with the extent of bone disease and survival in patients with multiple myeloma. Br J Haematol. 2003;123:106–109. doi: 10.1046/j.1365-2141.2003.04561.x. [DOI] [PubMed] [Google Scholar]
- 160.Vallet S, Raje N, Ishitsuka K, Hideshima T, Podar K, Chhetri S, Pozzi S, Breitkreutz I, Kiziltepe T, Yasui H, et al. MLN3897, a novel CCR1 inhibitor, impairs osteoclastogenesis and inhibits the interaction of multiple myeloma cells and osteoclasts. Blood. 2007;110:3744–3752. doi: 10.1182/blood-2007-05-093294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tak PP, Balanescu A, Tseluyko V, Bojin S, Drescher E, Dairaghi D, Miao S, Marchesin V, Jaen J, Schall TJ, et al. Chemokine receptor CCR1 antagonist CCX354-C treatment for rheumatoid arthritis: CARAT-2, a randomised, placebo controlled clinical trial. Ann Rheum Dis. 2013;72:337–344. doi: 10.1136/annrheumdis-2011-201605. [DOI] [PubMed] [Google Scholar]
- 162.Mundy GR, Elefteriou F. Boning up on ephrin signaling. Cell. 2006;126:441–443. doi: 10.1016/j.cell.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 163.Matsuo K, Otaki N. Bone cell interactions through Eph/ephrin: bone modeling, remodeling and associated diseases. Cell Adh Migr. 2012;6:148–156. doi: 10.4161/cam.20888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Irie N, Takada Y, Watanabe Y, Matsuzaki Y, Naruse C, Asano M, Iwakura Y, Suda T, Matsuo K. Bidirectional signaling through ephrinA2-EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J Biol Chem. 2009;284:14637–14644. doi: 10.1074/jbc.M807598200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Aronson LI, Davies FE. DangER: protein ovERload. Targeting protein degradation to treat myeloma. Haematologica. 2012;97:1119–1130. doi: 10.3324/haematol.2012.064923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Boot-Handford RP, Briggs MD. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res. 2010;339:197–211. doi: 10.1007/s00441-009-0877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tohmonda T, Miyauchi Y, Ghosh R, Yoda M, Uchikawa S, Takito J, Morioka H, Nakamura M, Iwawaki T, Chiba K, et al. The IRE1α-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep. 2011;12:451–457. doi: 10.1038/embor.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Saito A, Ochiai K, Kondo S, Tsumagari K, Murakami T, Cavener DR, Imaizumi K. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem. 2011;286:4809–4818. doi: 10.1074/jbc.M110.152900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jang WG, Kim EJ, Kim DK, Ryoo HM, Lee KB, Kim SH, Choi HS, Koh JT. BMP2 protein regulates osteocalcin expression via Runx2-mediated Atf6 gene transcription. J Biol Chem. 2012;287:905–915. doi: 10.1074/jbc.M111.253187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Xu G, Liu K, Anderson J, Patrene K, Lentzsch S, Roodman GD, Ouyang H. Expression of XBP1s in bone marrow stromal cells is critical for myeloma cell growth and osteoclast formation. Blood. 2012;119:4205–4214. doi: 10.1182/blood-2011-05-353300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA. 2003;100:9946–9951. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Terpos E, Sezer O, Croucher P, Dimopoulos MA. Myeloma bone disease and proteasome inhibition therapies. Blood. 2007;110:1098–1104. doi: 10.1182/blood-2007-03-067710. [DOI] [PubMed] [Google Scholar]
- 174.Delforge M, Terpos E, Richardson PG, Shpilberg O, Khuageva NK, Schlag R, Dimopoulos MA, Kropff M, Spicka I, Petrucci MT, et al. Fewer bone disease events, improvement in bone remodeling, and evidence of bone healing with bortezomib plus melphalan-prednisone vs. melphalan-prednisone in the phase III VISTA trial in multiple myeloma. Eur J Haematol. 2011;86:372–384. doi: 10.1111/j.1600-0609.2011.01599.x. [DOI] [PubMed] [Google Scholar]
- 175.Giuliani N, Morandi F, Tagliaferri S, Lazzaretti M, Bonomini S, Crugnola M, Mancini C, Martella E, Ferrari L, Tabilio A, et al. The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood. 2007;110:334–338. doi: 10.1182/blood-2006-11-059188. [DOI] [PubMed] [Google Scholar]
