

Abstract
The phosphorylation of the carboxy-terminal heptapeptide repeats of the largest subunit of RNA polymerase II (Pol II) controls several transcription-related events in eukaryotes. Trypanosomatids lack these typical repeats and display an unusual transcription control. RNA Pol II associates with the transcription site of the spliced leader (SL) RNA, which is used in the trans-splicing of all mRNAs transcribed on long polycistronic units. We found that Trypanosoma cruzi RNA Pol II associated with chromatin is highly phosphorylated. When transcription is inhibited by actinomycin D, the enzyme runs off from SL genes, remaining hyperphosphorylated and associated with polycistronic transcription units. Upon heat shock, the enzyme is dephosphorylated and remains associated with the chromatin. Transcription is partially inhibited with the accumulation of housekeeping precursor mRNAs, except for heat shock genes. DNA damage caused dephosphorylation and transcription arrest, with RNA Pol II dissociating from chromatin although staying at the SL. In the presence of calyculin A, the hyperphosphorylated form detached from chromatin, including the SL loci. These results indicate that in trypanosomes, the unusual RNA Pol II is phosphorylated during the transcription of SL and polycistronic operons. Different types of stresses modify its phosphorylation state, affecting pre-RNA processing.
INTRODUCTION
In higher eukaryotes, the synthesis of all mRNAs and some small nuclear RNAs is catalyzed by RNA polymerase II (Pol II). Rpb1, the largest subunit of RNA Pol II, contains a highly flexible structure at its C terminus, the carboxy-terminal domain (CTD). The CTD is unique in RNA Pol II and possesses multiple tandemly repeated heptapeptides with the consensus sequence YSPTSPS. This structure is essential for regulating different steps of gene expression, from transcription initiation to elongation and termination (1–3).
The CTD is the target of phosphorylations at serine, threonine, and tyrosine residues and glycosylation at serine residues (4). Serine 5 is phosphorylated (Ser5P) by transcription factor IIH (TFIIH)-associated kinase, and it remains phosphorylated while RNA Pol II transcribes the first few hundred nucleotides of genes. Ser5P serves as an acceptor site for RNA triphosphatase and guanylyltransferase, both of which are involved in the capping reaction (5). Phosphorylation of serine 2 (Ser2P) is related to transcription elongation, and it is achieved by the positive transcription elongation factor b (p-TEFb) complex (5–7). Ser2P is required for polyadenylation and transcription termination of mRNA genes (8, 9). While Ser5P and Ser2P modifications appear to be involved in the expression of many genes (1), Ser7 phosphorylation is important for the transcription of specific genes, such as human snRNA gene expression (10). Thus, the CTD works as a platform for the binding and release of several transcriptional regulatory proteins (11).
The number of CTD repeats varies from organism to organism, and it appears to be related to genome complexity; for example, 5 repeats are present in Plasmodium yoelii, 26 are present in Saccharomyces cerevisiae, and 52 are present in mammals. RNA Pol II of trypanosomatids, protozoa that cause serious human and animal diseases, lack the typical heptapeptide repeats. Among these organisms is Trypanosoma cruzi, the etiological agent of Chagas' disease (12), which affects several million people in Latin America and currently represents a world health challenge (13). Trypanosomatids diverged early in evolution and have peculiar ways of regulating gene expression, with functionally unrelated genes being transcribed polycistronically, generating pre-mRNAs that are processed by trans-splicing and polyadenylation. trans-splicing adds a 39-nucleotide capped miniexon to the 5′ termini of all mRNAs and originates from spliced leader (SL) RNA (14–18), which is transcribed from a defined promoter by RNA Pol II (19, 20). Immunolocalization of RNA Pol II in T. cruzi shows that most of the enzyme is concentrated in a region associated with the transcription of the SL RNA (21). So far, there is no evidence demonstrating differential regulation of RNA Pol II transcription of individual genes or gene clusters, but transcription is known to be globally downregulated in nondividing parasites, which are the infective forms of these organisms (22, 23). It has been postulated that transcription also initiates at the beginning of long polycistronic arrays, but the nature of the promoter at these regions is still elusive (24).
The largest subunit of RNA Pol II in Trypanosoma brucei is phosphorylated in the carboxy-terminal region, and this modification plays an essential role in transcription (25, 26). As in T. brucei, the T. cruzi RNA (TcRNA) Pol II Rbp1 appears as a doublet in SDS-PAGE gels (27), suggesting that it is also phosphorylated. The fast-migrating form in most organisms is known as subunit IIa, which has a hypophosphorylated CTD, whereas the slow-migrating form is known as subunit IIo, which has a hyperphosphorylated CTD (28–30). Subunit IIa preferentially associates with the preinitiation complex at the promoter, and the subunit IIo form is instead stably associated with the elongating regions of the DNA (5, 31, 32). Here we aimed to understand the role of phosphorylation of Rpb1 in gene transcription in trypanosomes. To this end, we studied the relationship between transcription and phosphorylation. Initially, we confirmed that TcRNA Pol II contains the hypophosphorylated and hyperphosphorylated forms. We then analyzed whether hyperphosphorylated TcRNA Pol II is associated with the DNA and whether it is present in the SL or in genes in general. We found that the phosphorylation state is largely modified upon heat shock (HS) or DNA damage. We propose that the phosphorylation of TcRNA Pol II in trypanosomes signals stress responses, which can affect enzyme associations with chromatin and play a role in transcription-related events.
MATERIALS AND METHODS
Parasite cultures and incubations.
Trypanosoma cruzi epimastigotes of strain Dm28c were cultivated in liver infusion tryptose medium supplemented with 10% fetal bovine serum, 25 μg/ml hemin, and 1.5% yeast extract at 28°C (33). Exponentially growing parasites ([1 to 3] × 107 cells/ml) were treated for 3 h with 50 μg/ml actinomycin D (Sigma-Aldrich), 100 to 500 μM 5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole (DRB; Sigma-Aldrich), and 50 μg/ml proflavine (Sigma-Aldrich) dissolved in water. To induce DNA damage, cells were incubated for the indicated times with 0.02% methyl methanesulfonate (MMS), 150 μg/ml proflavine, and 250 μM H2O2 (Merck). The following phosphatase and kinase inhibitors were used: 60 to 120 nM calyculin and 100 to 500 nM okadaic acid (LC Laboratories). For HS experiments, cells were maintained at 40°C for 2 h. After each treatment, cells were collected and centrifuged at 2,000 × g for 2 min, and the resulting pellet was washed with phosphate-buffered saline (PBS) and used for Western blotting or immunofluorescence. The following antibodies were used: affinity-purified anti-TcRNA Pol II antibody (21), anti-T. cruzi β-tubulin C-terminal sequence antibody, anti-eukaryotic initiation factor 5A (eIF5A) antibody (34), and anti-nonacetylated histone H4 N-terminal sequence antibody (35).
Preparation of chromatin-enriched extracts and phosphatase treatment.
Chromatin-enriched extracts were prepared by lysis of 2 × 107 parasites in 50 μl of ice-cold PBS containing 0.1% Triton X-100, 20 mM EDTA, 1 mM phenylmethanesulfonyl fluoride (PMSF), and protease (Complete EDTA-Free) and phosphatase (PhosSTOP) inhibitors (Roche). The cells were pipetted up and down for 2 min on ice. Lysates were centrifuged at 7,000 × g for 5 min at 4°C, and supernatants were then removed, immediately mixed with sample buffer containing SDS, and heated for 5 min at 95°C. Pellets (rich in chromatin) were washed with lysis buffer without Triton X-100 and processed as described above for the supernatants. For treatment with phosphatase, 1 × 107 parasites were suspended in 1 ml of ice-cold solution containing 0.5% saponin, 1 mM EDTA, 0.25 M sucrose, 3 mM CaCl2, 10 mM Tris-HCl (pH 7.4), 1 mM PMSF, and a protease inhibitor cocktail (Complete EDTA-Free). The cells were mixed by vortexing for 15 s, following incubation for 15 s on ice, during 10 min. Lysates were centrifuged at 7,000 × g for 10 min at 4°C, the supernatant was removed, and the pellet was washed with the same solution as that described above for the phosphatase treatment, without saponin. The final pellet was then treated with 15 units of calf intestinal alkaline phosphatase (CIAP; Promega) at room temperature for 15 min. Control samples were incubated in the absence of enzyme.
Immunoblotting.
