

Abstract
Two types of enzyme for oxidative cleavage of poly(cis-1,4-isoprene) are known. One is rubber oxygenase (RoxA) that is secreted by Xanthomonas sp. strain 35Y and a few other Gram-negative rubber-degrading bacteria during growth on polyisoprene. RoxA was studied in the past, and the recently solved structure showed a structural relationship to bacterial cytochrome c peroxidases (J. Seidel et al., Proc. Natl. Acad. Sci. U. S. A. 110:13833–13838, 2013, http://dx.doi.org/10.1073/pnas.1305560110). The other enzyme is latex-clearing protein (Lcp) that is secreted by rubber-degrading actinomycetes, but Lcp has not yet been purified. Here, we expressed Lcp of Streptomyces sp. strain K30 in a ΔroxA background of Xanthomonas sp. strain 35Y and purified native (untagged) Lcp. The specific activities of Lcp and RoxA were 0.70 and 0.48 U/mg, respectively. Lcp differed from RoxA in the absence of heme groups and other characteristics. Notably, Lcp degraded polyisoprene via endo-type cleavage to tetra-C20 and higher oligo-isoprenoids with aldehyde and keto end groups, whereas RoxA used an exo-type cleavage mechanism to give the main end product 12-oxo-4,8-dimethyltrideca-4,8-diene-1-al (ODTD). RoxA was able to cleave isolated Lcp-derived oligo-isoprenoid molecules to ODTD. Inhibitor studies, spectroscopic investigations and metal analysis gave no indication for the presence of iron, other metals, or cofactors in Lcp. Our results suggest that Lcp could be a member of the growing group of cofactor-independent oxygenases and differs in the cleavage mechanism from heme-dependent RoxA. In conclusion, RoxA and Lcp represent two different answers to the same biochemical problem, the cleavage of polyisoprene, a polymer that has carbon-carbon double bonds as the only functional groups for enzymatic attack.
INTRODUCTION
A large fraction of the organic biomass on earth is composed of biopolymers such as polysaccharides, polyamino acids (proteins), polyconiferylalcohols (lignins), polyhydroxyalkanoic acids (PHAs), and polyisoprenes (rubbers). Biopolymers are degraded by chemical, physical, and/or biochemical reactions. These mineralization processes are essential to keep the global carbon cycle running. Here, microorganisms fulfill a major role, and the collectivity of all microorganisms is able to degrade any naturally produced biopolymer. The first step in the degradation of biopolymers consists of the breakdown of the polymer to low-molecular-weight products and is often catalyzed by hydrolytic enzymes such as cellulases, proteases, lipases, polyesterases, and related hydrolases. However, polyisoprene, which is naturally produced by many plants (e.g., the rubber tree Hevea brasiliensis [natural rubber]) is a pure hydrocarbon, and therefore polyisoprene cannot be attacked by hydrolytic reactions. Instead, oxidative reactions are necessary for the primary attack of rubber. The fact that natural rubber can be biodegraded and used as a carbon and energy source by microorganisms has been known for more than 100 years, but it was only a decade ago that the first polyisoprene-specific rubber-degrading enzyme (rubber oxygenase [RoxA]) was isolated (1). RoxA is an extracellular dioxygenase secreted by Xanthomonas sp. strain 35Y during growth on poly(cis-1,4-isoprene) and cleaves rubber to one major product, 12-oxo-4,8-dimethyltrideca-4,8-diene-1-al (ODTD; C15 tri-isoprenoid). Mature RoxA is a 73-kDa protein with two heme centers that are covalently attached to RoxA via two heme-binding motifs (2). Electron paramagnetic resonance (EPR) measurements and analysis of the three-dimensional structure revealed that the N-terminal heme represents the active site and has a stably bound dioxygen molecule as an axial ligand (3–5). Recently, we identified functional RoxA orthologs in several soil and marine myxobacteria (6); in addition, Imai et al. isolated Rhizobacter gummiphilus NS21 as a novel Gram-negative rubber-degrading species (7, 8). However, the total number of known Gram-negative rubber-degrading species is still rather low. In contrast to this, the ability to degrade rubber is widespread among Gram-positive organisms, in particular among actinomycetes. Remarkably, RoxA orthologs were not found in any of the rubber-degrading Gram-positive organisms as far as the genomes of the species have been sequenced. Instead, multiple evidence exists that another protein, named the latex-clearing protein (Lcp), is responsible for the primary attack on polyisoprene by actinomycetes. Lcp was first described for Streptomyces sp. strain K30 (9), but orthologs of Lcp can be found in Gordonia westfalica and Gordonia polyisoprenivorans (10) and apparently are present in all rubber-degrading Gram-positive species that have been examined thus far (for reviews, see references 11 and 12).