- 176.Pennisi A, Li X, Ling W, Khan S, Zangari M, Yaccoby S. The proteasome inhibitor, bortezomib suppresses primary myeloma and stimulates bone formation in myelomatous and nonmyelomatous bones in vivo. Am J Hematol. 2009;84:6–14. doi: 10.1002/ajh.21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.von Metzler I, Krebbel H, Hecht M, Manz RA, Fleissner C, Mieth M, Kaiser M, Jakob C, Sterz J, Kleeberg L, et al. Bortezomib inhibits human osteoclastogenesis. Leukemia. 2007;21:2025–2034. doi: 10.1038/sj.leu.2404806. [DOI] [PubMed] [Google Scholar]
- 178.Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010;15:243–249. doi: 10.1016/j.drudis.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 179.Hurchla MA, Garcia-Gomez A, Hornick MC, Ocio EM, Li A, Blanco JF, Collins L, Kirk CJ, Piwnica-Worms D, Vij R, et al. The epoxyketone-based proteasome inhibitors carfilzomib and orally bioavailable oprozomib have anti-resorptive and bone-anabolic activity in addition to anti-myeloma effects. Leukemia. 2013;27:430–440. doi: 10.1038/leu.2012.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Garcia-Gomez A, Quwaider D, Canavese M, Ocio EM, Tian Z, Blanco JF, Berger AJ, Ortiz-de-Solorzano C, Hernández-Iglesias T, Martens AC, et al. Preclinical activity of the oral proteasome inhibitor MLN9708 in Myeloma bone disease. Clin Cancer Res. 2014;20:1542–1554. doi: 10.1158/1078-0432.CCR-13-1657. [DOI] [PubMed] [Google Scholar]
- 181.Nakamura S, Miki H, Kido S, Nakano A, Hiasa M, Oda A, Amou H, Watanabe K, Harada T, Fujii S, et al. Activating transcription factor 4, an ER stress mediator, is required for, but excessive ER stress suppresses osteoblastogenesis by bortezomib. Int J Hematol. 2013;98:66–73. doi: 10.1007/s12185-013-1367-z. [DOI] [PubMed] [Google Scholar]
- 182.Fitter S, Dewar AL, Kostakis P, To LB, Hughes TP, Roberts MM, Lynch K, Vernon-Roberts B, Zannettino AC. Long-term imatinib therapy promotes bone formation in CML patients. Blood. 2008;111:2538–2547. doi: 10.1182/blood-2007-07-104281. [DOI] [PubMed] [Google Scholar]
- 183.O’Sullivan S, Naot D, Callon K, Porteous F, Horne A, Wattie D, Watson M, Cornish J, Browett P, Grey A. Imatinib promotes osteoblast differentiation by inhibiting PDGFR signaling and inhibits osteoclastogenesis by both direct and stromal cell-dependent mechanisms. J Bone Miner Res. 2007;22:1679–1689. doi: 10.1359/jbmr.070719. [DOI] [PubMed] [Google Scholar]
- 184.Tibullo D, Barbagallo I, Giallongo C, La Cava P, Branca A, Conticello C, Stagno F, Chiarenza A, Palumbo GA, Di Raimondo F. Effects of second-generation tyrosine kinase inhibitors towards osteogenic differentiation of human mesenchymal cells of healthy donors. Hematol Oncol. 2012;30:27–33. doi: 10.1002/hon.988. [DOI] [PubMed] [Google Scholar]
- 185.Lee YC, Huang CF, Murshed M, Chu K, Araujo JC, Ye X, deCrombrugghe B, Yu-Lee LY, Gallick GE, Lin SH. Src family kinase/abl inhibitor dasatinib suppresses proliferation and enhances differentiation of osteoblasts. Oncogene. 2010;29:3196–3207. doi: 10.1038/onc.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Id Boufker H, Lagneaux L, Najar M, Piccart M, Ghanem G, Body JJ, Journé F. The Src inhibitor dasatinib accelerates the differentiation of human bone marrow-derived mesenchymal stromal cells into osteoblasts. BMC Cancer. 2010;10:298. doi: 10.1186/1471-2407-10-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Garcia-Gomez A, Ocio EM, Crusoe E, Santamaria C, Hernández-Campo P, Blanco JF, Sanchez-Guijo FM, Hernández-Iglesias T, Briñón JG, Fisac-Herrero RM, et al. Dasatinib as a bone-modifying agent: anabolic and anti-resorptive effects. PLoS One. 2012;7:e34914. doi: 10.1371/journal.pone.0034914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rastegar F, Shenaq D, Huang J, Zhang W, Zhang BQ, He BC, Chen L, Zuo GW, Luo Q, Shi Q, et al. Mesenchymal stem cells: Molecular characteristics and clinical applications. World J Stem Cells. 2010;2:67–80. doi: 10.4252/wjsc.v2.i4.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mitsiades CS, Mitsiades NS, Richardson PG, Munshi NC, Anderson KC. Multiple myeloma: a prototypic disease model for the characterization and therapeutic targeting of interactions between tumor cells and their local microenvironment. J Cell Biochem. 2007;101:950–968. doi: 10.1002/jcb.21213. [DOI] [PubMed] [Google Scholar]