Immunoblots were performed with extracts containing the equivalent of 1 × 107 parasites per lane submitted to electrophoresis on 6% SDS-polyacrylamide gels, with a 2-h transfer onto nitrocellulose membranes using established procedures. The membranes were stained with 0.3% Ponceau S in 10% trichloroacetic acid (TCA) and then treated with 5% nonfat milk in PBS containing 0.1% Tween 20 for 1 h. Anti-TcRNA Pol II (CTD) antibody was used at a 1:400 dilution, anti-histone H4 was used at a 1:1,000 dilution, anti-eIF5A was used at a 1:5,000 dilution, and anti-β-tubulin was used at a 1:50,000 dilution. The membranes were incubated overnight in 5% nonfat milk in PBS containing 0.1% Tween and washed in PBS–0.1% Tween, followed by 1 h of incubation with a 1:15,000 dilution of the peroxidase-labeled anti-rabbit or anti-mouse IgG antibody (Invitrogen). Bound antibodies were detected by ECL (Millipore) as recommended by the manufacturer.
Immunofluorescence analysis.
Parasites were washed in PBS and attached to glass slides pretreated with 0.01% polylysine. Alternatively, cells were lysed as described above for the experiments using CIAP and washed in PBS before attachment to glass slides. Slides were incubated with 2% paraformaldehyde in PBS at room temperature for 5 min and permeabilized for 5 min in PBS containing 0.1% Triton X-100. Fixed and permeabilized cells were washed with PBS, and the slides were incubated with anti-TcRNA Pol II antibodies (1:150) for 1 h at room temperature. The slides were washed with PBS, incubated with Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen) at a 1:1,500 dilution, washed once more, and mounted in Prolong Gold Antifade reagent (Invitrogen) in the presence of 10 μg of 4-6-diamidino-2-phenylindole (DAPI) per milliliter.
DNA damage detection.
DNA damage was quantified by PCR. Parasites (1 × 108) were collect by centrifugation (2,000 × g for 2 min) and washed with PBS, and the DNA was extracted by using a DNeasy Blood and Tissue kit (Qiagen). DNA from each extraction was used for PCR amplification as described previously (36). The assay was performed, and the amplification of the DNA from a treated sample was compared with the amplification of the undamaged control (37). The primers for the 9.9-kb genomic fragment of the nuclear DNA were Lnsense and Lnantisense. Small noncoding DNAs (snDNAs) (257 bp) were amplified by using the primers Snsense and Snantisense. All oligonucleotides used in this study are listed in Table S1 in the supplemental material. The PCR amplifications were performed under the same conditions as those described previously (37).
In vitro transcription.
Parasites were washed three times and resuspended to 7 × 106 parasites per ml in transcription buffer containing 80 U/ml of RNase inhibitor (Invitrogen) (21). These cells were then permeabilized with 50 μg/ml of lysolecithin (Sigma-Aldrich) for 2 min on ice, centrifuged, washed in transcription buffer, and resuspended to the same cell density in transcription buffer containing 2 mM ATP, 1 mM CTP, 1 mM GTP, 0.5 mM bromo-UTP (Br-UTP) (Invitrogen), 200 μg/ml of creatine kinase, and 50 mM creatine phosphate. The cells were incubated for 5 min at room temperature and then added to glass slides coated with polylysine, fixed for 15 min with 4% paraformaldehyde in PBS, and processed for immunofluorescence using 2.5 μg of mouse monoclonal anti-5-bromodeoxyridine (BrdU) antibody (Roche) per milliliter and anti-TcRNA Pol II diluted in 1% bovine serum albumin (BSA)–PBS. Bound antibodies were detected by using Alexa Fluor 594 goat anti-mouse IgG and Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen).
Image collection and analysis.
The slides were observed in an Olympus BX-61 microscope equipped with a 100× Plan Apo-oil objective (numerical aperture [NA], 1.4). Images were acquired at every 0.2 μm for each set of excitation/emission filters by using a Hamamatsu Orca R2 charge-coupled-device (CCD) camera. Blind deconvolution was performed by employing AutoQuant X2.2 software (Media Cybernetics). Except where indicated, the presented images correspond to the sum of deconvolved sections.
Quantitative reverse transcription-PCR (RT-PCR) and gene expression analysis.
Two micrograms of total RNA prepared by TRIzol extraction (Life Sciences) from parasite pellets was used for cDNA preparation using SuperScript III reverse transcriptase (Life Sciences). Quantitative PCR was performed by using Power SYBR green PCR master mix (Applied Biosystems) on the LineGene 9660 apparatus (Bioer). All reactions were run in triplicate from three independent experiments. For pre-mRNA amplifications, forward primers were based on the coding sequence (CDS) near the ATG codon, and reverse primers were based on a sequence localized 100 bp downstream of each forward primer. For mature mRNA, amplifications employed the SL sequence as the forward primer and the same reverse primers used for the reactions for pre-mRNA amplification. Primer pairs used for the analysis of mature mRNAs and pre-mRNAs of α-tubulin were SLFow/TubRev2 and PrecTubFow/TubRev2, respectively. The Hsp70 mature mRNA and pre-mRNA were amplified with primer pairs SLFow/Hsp70rev and Hsp70Fow/Hsp70rev, respectively. Similarly, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), heat shock protein (Hsp83), and TcJ2 protein were amplified with primer SLFow and the specific reverse primer for the mature RNA and with the specific forward and reverse primers for the pre-mRNA (see Table S1 in the supplemental material). The specificity of amplicons was evaluated by melting curve analysis and agarose gel electrophoresis. Standard curves for oligonucleotide pairs were obtained from serial dilutions of control cDNA samples, and the R2 value was 0.99 for both mature mRNA and pre-mRNA oligonucleotide pairs. Gene expression was determined by using absolute quantification of each amplicon in control and heat-shocked cells.
Expression and detection of XPB in the parasite.
XPB (TcCLB.510149.50) was amplified by PCR from T. cruzi CL Brener genomic DNA by using the oligonucleotides XPBFow and XPBHARev (see Table S1 in the supplemental material). The amplified fragments were cloned into the TOPO TA cloning vector (Invitrogen) and sequenced to confirm the correct sequence. The desired fragment was removed by digestion with XbaI and BamHI and inserted into the p33 vector (38), which was previously digested with the same enzymes. The p33 plasmid containing XPB-hemagglutinin (HA) (50 μg) was used to transfect T. cruzi epimastigotes by using Amaxa Nucleofector followed by selection with 0.2 mg/ml Geneticin G418 (Invitrogen). The expression of XPB-HA was confirmed by Western blotting using total extracts of transfected parasites with anti-HA antibody (Covance).
RESULTS
TcRNA Pol II is phosphorylated, and the hyperphosphorylated form is tightly associated with chromatin fractions.
The anti-TcRNA Pol II antibody specifically labeled two bands in immunoblots of total T. cruzi lysates (Fig. 1A). When parasites were lysed in nondenaturing buffers containing detergent, protease, and phosphatase inhibitors, the slow-migrating band remained in the insoluble fraction, which corresponded to proteins associated with the DNA (chromatin-enriched extract), as is the case for histone H4. In contrast, the soluble fraction, enriched in cytosolic proteins such as eukaryotic initiation factor 5A (eIF5A) and proteins that are not bound to chromatin, contained the fast-migrating band.
FIG 1.
