

Abstract
Substrates having appendages that pre-encode skeletal information (σ-elements) can be converted into products having distinct skeletons using a common set of reaction conditions. The sequential use of the Ugi 4CC-IMDA reaction, followed by allylation, hydrolysis, and acylation of a chiral amino alcohol appendage (σ-element), leads to substrates for a ROM/RCM or RCM reaction. The stereochemistry of the σ-element and not its constitution controls the outcome of the pathway selected. This work illustrates the potential of linking stereochemical control to the challenging problem of skeletal diversity.
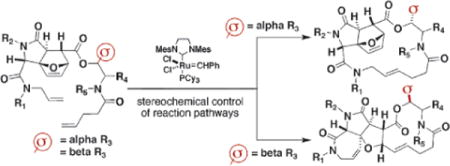
Diversity-oriented synthesis (DOS) aims to yield collections of products having many distinct molecular skeletons with controlled variation of stereochemistry in a small number (between three and five) of steps.1 The primary strategy for accomplishing skeletal diversity thus far has been to treat a substrate having potential for diverse reactivity with different reagents that produce different skeletons.2 A second strategy was recently demonstrated to generate skeletal diversity combinatorially.3 This strategy involves the conversion of a collection of substrates having different appendages that pre-encode skeletal information (named “σ-elements”) into products having distinct skeletons using a common set of reaction conditions. Here we report the characterization of a σ-element on the basis of stereochemistry rather than constitution. As such, it illustrates the potential of linking stereochemical control, for which many methods exist, to the challenging problem of skeletal diversity.
Tricyclic compounds resulting in “one pot” from consecutive Ugi and intramolecular Diels-Alder reactions4 can be readily converted to 4-pentenoic acid esters and amides (amide depicted in Figure 1). Related compounds have been found to be conformationally well-suited to undergo efficient 12-membered ring-forming reactions.5
Figure 1.
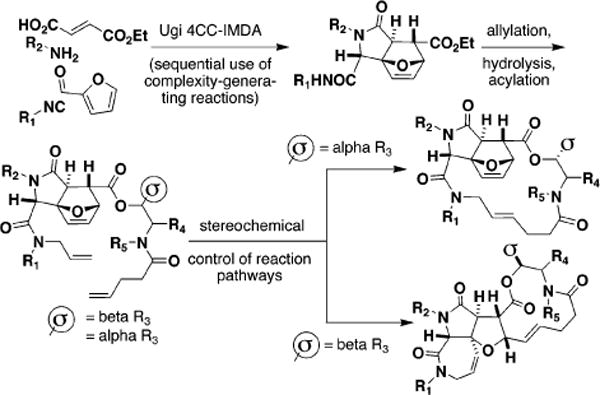
Previously, the sequential use of complexity-generating reactions, the Ugi 4CC–IMDA, was shown to produce complex molecules efficiently.4 Here we show that the orientation of a single substituent R3, denoted as a σ-element, leads to products having different skeletal arrays.
In this case, we envisioned forming such rings via ring-opening/ring-closing metathesis reactions,4b,6 although the indicated substrates are also candidates for 17-membered ring-forming metathesis reactions. We were attracted to this pathway for its potential to yield highly complex products having different skeletons efficiently. We found that a single stereocenter in the otherwise similar substrates dictates the resulting reaction pathway.
(±)-Tricyclic ester 1, available in one step from p-methoxybenzylamine, furfural, fumaric acid monoethyl ester, and benzyl isocyanide (MeOH, 48 h; 90% yield, 11:1 (structure of major diastereomer 1 ascertained by X-ray crystallography),4 was converted into the resolved acids (+)-4 and (−)-4 (Figure 2). Two intermediates in this process, 2 and 3, were first examined as tests of the proposed chemistry. Under the actions of a ruthenium-based catalyst7 in refluxing CH2Cl2 (4 h), 2 was converted into the fused macrocycle 5 (65%), while 3 was converted (4 h) to the bridged macrocycle 6 (87%, E/Z = 10:1; X-ray crystallography) (Figure 3). The pathway selectivity is uniformly high: in the reactions of 2 and 3, the product resulting from the alternative pathway was not observed to the limits of our detection (>100:1; HPLC-MS).
Figure 2.