- 190.Rabin N, Kyriakou C, Coulton L, Gallagher OM, Buckle C, Benjamin R, Singh N, Glassford J, Otsuki T, Nathwani AC, et al. A new xenograft model of myeloma bone disease demonstrating the efficacy of human mesenchymal stem cells expressing osteoprotegerin by lentiviral gene transfer. Leukemia. 2007;21:2181–2191. doi: 10.1038/sj.leu.2404814. [DOI] [PubMed] [Google Scholar]
- 191.Li X, Ling W, Khan S, Yaccoby S. Therapeutic effects of intrabone and systemic mesenchymal stem cell cytotherapy on myeloma bone disease and tumor growth. J Bone Miner Res. 2012;27:1635–1648. doi: 10.1002/jbmr.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li X, Ling W, Pennisi A, Wang Y, Khan S, Heidaran M, Pal A, Zhang X, He S, Zeitlin A, et al. Human placenta-derived adherent cells prevent bone loss, stimulate bone formation, and suppress growth of multiple myeloma in bone. Stem Cells. 2011;29:263–273. doi: 10.1002/stem.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhang G, Guo B, Wu H, Tang T, Zhang BT, Zheng L, He Y, Yang Z, Pan X, Chow H, et al. A delivery system targeting bone formation surfaces to facilitate RNAi-based anabolic therapy. Nat Med. 2012;18:307–314. doi: 10.1038/nm.2617. [DOI] [PubMed] [Google Scholar]
- 194.Arnulf B, Lecourt S, Soulier J, Ternaux B, Lacassagne MN, Crinquette A, Dessoly J, Sciaini AK, Benbunan M, Chomienne C, et al. Phenotypic and functional characterization of bone marrow mesenchymal stem cells derived from patients with multiple myeloma. Leukemia. 2007;21:158–163. doi: 10.1038/sj.leu.2404466. [DOI] [PubMed] [Google Scholar]
- 195.Garayoa M, Garcia JL, Santamaria C, Garcia-Gomez A, Blanco JF, Pandiella A, Hernández JM, Sanchez-Guijo FM, del Cañizo MC, Gutiérrez NC, et al. Mesenchymal stem cells from multiple myeloma patients display distinct genomic profile as compared with those from normal donors. Leukemia. 2009;23:1515–1527. doi: 10.1038/leu.2009.65. [DOI] [PubMed] [Google Scholar]
- 196.Wallace SR, Oken MM, Lunetta KL, Panoskaltsis-Mortari A, Masellis AM. Abnormalities of bone marrow mesenchymal cells in multiple myeloma patients. Cancer. 2001;91:1219–1230. doi: 10.1002/1097-0142(20010401)91:7<1219::aid-cncr1122>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 197.Markovina S, Callander NS, O’Connor SL, Xu G, Shi Y, Leith CP, Kim K, Trivedi P, Kim J, Hematti P, et al. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-kappaB activity in myeloma cells. Mol Cancer. 2010;9:176. doi: 10.1186/1476-4598-9-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.André T, Meuleman N, Stamatopoulos B, De Bruyn C, Pieters K, Bron D, Lagneaux L. Evidences of early senescence in multiple myeloma bone marrow mesenchymal stromal cells. PLoS One. 2013;8:e59756. doi: 10.1371/journal.pone.0059756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Zdzisińska B, Bojarska-Junak A, Dmoszyńska A, Kandefer-Szerszeń M. Abnormal cytokine production by bone marrow stromal cells of multiple myeloma patients in response to RPMI8226 myeloma cells. Arch Immunol Ther Exp (Warsz) 2008;56:207–221. doi: 10.1007/s00005-008-0022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wang X, Zhang Z, Yao C. Angiogenic activity of mesenchymal stem cells in multiple myeloma. Cancer Invest. 2011;29:37–41. doi: 10.3109/07357907.2010.496758. [DOI] [PubMed] [Google Scholar]
- 201.Zangari M, Terpos E, Zhan F, Tricot G. Impact of bortezomib on bone health in myeloma: a review of current evidence. Cancer Treat Rev. 2012;38:968–980. doi: 10.1016/j.ctrv.2011.12.007. [DOI] [PubMed] [Google Scholar]