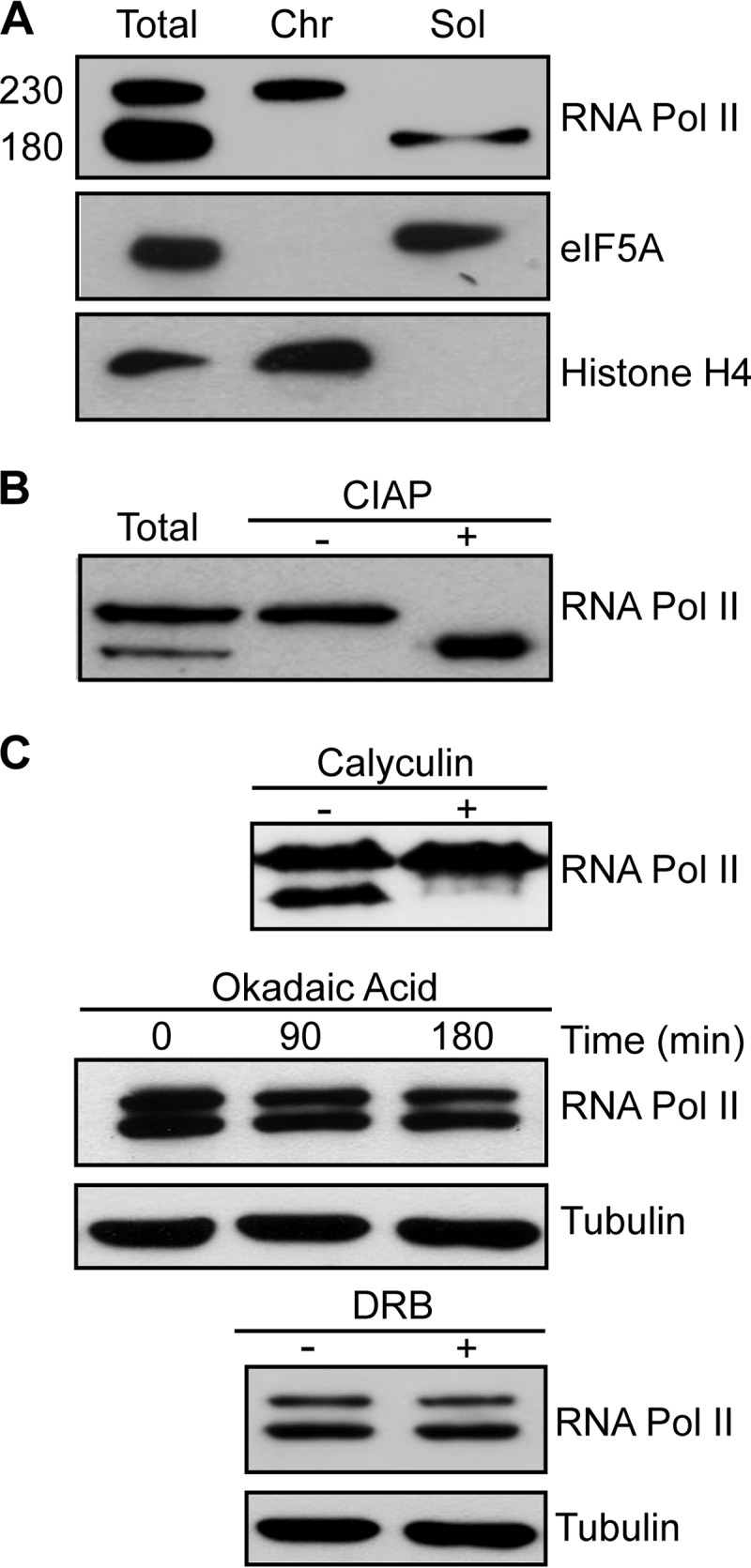
TcRNA Pol II is phosphorylated and tightly associated with chromatin. (A) Western blot of T. cruzi epimastigote pellets dissolved and boiled in SDS-PAGE sample buffer (Total) or previously lysed under nondenaturing conditions for the separation of chromatin (Chr) and soluble (Sol) fractions, before addition of sample buffer. The molecular mass of TcRNA Pol II is shown at left (kDa). (B) Western blot of parasite lysates (Total) or of the chromatin fraction treated without (−) or with (+) CIAP. (C) Western blots of total lysates of control cells (−) or cells pretreated (+) with 90 nM calyculin (2 h), 500 nM okadaic acid for the indicated times, or 500 μM DRB (3 h). The blots were probed with anti-TcRNA Pol II, anti-eIF5A, anti-histone H4, and anti-β-tubulin.
The 180-kDa band was the expected size for the largest RNA Pol II subunit, while the 230-kDa band might correspond to the phosphorylated form of Rpb1, as found previously for T. brucei (26). To determine whether the presence of these two forms was due to differences in the level of protein phosphorylation, extracts enriched for proteins associated with DNA were treated with alkaline phosphatase (CIAP). In samples incubated with CIAP, the 230-kDa band disappeared, and the 180-kDa band became more apparent (Fig. 1B). Likewise, treatment of live parasites with 90 nM calyculin A, a general protein phosphatase inhibitor, caused the fast-migrating band to disappear (Fig. 1C). The same effect was not observed for parasites incubated with okadaic acid (another phosphatase inhibitor) at concentrations of up to 500 nM and DRB (a specific inhibitor of some cyclin-dependent kinases [CDKs], including the kinase present in the p-TEFb complex) (Fig. 1C). Taken together, these results demonstrate that phosphorylated Rpb1 is strongly associated with DNA and that specific protein phosphatases are involved in dephosphorylation of the enzyme.
Inhibition of transcription by intercalating agents does not affect TcRNA Pol II phosphorylation levels.
The tight association of the phosphorylated form of TcRNA Pol II with chromatin suggests that it could correspond to the elongating enzyme, as shown for Rbp1 proteins of several organisms that undergo phosphorylation of the CTD heptapeptide. Therefore, we investigated whether transcription inhibitors could affect TcRNA Pol II phosphorylation and its association with chromatin. The parasites were incubated with actinomycin D, a compound that prevents initiation and elongation by binding to DNA (39). In T. cruzi, no dephosphorylation was observed within 3 h of incubation (Fig. 2A), while transcription was markedly reduced, as judged by Br-UTP incorporation (Fig. 2B). Interestingly, the phosphorylated form of TcRNA Pol II remained associated with chromatin after these treatments (Fig. 2C). When the RNA Pol II distribution in the nucleus was evaluated for parasites treated with actinomycin D, we observed an accumulation of the protein in several spots throughout the nucleus (Fig. 2D). We have previously shown that the major spot of RNA Pol II localization corresponds to the site of SL gene transcription, which disappears following actinomycin D treatment (21). Consequently, the current results suggest that hyperphosphorylated TcRNA Pol II is trapped in long polycistronic regions and not in the SL RNA gene spot and that TcRNA Pol II is phosphorylated in non-SL transcription sites. Hyperphosphorylated TcRNA Pol II is also at the SL transcription sites in untreated cells, as demonstrated in experiments detecting the enzyme associated with the SL body after detergent extraction (see Fig. S1A in the supplemental material), a treatment that ensures that most of the insoluble enzyme corresponds to the hyperphosphorylated form (see Fig. S1B in the supplemental material).
FIG 2.

TcRNA Pol II phosphorylation and association with chromatin are maintained in cells treated with actinomycin D. (A) Epimastigotes were treated with 50 μg/ml of actinomycin D in dimethyl sulfoxide (DMSO) for the indicated periods of time, washed and dissolved in SDS-PAGE sample buffer, and processed for Western blotting using anti-TcRNA Pol II and anti-β-tubulin antibodies. (B) Cells treated with actinomycin D were also washed and fractionated under nondenaturing conditions before Western blotting using anti-TcRNA Pol II antibodies. (C and D) Treated and untreated cells were also washed, attached to glass slides, and processed for in vitro transcription using Br-UTP (C) or for immunofluorescence using anti-TcRNA Pol II antibodies (D). The images in the figure are deconvolved sections of DAPI (blue), Br-UTP (red), or TcRNA Pol II (green) labeling. The fluorescence images correspond to the portions of differential interference contrast (DIC) images delimited by the dashed lines. k, kinetoplast; N, nucleus. Bar = 5 μm.
Heat shock causes TcRNA Pol II dephosphorylation without dissociation from chromatin.
Genome-wide downregulation of transcription occurs in many organisms with HS (40). In higher eukaryotes, the largest subunit of RNA Pol II becomes more phosphorylated, and it is released from DNA (41–43). Therefore, we investigated how HS could affect TcRNA Pol II phosphorylation. As shown in Fig. 3A, parasites incubated at 40°C exhibited a progressive dephosphorylation of TcRNA Pol II, with most of the enzyme being dephosphorylated after 2 h. Dephosphorylation did not occur when calyculin A was added to the medium during HS (Fig. 3B). Importantly, upon HS, parasites remained fully motile, and although they exhibited a growth delay, they fully recovered after 96 h (Fig. 3C). However, in contrast to control cells, the hypophosphorylated form of TcRNA Pol II remained associated with chromatin (Fig. 3D). Furthermore, no significant dispersion was observed compared to nonstressed cells (Fig. 3E), even with a 3.5-fold decrease in transcription levels, as assessed by Br-UTP incorporation (Fig. 3F). Although this transcription determination is subjected to variations, the difference was large enough to evidence a significant decrease in transcription levels.
FIG 3.

Heat shock causes TcRNA Pol II dephosphorylation. The enzyme remains attached to chromatin, and transcription is partially active. (A and B) Western blots of epimastigotes preincubated for the indicated periods of time at 40°C in the absence (A) and presence (B) of 50 nM calyculin A. The Western blots were probed with anti-TcRNA Pol II and anti-β-tubulin antibodies, as indicated. (C) Growth of epimastigotes submitted to HS. (D) Western blot of total and chromatin-associated (Chr) fractions of control cells and epimastigotes submitted to HS for 2 h. (E and F) The same cells were fixed, permeabilized, and processed for immunofluorescence using anti-TcRNA Pol II antibodies (E) or permeabilized and incubated with Br-UTP (F), following labeling with anti-BrdU as described in Materials and Methods. The images show DAPI, anti-TcRNA Pol II (green), or anti-Br-UTP (BrU) (red) staining, and the merged images and differential interference contrast images indicated by the dashed lines are the regions corresponding to the fluorescent images. k, kinetoplast; N, nucleus. Bar = 2 μm. The white numbers in Br-UTP images correspond to the means ± standard deviations of the red channel pixel intensity (n = 100). (G and H) Relative abundances (means ± standard deviations; n = 3) of the mature mRNAs (processed) (G) and the precursor mRNAs (precursor) (H) of the indicated genes. Asterisks indicate a P value of <0.01, for comparisons between the control and HS samples for mature and precursor tubulin, and a P value of <0.05 for the GAPDH gene, calculated by the Student t test.