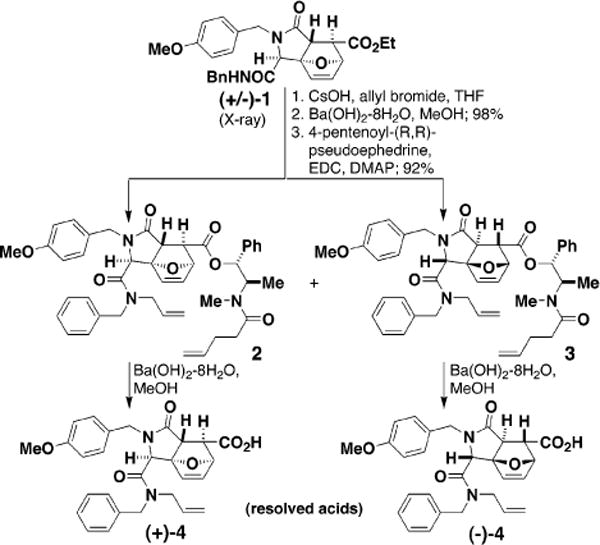
Racemic tricycle 1 was acylated with 4-pentenoyl-(R,R)-pseudoephedrine, yielding diastereomers that were purified using silica gel chromatography. Following saponification, the resolved acids (+)-4 and (−)-4 were used in subsequent experiments.
Figure 3.
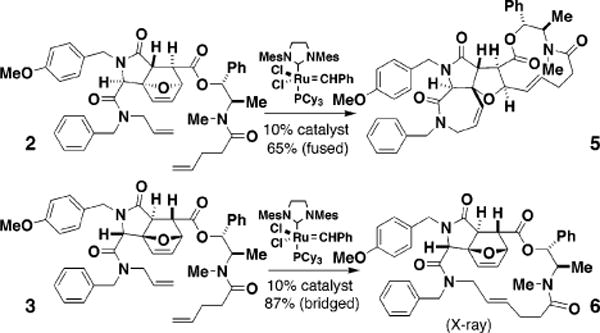
Two examples of N–Me pentenamides illustrating the role of stereochemistry in determining the direction followed in ring-closing vs ring-opening/ring-closing reactions.
To harness the potential of these reactions in DOS, we sought to understand the structural basis of the pathway selectivity. A systematic study of ring-closing reactions of substrates prepared from (+)-4 and (−)-4 and bearing chiral amino alcohols that are structurally reminiscent of pseudoephedrine was performed. The six examples of substrates having an NH in place of the N-Me group are shown in Figure 4. Substrates 7 and 9 illustrate the dominant role of the amino alcohol-derived side chain (compare to 2 and 3). 11 and 13 illustrate the dominant role played by the stereochemistry of the carbon adjacent to the ester rather than the amide of the amino alcohol moiety (compare to 7 and 9). The simpler substrates 15 and 17 reinforce this point and reveal that the stereochemistry and not the nature of the substituent (Ph vs Me) controls the reaction pathway.8
Figure 4.
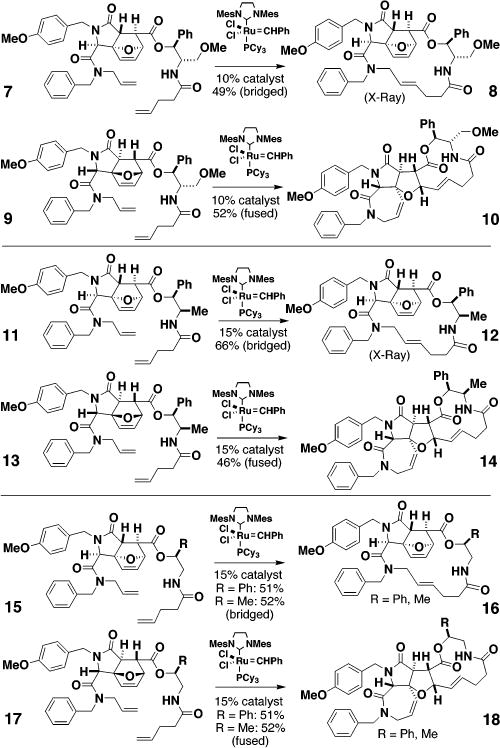
Six examples of NH pentenamides illustrating the role of a single stereogenic center in determining the direction followed in ring-closing vs ring-opening/ring-closing reactions.
Finally, 19 and 21, which have the orientation of their amino alcohol moieties reversed (Figure 5), reveal that the location of ester and amide groups does not override the stereocontrol element (though increased yields are observed in the latter orientation).9 This study suggests ways to exploit the influence of stereocenters on alternative reaction pathways and, more generally, to plan short reaction sequences that lead to complex small molecules having different skeletons.
Figure 5.