Lcps are not related to RoxAs. For instance, Lcps (∼40 kDa) and RoxAs (∼73 kDa) differ in molecular mass, and Lcp sequences do not have a binding motif for covalent attachment of heme to the protein or other motifs that could be indicative for the binding of metal ions such as a cupin fold (13). This indicates that Lcps could be members of the family of cofactor-independent oxygenases (14, 15). Unfortunately, Lcp—similarly to RoxA—is difficult to express in quantities sufficient for protein purification in wild-type strains, and the purification of Lcp in active form has not yet been described. In the present study, we expressed functional Lcp by chromosomal anchoring of lcp in a Xanthomonas ΔroxA background. We developed a purification procedure for native Lcp and determined the biochemical properties of this novel type of extracellular rubber oxygenase. Comparison of the properties of RoxA and Lcp showed that both enzymes oxidatively cleaved rubber by different mechanisms.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Table 1 shows the strains and plasmids used in the present study. Xanthomonas sp. 35Y and mutant strains derived from the wild type were grown in lysogeny broth (LB) or in modified LB with a reduced concentration of yeast extract (10 g of tryptone, 5 g of NaCl, and 0.25 g of yeast extract/liter). To demonstrate clearing zones on opaque latex agar, Xanthomonas strains were grown on Tsuchii's mineral salts medium with 0.25% (vol/vol) purified rubber latex and 1.5% (wt/vol) agar at 30°C for 1 to 3 weeks (16). Polyisoprene latex (0.25% [wt/vol]) was kindly provided by Weber and Schaer (Hamburg, Germany) and was used after three washing steps in 0.1% (wt/vol) Nonidet P-40. For the purification of recombinant Lcp, the Xanthomonas ΔroxA-attB strain with the chromosomally integrated pNH1::lcp plasmid was grown in modified LB medium (12 individual 1-liter cultures, each in a 3-liter Erlenmeyer flask) supplemented with 0.1% (wt/vol) l-rhamnose for 3 to 4 days at room temperature by continuous shaking. Cells were harvested (4°C) by centrifugation, and Lcp was purified from cell-free culture fluid as described below.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| JM109 | Plasmid storage | |
| XL1-Blue | QuikChange transformation strain | Stratagene |
| Xanthomonas sp. | ||
| 35Y | Growth on poly(cis-1,4-isoprene) latex, clearing zone formation | 16 |
| 35Y-CM | Cmr mutant of 35Y | 18 |
| 35Y ΔroxA-attB (SN3727) | Chromosomal deletion of roxA in Xanthomonas sp. 35Y-CM, attB at former roxA site; no clearing zone formation on latex agar | 4 |
| 35Y ΔroxA-attB pNH1-roxA-attP in chromosome (SN4230) | Expression of RoxA from rhamnose promoter in Xanthomonas sp. 35Y-CM; Kmr Cmr; clearing zone formation in the presence of rhamnose plus latex | 4 |
| 35Y ΔroxA-attB pNH1-lcp-attP in chromosome (SN5343) | Expression of Lcp from rhamnose promoter in Xanthomonas sp. 35Y-CM; Kmr Cmr; positive fuchsin staining on latex overlay agar | This study |
| Plasmids | ||
| pMK::lcp (SN5314) | GeneArt vector, source of lcp; Kmr | This study |
| pUC9::lcp (SN5339) | Cloning vector for lcp; Apr | This study |
| pNH1-roxA-attP (SN4230) | Coding sequence of roxA under rhamnose promoter control, attP site; Kmr | 4 |
| pNH1-lcp-attP (SN5341) | Coding sequence of lcp under rhamnose promoter control, attP site; Kmr | This study |
Cmr, chloramphenicol resistance; Apr, ampicillin resistance; Kmr, kanamycin resistance.
Cloning of the Streptomyces sp. K30 lcp gene and expression of Lcp in Xanthomonas sp.
A DNA sequence that coded for the complete 407 amino acids of Lcp (including the Tat signal peptide) of Streptomyces sp. K30 (GenBank accession number AY387589) (17) was synthesized by GeneArt (Darmstadt, Germany). The codon usage was adapted to that of Xanthomonas sp. 35Y to enhance the probability of a high expression level in Xanthomonas sp. 35Y. The lcp gene was cloned via single NdeI und SacI sites into the pNH1 vector (Table 1), yielding pNH1::lcp, and transformed into E. coli S17-1. The pNH1::lcp construct was conjugatively transferred to the Xanthomonas ΔroxA-attB strain as described previously for pNH1::roxA (4, 18). Integration of lcp into the chromosome at the former roxA site via Phi31 integrase-mediated recombination was verified by PCR and DNA sequencing. Figure 1 shows the map and relevant characteristics of the pNH1::lcp plasmid.