In T. brucei, HS promotes accumulation of mRNA precursors except for those of the heat shock proteins (Hsps), possibly due to defects in the trans-splicing reaction (44). Using T. cruzi epimastigotes submitted to temperatures of 40°C for 2 h, Nazer and collaborators (45, 46) found that most mRNA accumulated in the nucleus, except for mRNAs corresponding to Hsp, which were exported to the cytosol. Therefore, we searched for the presence of the processed precursor of each of the following genes before and after HS by quantitative real-time PCR: α-tubulin, GAPDH, Hsp83, Hsp70, and Tcj2. As shown in Fig. 3G, as expected, there was a decrease in the mRNA amplified with a primer for the coding region of α-tubulin and GAPDH with the SL, while the levels of precursor mRNA were not affected when a pair of primers that recognizes the beginning of the coding sequence of the α-tubulin and GAPDH genes was used (Fig. 3G). In contrast, the levels of precursor and processed Hsp70, Hsp83, and Tcj2 remained unaffected (Fig. 3H). These results suggest that HS, by causing hypophosphorylation of TcRNA Pol II, produces inefficient trans-splicing of housekeeping genes but not of HS proteins.
DNA damage leads to TcRNA Pol II dephosphorylation.
It has been shown that agents that cause DNA damage affect phosphorylation of the largest subunit of RNA Pol II in several organisms (47–50). Therefore, we investigated whether DNA-damaging agents could also affect TcRNA Pol II phosphorylation and transcription. After incubation of parasites with 150 μg/ml of proflavine, 250 μM hydrogen peroxide (H2O2) for 3 h, or 0.02% MMS for 24 h, cellular DNA was extracted and analyzed by PCR using primers that amplified short (250 bp) and long (9.9 kb) nuclear DNA fragments from the same region for estimation of DNA breaks. Proflavine, H2O2, and MMS caused DNA damage at the concentrations used (Fig. 4A), but no significant damage was detected after actinomycin D incubation (see Fig. S2C in the supplemental material) or HS treatment (data not shown). H2O2 and proflavine also impaired cellular growth but did not kill the parasites, which were able to reestablish normal growth (Fig. 4B and C). In contrast, parasites treated with MMS remained fully motile but did not recover growth (Fig. 4C). Notably, the levels of the hyperphosphorylated TcRNA Pol II markedly decreased after these treatments, while levels of the hypophosphorylated form increased, except in the case of H2O2, which also caused TcRNA Pol II degradation (Fig. 4D). Moreover, in all these treatments, only hyperphosphorylated TcRNA Pol II remained associated with chromatin (Fig. 4E). The addition of calyculin A prevented proflavine-induced dephosphorylation (Fig. 4F), indicating that dephosphorylation caused by proflavine depends on a phosphatase. In contrast, no dephosphorylation of TcRNA Pol II was observed in cells exposed to gamma irradiation (500 Gy) (see Fig. S2A in the supplemental material), which was enough to produce growth arrest, with further recovery after 3 to 4 days (see Fig. S2B in the supplemental material). We also observed that high doses of UV light did not affect growth or the TcRNA Pol II phosphorylation state (see Fig. S2A in the supplemental material). Dephosphorylation would probably require extensive DNA breaks, or particular modifications, which are not produced by gamma irradiation or UV light (see Fig. S2C in the supplemental material).
FIG 4.

DNA damage leads to TcRNA Pol II dephosphorylation. (A) The relative amount of breaks was measured by agarose gel electrophoresis and ethidium bromide staining of PCR products (9.9 kb and 250 bp) of DNA extracted from control cells (C) or cells treated for 3 h with 250 μM H2O2, 150 μg/ml of proflavine (Pro), or 0.02% MMS for 24 h in medium. (B and C) Growth of parasites after treatments. (D to F) Western blots using anti-TcRNA Pol II antibodies for experiments with control parasites (C) and parasites treated with the genotoxic components as described above for panel A (D), total lysates or the chromatin-associated fraction (Chr) of control parasites or parasites treated with 150 μg/ml proflavine (E), and total lysates of control parasites or parasites treated with proflavine (150 μg/ml) previously treated for 2 h without (−) or with (+) 50 nM calyculin A (F).
Genotoxic stress results in global transcription arrest, with a minor effect on RNA Pol II distribution.
In cells treated with H2O2, proflavine, or MMS, a 10-fold decrease in global transcription was also observed in comparison to control cells (Fig. 5A). Quantitative analyses of these differences are shown in Fig. 5 (white numbers). In contrast to the parasites treated with actinomycin D, the parasites treated with the DNA-damaging agents did not show relocalization of RNA Pol II, which remained associated with the single major spot corresponding to the SL RNA gene, as in control cells (Fig. 5B). Quantitative analyses showed no significant redistribution of fluorescence based on the fluorescence ratio between the major spot and the total nucleus (Fig. 5B, white numbers).
FIG 5.

Inhibition of transcription by DNA damage causes decreased transcription but not RNA Pol II dispersion. Parasites were treated with dimethyl sulfoxide (control), proflavine, H2O2, and MMS as described in the legend of Fig. 4. The incorporation of Br-UTP in permeable cells (A) and RNA Pol II labeling in fixed and permeabilized cells (B) were performed as described in Materials and Methods. The panels show DAPI, antibody, and differential interference contrast images, with insets indicating the positions of the fluorescent images. k, kinetoplast; N, nucleus. Bar = 3 μm. The numbers in the Br-UTP images indicate the means ± standard deviations of the red channel pixel intensity (n = 100), and the numbers in the TcRNA Pol II images indicate the percentages of pixels associated with the SL structure compared to total pixels in the green channel (n = 100) for each situation.
Inhibition of dephosphorylation causes dispersion of RNA Pol II and protects proteasome-dependent enzyme degradation.
To determine whether dephosphorylation of TcRNA Pol II would cause it to be released from chromatin, cells were treated with calyculin A and probed for the presence of phosphorylated TcRNA Pol II in chromatin and soluble fractions. As shown in Fig. 6A, calyculin A treatment prevented dephosphorylation, and the hyperphosphorylated form of TcRNA Pol II was found to be dissociated from chromatin (Fig. 6A). Moreover, immunofluorescence analysis showed that TcRNA Pol II in cells treated only with calyculin A was dissociated from the SL and dispersed to the other parts of the nucleus (Fig. 6B), as the levels of enzyme were maintained in calyculin A-treated parasites (Fig. 1C). Quantitative analyses of the amount of RNA Pol II in the SL RNA spot confirmed the dispersion (Fig. 6C), suggesting that dephosphorylation could be required for associations for a new cycle of initiation, mainly in the SL structure. As we systematically observed less of the hypophosphorylated form in the soluble fraction, even in the presence of several protease and phosphatase inhibitors, we conjectured whether it could be related to proteasome degradation, already primed during extract preparation. Therefore, we treated live cells with proteasome inhibitors (MG132 and lactacystin). As shown in Fig. 6D, this treatment caused a more efficient recovery of hypophosphorylated TcRbp1 without changes in the hyperphosphorylated form, denoting that phosphorylation can also prevent TcRNA Pol II degradation in live cells.
FIG 6.

Effects of calyculin A and proteasome inhibitors on TcRNA Pol II phosphorylation. (A) Control cells or cells incubated with 50 nM calyculin A for 2 h were processed for Western blotting using total lysates. The chromatin-associated (Chr) and soluble (Sol) fractions were probed with anti-TcRNA Pol II antibodies. (B) Immunofluorescence labeling with anti-TcRNA Pol II antibodies and DAPI staining of control or calyculin A-treated cells (k, kinetoplast; N, nucleus). Bar = 2 μm. (C) Amount of TcRNA Pol II labeling (means ± standard deviations; n = 30) associated with the SL relative to the other areas of the nucleus in control and calyculin-treated samples. (D) Cells were treated with 10 μM MG132 and 10 μM lactacystin for 2 h, and the total, chromatin-associated, and soluble fractions were analyzed by Western blotting using anti-TcRNA Pol II.
TFIIH is less associated with RNA Pol II in cells treated with proflavine.