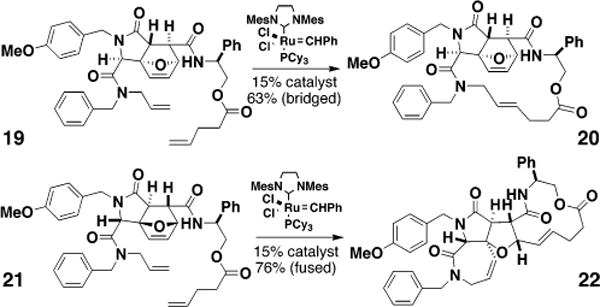
Two examples of pentenoate esters illustrating the independence of amino alcohol orientation in determining the direction followed in ring-closing vs ring-opening/ring-closing reactions.
Encouragingly, catalysts that induce high enantioselectivity in the initial 4CC reaction have been developed, and the macrocyclizations described herein have been determined to proceed efficiently on macrobead supports (unpublished results of P.R.A.). Finally, we note that this study also illustrates how DOS research can uncover new and subtle facets of reactivity in organic chemistry.
Supplementary Material
Acknowledgments
We thank Dr. Richard Staples for performing X-ray crystallographic analysis. J.K.S. acknowledges predoctoral fellowships from the National Science Foundation and the ACS Division of Organic Chemistry, Pharmacia & Upjohn. D.L. and S.L.S. are Research Associate and Investigator, respectively, at the Howard Hughes Medical Institute at Harvard University.
Footnotes
Supporting Information Available: Representative experimental procedure and characterization data and an X-ray cryatallographic file (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Schreiber SL. Science. 2000;287:1964–1968. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]; (b) Burke MD, Schreiber SL. Angew Chem, Int Ed. doi: 10.1002/anie.200300626. in press. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber SL. Chem Eng News. 2003;81:51–61. [Google Scholar]
- 3.Burke MD, Berger EM, Schreiber SL. Science. in press. [Google Scholar]
- 4.(a) Paulvannan K. Tetrahedron Lett. 1999;40:1851–1854. [Google Scholar]; (b) Lee D, Sello JK, Schreiber SL. Org Lett. 2000;2:709–712. doi: 10.1021/ol005574n. [DOI] [PubMed] [Google Scholar]
- 5.Lee D, Sello JK, Schreiber SL. J Am Chem Soc. 1999;121:10648–10649. [Google Scholar]
- 6.(a) Zuercher WJ, Hashimoto M, Grubbs RH. J Am Chem Soc. 1996;118:6634–6640. [Google Scholar]; (b) Zuercher WJ, Scholl M, Grubbs RH. J Org Chem. 1998;63:4291–4298. [Google Scholar]; (c) Stragies R, Blechert S. Tetrahedron. 1999;55:8179–8188. [Google Scholar]; (d) Arjona O, Csákÿ AG, Murcia MC, Plumet J. J Org Chem. 1999;64:9739–9741. [Google Scholar]; (e) Weatherhead GS, Ford JG, Alexanian EJ, Schrock RR, Hoveyda AH. J Am Chem Soc. 2000;122:1828–1829. [Google Scholar]
- 7.Tricyclohexylphosphine[1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimi-dazol-2-ylidene] [benzylidene]ruthenium(IV) dichloride:; (a) Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]; (b) Chatterjee AK, Grubbs RH. Org Lett. 1999;1:1751–1753. doi: 10.1021/ol991023p. [DOI] [PubMed] [Google Scholar]; (c) Huang JK, Stevens ED, Nolan SP, Petersen JL. J Am Chem Soc. 1999;121:2674–2678. [Google Scholar]; (d) Huang JE, Schanz H-J, Stevens ED, Nolan SP. Organometallics. 1999;18:5375–5380. [Google Scholar]
- 8.Remote stereocenters have been reported to impact the stereoselectivity of ring-closing metathesis reactions (; Meng D, Su D-S, Balog A, Bertinato P, Sorensen EJ, Danishefsky SJ, Zheng Y-H, Chou T-C, He L, Horwitz SB. J Am Chem Soc. 1997;119:2733–2734. [Google Scholar]; ) and the diastereoselectivity of other types of ring-closing reactions (review: Tokoroyama, T. Chem. Eur. J. 1998, 4, 2397–2404).
- 9.(a) Fürstner A. Top Organomet Chem. 1998;1:37–72. [Google Scholar]; (b) Fürstner A. Angew Chem, Int Ed. 2000;39:3012–3043. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.