FIG 1.
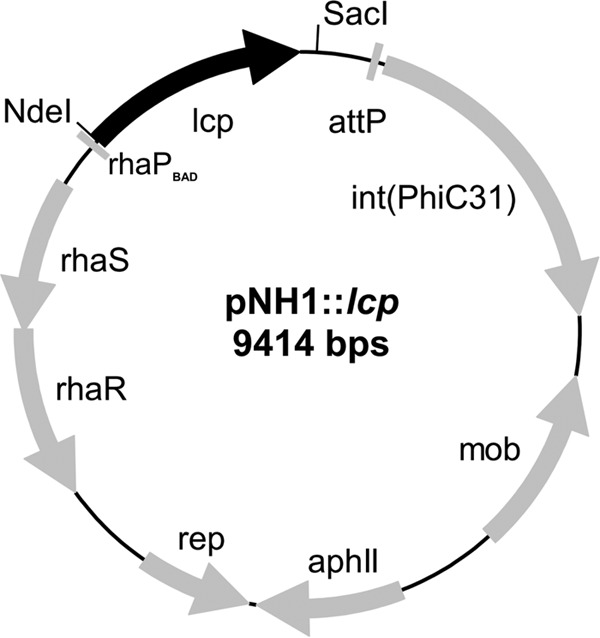
Map of the pNH1::lcp plasmid. The genes responsible for replication (rep), conjugative transfer (mob), chromosomal integration via attB/attP recombination [int(PhiC31)], l-rhamnose control (rhaS, rhaR), the selective marker (aphII), the rhamnose promoter (rhaPBAD) and latex clearing protein (lcp) are indicated.
Purification of recombinant RoxA and Lcp.
RoxA was purified from the Xanthomonas ΔroxA strain with chromosomally integrated roxA plasmid (pNH1::roxA) under rhamnose control as described previously (3, 4). Lcp was purified at room temperature using an ÄKTA fast-performance liquid chromatographic system (GE Healthcare, Uppsala, Sweden). In brief, the combined cell-free supernatants of 12 parallel 1-liter cultures of the Xanthomonas ΔroxA strain with integrated pNH1::lcp plasmid were concentrated and buffer exchanged against 20 mM Tris-HCl (pH 8) via cross-flow ultrafiltration (Amersham KVICKLAB cartridge, 10-kDa cutoff). The concentrate was then applied to a Q-Sepharose Fast Flow column (Q-FF 50/11, 210-ml bed volume) that had been equilibrated with 20 mM Tris-HCl (pH 8) at a flow rate of 8 ml/min. Lcp was bound to the column and—after a washing step with 1 volume of equilibration buffer—eluted by applying a step NaCl gradient (0 to 120 mM) in equilibration buffer. Lcp-containing fractions were identified by activity staining of selected fractions with Schiff reagent (see below) and by SDS-PAGE, combined and concentrated via ultrafiltration. A second chromatographic step was performed on a hydroxyapatite column. To this end, the Q-Sepharose pool was rebuffered by size-exclusion chromatography on a HiPrep Sephadex G25 26/10 column (bed volume, 53 ml) with potassium phosphate buffer (1 mM, pH 6.0) and subsequently loaded on a hydroxyapatite column (CHT5-I; bed volume, 20 ml) that had been equilibrated with the same buffer. Lcp was eluted with a linear gradient of 1 to 200 mM potassium phosphate (pH 6.0) at ∼75 mM. Lcp fractions were pooled, concentrated via ultrafiltration (10-kDa cutoff), and stored on ice for up to 2 weeks or frozen (−70°C) for long-term storage.
Assay of Lcp.