In T. brucei, transcription initiation depends on the presence of the unusual TFIIH, which lacks the kinase domain of the classical cyclin-dependent kinase 7 (CDK7) (51). The phosphorylation of the largest subunit of RNA Pol II is dependent on CDC2-related kinase 9 (CRK9) (52), suggesting that initiation is not dependent on phosphorylation. Therefore, we analyzed the distribution of TFIH using a tagged version of the XPB protein, a component of the complex in control and proflavine-treated T. cruzi parasites. We considered three situations: (i) when Pol II colocalized with XPB in the SL, (ii) when colocalization was partial, and (iii) when there was no colocalization with the SL. As shown in Fig. 7A, in 77% of the cells, TcRNA Pol II in the SL RNA spot colocalized with the strongest spot of XPB-HA in normally growing cells. In contrast, this colocalization was reduced to one-half after 3 h of proflavine treatment. As the labeling pattern of RNA Pol II is maintained, i.e., most of the enzyme is found in the major SL spot, this suggests that although hypophosphorylated, TcRNA Pol II is localized at the putative initiation sites without fully assembled initiation complexes.
FIG 7.

XPB colocalization with RNA Pol II in the SL structure is decreased in proflavine-treated T. cruzi parasites. Epimastigotes stably transfected with plasmid p33 containing T. cruzi XBP fused the HA epitope at the C terminus were incubated in the absence (A) and presence (B) of 150 μg of proflavine per ml for 3 h. The cells were washed, fixed, permeabilized, and processed for immunofluorescence using anti-TcRNA Pol II and anti-HA antibodies. The figures show the merged fluorescence images and the image corresponding to the deconvolved section for each antibody. The three representative colocalization patterns found are, from top to bottom, colocalization of RNA Pol II with the strongest spot of XPB, colocalization of RNA Pol II but not with the strongest spot of XPB, and no colocalization with the strongest spot of XPB. The percentage of each colocalization pattern found in 100 images is shown on the left. k, kinetoplast; N, nucleus. Bar = 1.5 μm.
DISCUSSION
In this study, we demonstrated that the largest RNA Pol II subunit of T. cruzi, which lacks typical heptapeptides, is present in two forms: a hyperphosphorylated form, connected mainly to chromatin in the SL-transcribed loci, and a hypophosphorylated form, which can be dissociated from the DNA and released in a soluble form upon cell lysis in the presence of detergents. This conclusion is supported by their presence as two bands in SDS-PAGE gels. The slow-migrating band is associated with chromatin, and it can be converted into the fast-migrating band by phosphatase treatment of cellular extracts. In addition, cell treatment with calyculin A, but not okadaic acid, increased the levels of the hyperphosphorylated form, indicating that PP1-like phosphatases participate in the maintenance of the phosphorylation levels of TcRNA Pol II. Interestingly, three bands are seen in cells treated with calyculin A (Fig. 3B, 4F, and 6A), suggesting the existence of an intermediate phosphorylated form. Thus, either of the two phosphatases (one calyculin sensitive and the other not) is operating. We cannot exclude the possibility that calyculin activates one of the polymerase kinases. There are two main phosphatases implicated in the removal of phosphates from eukaryotic CTDs, Fcp-1 and Ssu72, and both are highly conserved (53). Ssu72 is responsible for removal of the phosphate from Ser5, thus facilitating the transition of RNA Pol II from the initiation phase to the elongation phase (54, 55). Ssu72 also dephosphorylates Ser7 at the end of transcription (11). Fcp-1, on the other hand, preferentially removes the phosphate from Ser2 at the end of transcription, leaving RNA Pol II ready to begin a new cycle of transcription (1, 56). Because Fcp-1 is resistant to okadaic acid (57) and several genes for Fcp-1 are detected in trypanosomes (58), one of these enzymes could be involved in TcRNA Pol II dephosphorylation. In addition, an enzyme from the RPA2 family of CTD phosphatases that dephosphorylates Ser7 (59) is found in the trypanosome genome (TcCLB.511367.310). Notably, the results obtained for T. cruzi differ from what is observed in HeLa cells, in which okadaic acid inhibits the dephosphorylation of the largest subunit of RNA Pol II (60), indicating that a more restricted set of phosphatases is involved in the dephosphorylation of the largest subunit of RNA Pol II in trypanosomes. Notwithstanding, the pattern of bands of TcRNA Pol II in cells treated with calyculin A is suggestive of several phosphorylating reactions. Accordingly, several serine and threonine residues located at the C terminus of T. brucei Rbp1 are required for viability, probably as targets of phosphorylation (26). In addition, we cannot presently exclude that phosphorylation also occurs at sites other than the CTD.
After transcription arrest triggered by the intercalation of actinomycin D into DNA, the hyperphosphorylated form of TcRNA Pol II remained bound to chromatin, mainly at the polycistronic loci, a conclusion based on the dispersion of TcRNA Pol II toward the nuclear periphery, as observed previously (21). This result clearly demonstrates that TcRNA Pol II can also be phosphorylated in non-SL transcribed genes. Moreover, the accumulation of RNA Pol II in gene arrays of distinct chromosomes is compatible with the polycistronic transcription and stalling of the polymerase in long transcription units, as opposed to being located within the short transcription units of the SL genes. In mammalian cells, actinomycin increases the level of phosphorylation of the largest subunit of RNA Pol II (61), and this is interpreted as being a consequence of an increase in the elongating complex activity (39). However, we found that TcRNA Pol II phosphorylation was not affected by DRB, an inhibitor that blocks productive transcription by preventing phosphorylation at Ser2 in the CTD heptapeptide in several eukaryotes (39). As knockdown of CRK9 decreases the level of phosphorylated Rpb1 in T. brucei (52), we presume that DRB, which affects T. cruzi growth (62), is not able to inhibit the kinases involved in Rpb1 phosphorylation in trypanosomatids.
We also observed that in T. cruzi, HS caused the total dephosphorylation of TcRNA Pol II, with partial inhibition of transcription and accumulation of unprocessed mRNA. The fact that, upon HS, hypophosphorylated TcRNA Pol II remained tightly associated with chromatin clearly indicates that dephosphorylation does not cause RNA Pol II to be released, with hypophosphorylated Rpb1 still elongating, as previously shown for T. brucei (52). In addition, after HS, the distribution of T. cruzi RNA Pol II in the nucleus was minimally affected, with a significant part of the enzyme remaining in the major SL spot. This is compatible with previous observations showing that HS does not fully inhibit SL transcription (63) and with the work of Badjatia and collaborators (52), who showed that knockdown of CRK9 in T. brucei, the only kinase involved in Rpb1 phosphorylation, causes incomplete cap4 formation. Accordingly, after HS, we detected decreased levels of processed mRNAs of α-tubulin and GAPDH without a decrease in the pre-mRNA levels. In contrast, the levels of processed Hsp70, Hsp83, and Tcj2 mRNAs were unchanged, indicating that they were less (or not) affected by the absence of cap4. We therefore speculated that HS could activate a specific phosphatase, which would cause incomplete capping of the SL RNA, affecting the trans-splicing reaction and leading to the accumulation of mRNA precursors, except for the HS-induced genes. This idea agrees with early observations that HS preferentially allows the trans-splicing of Hsp (44) and with recent findings showing the bulk of pre-mRNA accumulation in the nucleolus, with the exception of Hsp in T. cruzi (45). This also explains why translation is decreased independent of eIF2 phosphorylation with accumulation of stress granules in T. brucei (64). Indeed, in T. brucei, HS does not produce an accumulation of mRNA in the nucleolus (45). In mammalian cells, HS causes a decrease in the activity of the TFIIH-associated kinase, which is related to poor transcription initiation (42). It also decreases the activity of the specific CTD phosphatase, resulting in increased phosphorylation levels (41). In Drosophila melanogaster, HS causes phosphorylation of Rpb1 on HS promoters (65). These events do not seem to occur in trypanosomes, which regulate HS proteins posttranscriptionally (22), explaining why T. cruzi RNA Pol II is not released from chromatin upon HS, as found for mammalian RNA Pol II (43). We cannot fully exclude the possibility that TcRNA Pol II dephosphorylation is a secondary effect of the RNA processing defect in heat shock-treated cells. However, this is unlikely, as dephosphorylation is seen 30 min after HS.
We also found that some genotoxic agents caused TcRNA Pol II dephosphorylation. In these cases, the enzyme was released from the DNA but remained associated with the SL locus and probably other initiation sites. The fact that proflavine, an intercalating agent, caused DNA breaks is compatible with its capacity to promote helix torsion followed by DNA breaks produced by elongating polymerases (66, 67). Similarly, H2O2 and MMS introduce modifications in the DNA that could lead to breaks by elongating polymerases. Thus, dephosphorylation of TcRNA Pol II could be a direct consequence of DNA damage. Alternatively, dephosphorylation could result from the activation of signaling machinery upon DNA breaks and growth arrest. The fact that gamma irradiation also caused growth arrest, but with much fewer DNA breaks and essentially no dephosphorylation of TcRNA Pol II, argues against general stress response signaling. In fact, our results showing that calyculin prevented dephosphorylation in the presence of proflavine support the notion that specific phosphatases are activated depending on the type of DNA damage. Calyculin A, for example, prevented TcRNA Pol II dephosphorylation induced only by proflavine and not by H2O2. Thus, TcRNA Pol II could sense DNA alterations, being released and/or signaling for repair, which is quite robust in trypanosomes (68). In higher eukaryotes, phosphorylation of the CTD increases in response to DNA damage induced by MMS, specifically at Ser2 (47, 48). Moreover, a phosphorylated CTD is required for binding of several damage response elements to allow repair and survival, maintaining the integrity of the genome (50).
RNA Pol II is ubiquitinated when eukaryotic cells are exposed to genotoxic agents (69, 70). This ubiquitination is dependent on CTD phosphorylation (71). The fact that proteasome inhibitors did not affect the levels of TcRNA Pol II phosphorylation in the presence of DNA-damaging agents (not shown) supports the notion that different signaling events occur in trypanosomes and that, at least for DNA damage, the decrease in the levels of phosphorylated enzyme is not due to ubiquitination of Rbp1. Interestingly, we provide evidence that the hypophosphorylated enzyme was degraded in nuclear extracts, given that inhibition of dephosphorylation with calyculin A prevented degradation. These facts suggest that the hypophosphorylated form of the enzyme could be targeted for degradation. Somehow, proteasome activity might be involved, because treatment of cells with proteasome inhibitors prevents degradation after lysis. Perhaps, accumulation of an unknown factor helps to stabilize RNA Pol II in trypanosomes.
RNA Pol II must be recycled for a new round of transcription initiation. For this, in most eukaryotic cells, the CTD of the enzyme must be dephosphorylated by specific phosphatases (72). In eukaryotes, RNA Pol II is recruited to a preinitiation complex at gene promoters (5, 31, 32, 73). For transcription initiation, Ser5 residues are phosphorylated by TFIIH-associated kinase (CDK7), and their level of phosphorylation remains high as RNA Pol II transcribes the first few hundred nucleotides of the gene (5). This phosphorylation is important for clamping RNA Pol II to the DNA and forming a stable complex with chromatin. Since this stable complex is observed in trypanosome RNA Pol II, it would be reasonable to presume an initial phosphorylation event for transcription initiation in these organisms. The fact that hypophosphorylated TcRNA Pol II is found in the SL structure after DNA damage may indicate that it binds to transcription initiation sites after dephosphorylation in T. cruzi, a typical feature of eukaryotic RNA Pol II. However, the CDK7-related kinase has not been detected in the TFIIH complex of T. brucei (51), and cells remain viable in the absence of the paralogous kinases, although other similar kinases are essential (52). We found that the XPB helicase, a component of TFIIH of trypanosomes (51), colocalizes with RNA Pol II in T. cruzi and that this colocalization is partially lost upon genotoxic stress. Because most of the RNA Pol II was still present at the SL RNA site, as judged by immunofluorescence, we inferred that initiation has not begun in these cells due to high phosphatase activity, acting on RNA Pol II or indirectly on other initiation factors. In addition to acting in transcription initiation, TFIIH, including the XBP helicase, has been implicated in DNA repair (74). However, in T. brucei, there are two divergent forms of XBP. One is involved in DNA repair, while the other is apparently active in transcription initiation (75), which corresponds to the form which we used for a tag. Thus, we infer that the dissociation of XBP from RNA Pol II in the presence of proflavine is not due to recruitment of TFIIH to repairing sites and most likely corresponds to a mechanism involved in transcription control of trypanosomes.
The phosphorylation sites in the T. cruzi CTD are unknown, and despite the lack of canonical heptapeptide repeats and little amino acid similarity to CTDs of other organisms, alignments showed that serines, tyrosines, and prolines are well conserved. Most phosphorylated sites detected by phosphoproteomic analysis (76) of the T. brucei CTD, as defined previously by Das and Bellofatto (26), are conserved in T. cruzi. However, these sites were not found by T. cruzi phosphoproteomic analysis (77). We therefore postulate that TcRNA Pol II dephosphorylation is required for a new round of association of the enzyme with transcription initiation sites, while phosphorylation controls enzyme stability when it is not bound to chromatin. In addition, the phosphorylation state during elongation under normal growing conditions provides adequate cap formation and mRNA processing of the SL RNA. Upon HS, or perhaps other stresses, the enzyme is dephosphorylated, facilitating the processing of stress response genes. These stresses occur during the T. cruzi life cycle and may have an important role in controlling gene expression. These stresses include changes in temperature and exposure to oxidative stress when the parasite infects a mammalian host, with transcription being largely modulated (23). In addition, due to the large amount of polycistronic transcription found in trypanosomes, it is reasonable that TcRNA Pol II phosphorylation could act as a sensor of DNA damage for further repair. This simple mechanism might represent an early stage of transcription control during evolution.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo-FAPESP (grants 2011/51973-3 to S.S., 2012/09327-0 to A.A.R., and 2012/09403-8 to N.S.M.) and by the Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq (grant 477143/2011-3 to S.S., Instituto Nacional de Ciência e Tecnologia de Vacinas), Brazil.
We thank Claudio Rogério Oliveira and Claudeci Medeiros for technical help.
Footnotes
Published ahead of print 9 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00066-14.
REFERENCES
- 1.Egloff S, Dienstbier M, Murphy S. 2012. Updating the RNA polymerase CTD code: adding gene-specific layers. Trends Genet. 28:333–341. 10.1016/j.tig.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 2.Chapman RD, Heidemann M, Hintermair C, Eick D. 2008. Molecular evolution of the RNA polymerase II CTD. Trends Genet. 24:289–296. 10.1016/j.tig.2008.03.010 [DOI] [PubMed] [Google Scholar]
- 3.Liu P, Kenney JM, Stiller JW, Greenleaf AL. 2010. Genetic organization, length conservation, and evolution of RNA polymerase II carboxyl-terminal domain. Mol. Biol. Evol. 27:2628–2641. 10.1093/molbev/msq151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egloff S, Murphy S. 2008. Cracking the RNA polymerase II CTD code. Trends Genet. 24:280–288. 10.1016/j.tig.2008.03.008 [DOI] [PubMed] [Google Scholar]
- 5.Komarnitsky P, Cho EJ, Buratowski S. 2000. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14:2452–2460. 10.1101/gad.824700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marshall NF, Peng J, Xie Z, Price DH. 1996. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 271:27176–27183. 10.1074/jbc.271.43.27176 [DOI] [PubMed] [Google Scholar]
- 7.Ramanathan Y, Rajpara SM, Reza SM, Lees E, Shuman S, Mathews MB, Pe'ery T. 2001. Three RNA polymerase II carboxyl-terminal domain kinases display distinct substrate preferences. J. Biol. Chem. 276:10913–10920. 10.1074/jbc.M010975200 [DOI] [PubMed] [Google Scholar]
- 8.Licatalosi DD, Geiger G, Minet M, Schroeder S, Cilli K, McNeil JB, Bentley DL. 2002. Functional interaction of yeast pre-mRNA 3′ end processing factors with RNA polymerase II. Mol. Cell 9:1101–1111. 10.1016/S1097-2765(02)00518-X [DOI] [PubMed] [Google Scholar]
- 9.Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S. 2004. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432:517–522. 10.1038/nature03041 [DOI] [PubMed] [Google Scholar]
- 10.Egloff S, O'Reilly D, Chapman RD, Taylor A, Tanzhaus K, Pitts L, Eick D, Murphy S. 2007. Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science 318:1777–1779. 10.1126/science.1145989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bataille AR, Jeronimo C, Jacques PE, Laramee L, Fortin ME, Forest A, Bergeron M, Hanes SD, Robert F. 2012. A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol. Cell 45:158–170. 10.1016/j.molcel.2011.11.024 [DOI] [PubMed] [Google Scholar]
- 12.Souza W. 2009. Structural organization of Trypanosoma cruzi. Mem. Inst. Oswaldo Cruz 104(Suppl 1):S89–S100. 10.1590/S0074-02762009000900014 [DOI] [PubMed] [Google Scholar]
- 13.Coura JR, Vinas PA. 2010. Chagas disease: a new worldwide challenge. Nature 465:S6–S7. 10.1038/nature09221 [DOI] [PubMed] [Google Scholar]
- 14.Johnson PJ, Kooter JM, Borst P. 1987. Inactivation of transcription by UV irradiation of T. brucei provides evidence for a multicistronic transcription unit including a VSG gene. Cell 51:273–281. 10.1016/0092-8674(87)90154-1 [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Calvillo S, Yan S, Nguyen D, Fox M, Stuart K, Myler PJ. 2003. Transcription of Leishmania major Friedlin chromosome 1 initiates in both directions within a single region. Mol. Cell 11:1291–1299. 10.1016/S1097-2765(03)00143-6 [DOI] [PubMed] [Google Scholar]
- 16.Campbell DA, Thomas S, Sturm NR. 2003. Transcription in kinetoplastid protozoa: why be normal? Microbes Infect. 5:1231–1240. 10.1016/j.micinf.2003.09.005 [DOI] [PubMed] [Google Scholar]
- 17.Clayton CE. 2002. Life without transcriptional control? From fly to man and back again. EMBO J. 21:1881–1888. 10.1093/emboj/21.8.1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parsons M, Nelson RG, Watkins KP, Agabian N. 1984. Trypanosome mRNAs share a common 5′ spliced leader sequence. Cell 38:309–316. 10.1016/0092-8674(84)90552-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nunes LR, Carvalho MR, Shakarian AM, Buck GA. 1997. The transcription promoter of the spliced leader gene from Trypanosoma cruzi. Gene 188:157–168. 10.1016/S0378-1119(96)00726-3 [DOI] [PubMed] [Google Scholar]
- 20.Gilinger G, Bellofatto V. 2001. Trypanosome spliced leader RNA genes contain the first identified RNA polymerase II gene promoter in these organisms. Nucleic Acids Res. 29:1556–1564. 10.1093/nar/29.7.1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dossin FM, Schenkman S. 2005. Actively transcribing RNA polymerase II concentrates on spliced leader genes in the nucleus of Trypanosoma cruzi. Eukaryot. Cell 4:960–970. 10.1128/EC.4.5.960-970.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clayton C, Shapira M. 2007. Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol. Biochem. Parasitol. 156:93–101. 10.1016/j.molbiopara.2007.07.007 [DOI] [PubMed] [Google Scholar]
- 23.Moretti NS, Schenkman S. 2013. Chromatin modifications in trypanosomes due to stress. Cell. Microbiol. 15:709–717. 10.1111/cmi.12111 [DOI] [PubMed] [Google Scholar]
- 24.Ruan JP, Arhin GK, Ullu E, Tschudi C. 2004. Functional characterization of a Trypanosoma brucei TATA-binding protein-related factor points to a universal regulator of transcription in trypanosomes. Mol. Cell. Biol. 24:9610–9618. 10.1128/MCB.24.21.9610-9618.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapman AB, Agabian N. 1994. Trypanosoma brucei RNA polymerase II is phosphorylated in the absence of carboxyl-terminal domain heptapeptide repeats. J. Biol. Chem. 269:4754–4760 [PubMed] [Google Scholar]
- 26.Das A, Bellofatto V. 2009. The non-canonical CTD of RNAP-II is essential for productive RNA synthesis in Trypanosoma brucei. PLoS One 4:e6959. 10.1371/journal.pone.0006959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferreira LRP, Ramos FM, Ramos TC, Freymuller E, Schenkman S. 2008. Active transcription and ultrastructural changes during Trypanosoma cruzi metacyclogenesis. An. Acad. Bras. Cienc. 80:157–166. 10.1590/S0001-37652008000100011 [DOI] [PubMed] [Google Scholar]
- 28.Schwartz LB, Roeder RG. 1975. Purification and subunit structure of deoxyribonucleic acid-dependent ribonucleic acid polymerase II from the mouse plasmacytoma, MOPC 315. J. Biol. Chem. 250:3221–3228 [PubMed] [Google Scholar]
- 29.Kim WY, Dahmus ME. 1986. Immunochemical analysis of mammalian RNA polymerase II subspecies. Stability and relative in vivo concentration. J. Biol. Chem. 261:14219–14225 [PubMed] [Google Scholar]
- 30.Chesnut JD, Stephens JH, Dahmus ME. 1992. The interaction of RNA polymerase II with the adenovirus-2 major late promoter is precluded by phosphorylation of the C-terminal domain of subunit IIa. J. Biol. Chem. 267:10500–10506 [PubMed] [Google Scholar]
- 31.Laybourn PJ, Dahmus ME. 1989. Transcription-dependent structural changes in the C-terminal domain of mammalian RNA polymerase subunit IIa/o. J. Biol. Chem. 264:6693–6698 [PubMed] [Google Scholar]
- 32.Lu H, Flores O, Weinmann R, Reinberg D. 1991. The nonphosphorylated form of RNA polymerase II preferentially associates with the preinitiation complex. Proc. Natl. Acad. Sci. U. S. A. 88:10004–10008. 10.1073/pnas.88.22.10004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Contreras VT, Araujo-Jorge TC, Bonaldo MC, Thomaz N, Barbosa HS, Meirelles MNSL, Goldenberg S. 1988. Biological aspects of the Dm 28c clone of Trypanosoma cruzi after metacyclogenesis in chemically defined media. Mem. Inst. Oswaldo Cruz 83:123–133. 10.1590/S0074-02761988000100016 [DOI] [PubMed] [Google Scholar]
- 34.Chung J, Rocha AA, Tonelli RR, Castilho BA, Schenkman S. 2013. Eukaryotic initiation factor 5A dephosphorylation is required for translational arrest in stationary phase cells. Biochem. J. 451:257–267. 10.1042/BJ20121553 [DOI] [PubMed] [Google Scholar]
- 35.Nardelli SC, da Cunha JP, Motta MC, Schenkman S. 2009. Distinct acetylation of Trypanosoma cruzi histone H4 during cell cycle, parasite differentiation, and after DNA damage. Chromosoma 118:487–499. 10.1007/s00412-009-0213-9 [DOI] [PubMed] [Google Scholar]
- 36.Santos JH, Meyer JN, Mandavilli BS, Van Houten B. 2006. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 314:183–199. 10.1385/1-59259-973-7:183 [DOI] [PubMed] [Google Scholar]
- 37.Cabrera G, Barria C, Fernandez C, Sepulveda S, Valenzuela L, Kemmerling U, Galanti N. 2011. DNA repair BER pathway inhibition increases cell death caused by oxidative DNA damage in Trypanosoma cruzi. J. Cell. Biochem. 112:2189–2199. 10.1002/jcb.23138 [DOI] [PubMed] [Google Scholar]
- 38.Ramirez M, Yamauchi L, de Freitas L, Uemura H, Schenkman S. 2000. The use of the green fluorescent protein to monitor and improve transfection in Trypanosoma cruzi. Mol. Biochem. Parasitol. 111:235–240. 10.1016/S0166-6851(00)00309-1 [DOI] [PubMed] [Google Scholar]
- 39.Bensaude O. 2011. Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity? Transcription 2:103–108. 10.4161/trns.2.3.16172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teves SS, Henikoff S. 2011. Heat shock reduces stalled RNA polymerase II and nucleosome turnover genome-wide. Genes Dev. 25:2387–2397. 10.1101/gad.177675.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dubois MF, Marshall NF, Nguyen VT, Dahmus GK, Bonnet F, Dahmus ME, Bensaude O. 1999. Heat shock of HeLa cells inactivates a nuclear protein phosphatase specific for dephosphorylation of the C-terminal domain of RNA polymerase II. Nucleic Acids Res. 27:1338–1344. 10.1093/nar/27.5.1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dubois MF, Vincent M, Vigneron M, Adamczewski J, Egly JM, Bensaude O. 1997. Heat-shock inactivation of the TFIIH-associated kinase and change in the phosphorylation sites on the C-terminal domain of RNA polymerase II. Nucleic Acids Res. 25:694–700. 10.1093/nar/25.4.694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hieda M, Winstanley H, Maini P, Iborra FJ, Cook PR. 2005. Different populations of RNA polymerase II in living mammalian cells. Chromosome Res. 13:135–144. 10.1007/s10577-005-7720-1 [DOI] [PubMed] [Google Scholar]
- 44.Muhich ML, Hsu MP, Boothroyd JC. 1989. Heat-shock disruption of trans-splicing in trypanosomes: effect on Hsp70, Hsp85 and tubulin mRNA synthesis. Gene 82:169–175. 10.1016/0378-1119(89)90042-5 [DOI] [PubMed] [Google Scholar]
- 45.Nazer E, Verdun RE, Sanchez DO. 2012. Severe heat shock induces nucleolar accumulation of mRNAs in Trypanosoma cruzi. PLoS One 7:e43715. 10.1371/journal.pone.0043715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nazer E, Verdun RE, Sanchez DO. 2011. Nucleolar localization of RNA binding proteins induced by actinomycin D and heat shock in Trypanosoma cruzi. PLoS One 6:e19920. 10.1371/journal.pone.0019920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heo JH, Jeong SJ, Seol JW, Kim HJ, Han JW, Lee HW, Cho EJ. 2004. Differential regulation of gene expression by RNA polymerase II in response to DNA damage. Biochem. Biophys. Res. Commun. 325:892–898. 10.1016/j.bbrc.2004.10.101 [DOI] [PubMed] [Google Scholar]
- 48.Jeong SJ, Kim HJ, Yang YJ, Seol JH, Jung BY, Han JW, Lee HW, Cho EJ. 2005. Role of RNA polymerase II carboxy terminal domain phosphorylation in DNA damage response. J. Microbiol. 43:516–522 http://www.msk.or.kr/jsp/view_old_journalD.jsp?paperSeq=2296 [PubMed] [Google Scholar]
- 49.Wong JM, Ingles CJ. 2001. A compromised yeast RNA polymerase II enhances UV sensitivity in the absence of global genome nucleotide excision repair. Mol. Gen. Genet. 264:842–851. 10.1007/s004380000374 [DOI] [PubMed] [Google Scholar]
- 50.Winsor TS, Bartkowiak B, Bennett CB, Greenleaf AL. 2013. A DNA damage response system associated with the phosphoCTD of elongating RNA polymerase II. PLoS One 8:e60909. 10.1371/journal.pone.0060909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JH, Jung HS, Gunzl A. 2009. Transcriptionally active TFIIH of the early-diverged eukaryote Trypanosoma brucei harbors two novel core subunits but not a cyclin-activating kinase complex. Nucleic Acids Res. 37:3811–3820. 10.1093/nar/gkp236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Badjatia N, Ambrosio DL, Lee JH, Gunzl A. 2013. Trypanosome cdc2-related kinase 9 controls spliced leader RNA cap4 methylation and phosphorylation of RNA polymerase II subunit RPB1. Mol. Cell. Biol. 33:1965–1975. 10.1128/MCB.00156-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meinhart A, Kamenski T, Hoeppner S, Baumli S, Cramer P. 2005. A structural perspective of CTD function. Genes Dev. 19:1401–1415. 10.1101/gad.1318105 [DOI] [PubMed] [Google Scholar]
- 54.Krishnamurthy S, He X, Reyes-Reyes M, Moore C, Hampsey M. 2004. Ssu72 is an RNA polymerase II CTD phosphatase. Mol. Cell 14:387–394. 10.1016/S1097-2765(04)00235-7 [DOI] [PubMed] [Google Scholar]
- 55.Reyes-Reyes M, Hampsey M. 2007. Role for the Ssu72 C-terminal domain phosphatase in RNA polymerase II transcription elongation. Mol. Cell. Biol. 27:926–936. 10.1128/MCB.01361-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hausmann S, Shuman S. 2002. Characterization of the CTD phosphatase Fcp1 from fission yeast. Preferential dephosphorylation of serine 2 versus serine 5. J. Biol. Chem. 277:21213–21220. 10.1074/jbc.M202056200 [DOI] [PubMed] [Google Scholar]
- 57.Chambers RS, Dahmus ME. 1994. Purification and characterization of a phosphatase from HeLa cells which dephosphorylates the C-terminal domain of RNA polymerase II. J. Biol. Chem. 269:26243–26248 [PubMed] [Google Scholar]
- 58.Szoor B. 2010. Trypanosomatid protein phosphatases. Mol. Biochem. Parasitol. 173:53–63. 10.1016/j.molbiopara.2010.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mosley AL, Pattenden SG, Carey M, Venkatesh S, Gilmore JM, Florens L, Workman JL, Washburn MP. 2009. Rtr1 is a CTD phosphatase that regulates RNA polymerase II during the transition from serine 5 to serine 2 phosphorylation. Mol. Cell 34:168–178. 10.1016/j.molcel.2009.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dubois MF, Bellier S, Seo SJ, Bensaude O. 1994. Phosphorylation of the RNA polymerase II largest subunit during heat shock and inhibition of transcription in HeLa cells. J. Cell. Physiol. 158:417–426. 10.1002/jcp.1041580305 [DOI] [PubMed] [Google Scholar]
- 61.Dubois MF, Nguyen VT, Bellier S, Bensaude O. 1994. Inhibitors of transcription such as 5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole and isoquinoline sulfonamide derivatives (H-8 and H-7) promote dephosphorylation of the carboxyl-terminal domain of RNA polymerase II largest subunit. J. Biol. Chem. 269:13331–13336 [PubMed] [Google Scholar]
- 62.Augustine SAJ, Kleshchenko YY, Nde PN, Pratap S, Ager EA, Burns JM, Jr, Lima MF, Villalta F. 2006. Molecular cloning of a Trypanosoma cruzi cell surface casein kinase II substrate, Tc-1, involved in cellular infection. Infect. Immun. 74:3922–3929. 10.1128/IAI.00045-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee MGS. 1995. Heat shock does not increase the transcriptional efficiency of the Hsp 70 genes of Trypanosoma brucei. Exp. Parasitol. 81:608–613. 10.1006/expr.1995.1156 [DOI] [PubMed] [Google Scholar]
- 64.Kramer S, Queiroz R, Ellis L, Webb H, Hoheisel JD, Clayton C, Carrington M. 2008. Heat shock causes a decrease in polysomes and the appearance of stress granules in trypanosomes independently of eIF2α phosphorylation at Thr169. J. Cell Sci. 121:3002–3014. 10.1242/jcs.031823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ni Z, Schwartz BE, Werner J, Suarez JR, Lis JT. 2004. Coordination of transcription, RNA processing, and surveillance by P-TEFb kinase on heat shock genes. Mol. Cell 13:55–65. 10.1016/S1097-2765(03)00526-4 [DOI] [PubMed] [Google Scholar]
- 66.Tang P, Juang CL, Harbison GS. 1990. Intercalation complex of proflavine with DNA: structure and dynamics by solid-state NMR. Science 249:70–72. 10.1126/science.2367853 [DOI] [PubMed] [Google Scholar]
- 67.Aslanoglu M. 2006. Electrochemical and spectroscopic studies of the interaction of proflavine with DNA. Anal. Sci. 22:439–443. 10.2116/analsci.22.439 [DOI] [PubMed] [Google Scholar]
- 68.Passos-Silva DG, Rajao MA, Nascimento de Aguiar PH, Vieira-da-Rocha JP, Machado CR, Furtado C. 2010. Overview of DNA repair in Trypanosoma cruzi, Trypanosoma brucei, and Leishmania major. J. Nucleic Acids 2010:840768. 10.4061/2010/840768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bregman DB, Halaban R, van Gool AJ, Henning KA, Friedberg EC, Warren SL. 1996. UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. U. S. A. 93:11586–11590. 10.1073/pnas.93.21.11586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luo Z, Zheng J, Lu Y, Bregman DB. 2001. Ultraviolet radiation alters the phosphorylation of RNA polymerase II large subunit and accelerates its proteasome-dependent degradation. Mutat. Res. 486:259–274. 10.1016/S0921-8777(01)00097-0 [DOI] [PubMed] [Google Scholar]
- 71.Somesh BP, Reid J, Liu WF, Sogaard TM, Erdjument-Bromage H, Tempst P, Svejstrup JQ. 2005. Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell 121:913–923. 10.1016/j.cell.2005.04.010 [DOI] [PubMed] [Google Scholar]
- 72.Cho H, Kim TK, Mancebo H, Lane WS, Flores O, Reinberg D. 1999. A protein phosphatase functions to recycle RNA polymerase II. Genes Dev. 13:1540–1552. 10.1101/gad.13.12.1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ranuncolo SM, Ghosh S, Hanover JA, Hart GW, Lewis BA. 2012. Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo. J. Biol. Chem. 287:23549–23561. 10.1074/jbc.M111.330910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Compe E, Egly J-M. 2012. TFIIH: when transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 13:343–354. 10.1038/nrm3350 [DOI] [PubMed] [Google Scholar]
- 75.Badjatia N, Nguyen TN, Lee JH, Günzl A. 2013. Trypanosoma brucei harbors a divergent XPB helicase paralog that is specialized in nucleotide excision repair and conserved among kinetoplastid organisms. Mol. Microbiol. 90:1293–1308. 10.1111/mmi.12435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nett IRE, Martin DMA, Miranda-Saavedra D, Lamont D, Barber JD, Mehlert A, Ferguson MAJ. 2009. The phosphoproteome of bloodstream form Trypanosoma brucei, causative agent of African sleeping sickness. Mol. Cell. Proteomics 8:1527–1538. 10.1074/mcp.M800556-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Godoy LMF, Marchini FK, Pavoni DP, Rampazzo RCP, Probst CM, Goldenberg S, Krieger MA. 2012. Quantitative proteomics of Trypanosoma cruzi during metacyclogenesis. Proteomics 12:2694–2703. 10.1002/pmic.201200078 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.