Lcp activity was assayed in several ways. (i) For rapid estimation of Lcp activity in fractions after a chromatographic step, 25 μl of the respective fraction was incubated in 0.25 ml of assay buffer (100 mM potassium phosphate buffer [pH 7]) containing 0.2% polyisoprene latex at room temperature for 1 h. The presence of polyisoprene cleavage products was semiquantitatively indicated by the intensity of the developed purple color after the addition of 25 μl of a fuchsin solution (2 g of fuchsin dissolved in 50 ml of glacial acetic acid, 10 g of Na2S2O5, 100 ml of 0.1 N HCl, and 50 ml of H2O) to the assay mixture, as described previously by Yikmis and Steinbüchel (19). (ii) Lcp activity was also determined by high-pressure liquid chromatography (HPLC) detection of cleavage products. The reaction mixture contained 100 μl of purified Lcp (1 to 5 μg/ml), rubber latex (0.2% [wt/vol] emulsion), and potassium phosphate buffer (100 mM; pH 7.0) in a total volume of 1 ml. The reaction was performed at room temperature, at 30°C, or at 37°C (as indicated) for 2 h in a 15-ml Falcon tube. The mixture was extracted with ethyl acetate (1.5 ml); 1 ml of the mixture was dried, dissolved in 100 μl of methanol, and then subjected to HPLC analysis. An RP8 HPLC column (12 by 4 mm, 5-μm particle size) was operated at 0.7 ml/min with water (A) and methanol (B) as mobile phases. The concentration of B was increased from 50% (vol/vol) to 100% (vol/vol) within 15 min; products were detected at 210 nm. Purified ODTD was used as a standard and eluted after 15.3 min. Lcp-derived cleavage products eluted after ODTD (18.8 min and later). Mixtures without Lcp or with heat-inactivated Lcp (10 min 95°C) served as controls. (iii) Lcp activity was also determined by monitoring the consumption of molecular oxygen in a YSI model 5300 biological oxygen monitor (Scientific Division, Yellow Springs Instrument Co., Inc., Yellow Springs, OH) equipped with an oxygen electrode. (iv) Lcp activity in vivo was also demonstrated by the cultivation of recombinant Xanthomonas ΔroxA::attB strains harboring chromosomally anchored lcp under rhamnose control on mineral salts agar plates with an opaque overlay agar (∼8 ml) of polyisoprene latex (0.25% [wt/vol] in agar) and 0.1% (wt/vol) l-rhamnose prepared as described previously (6). Lcp-expressing strains formed (after a prolonged incubation period at room temperature) a small zone around arising colonies that could be stained with a solution of Schiff's reagent (for the composition, see above) and indicated the formation of keto-/aldehyde group-containing cleavage products. The colonies of a Xanthomonas ΔroxA clone (negative control) did not form any halos. Purified RoxA was assayed as described previously (3, 4) or by the methods described for Lcp above.
HPLC-mass spectrometry (MS).
Lcp-catalyzed polyisoprene cleavage products were separated on an RP8 HPLC column (12 by 4 mm; 5-μm particle size; flow rate, 0.5 ml/min) that was directly coupled with an Agilent 6130 series quadrupole LC/MS system (buffer A, water containing 0.1% [vol/vol] formic acid; buffer B, methanol, HPLC grade; stating condition 50% B, gradient to 100% B within 21 min).
Inhibitor studies and stability assays. (i) Inhibitor assays.
To examine the effect of potential inhibitors on Lcp activity, 1 mM concentrations of each test compound (unless stated otherwise) were added to the activity assay described for assay ii in the “Assay of Lcp” section above before the reaction was started by the addition of Lcp.
(ii) Stability assays.
RoxA or Lcp were diluted in potassium phosphate buffer (100 mM, pH 7) to a final concentration of 0.05 mg/ml. The respective enzyme solutions were incubated at room temperature or at 37°C for up to 22 h. After the indicated time intervals, 100-μl portions of RoxA or Lcp were used for activity assay as described for assay ii in the “Assay of Lcp” section above. The activity assay mixture was incubated for 2 h at room temperature, at 30°C, or at 37°C. For each time interval, the activity of the enzymes was determined by integration of the corresponding peak after HPLC analysis (RoxA, ODTD peak at 15.3 min; Lcp, C35 oligo-isoprenoid peak at 22.9 min).
Other techniques.
The protein concentration was determined by the bicinchoninic acid (BCA) method using a BCA kit. The concentrations of purified Lcp samples were also determined from the molar extinction coefficients of Lcp at 280 nm (ε280 = 68,000 M−1 cm−1). Heme staining for pseudoperoxidase activity was conducted as described previously (3) after the separation of proteins via nonreducing SDS-PAGE. The metal content of purified Lcp was determined using inductively coupled plasma-MS (ICP-MS) by the Spuren-Analytisches Laboratorium Dr. Baumann (Germany).
RESULTS
Construction of a Xanthomonas sp. strain for stable expression of recombinant Lcp from Streptomyces sp. K30.
All attempts to express Xanthomonas RoxA in substantial amounts in recombinant Escherichia coli even in the presence of cytochrome maturation genes or in other heterologous hosts were unsuccessful (for details, see our previous report [4]). We assumed that the synthesis of functional RoxA requires the presence of some kind of maturation system that is absent or is not correctly working in E. coli. Therefore, expression of RoxA was performed by homologous expression in a ΔroxA background. A polyisoprene-independent induction of RoxA expression was achieved by anchoring the roxA gene into the chromosome under the control of a l-rhamnose-inducible promoter. This enabled us to shorten the cultivation time of the expression strain from ∼2 weeks in the presence of natural rubber latex to 3 to 4 days in the presence of l-rhamnose.
Substantial expression of active Lcp from Streptomyces sp. K30 in heterologous hosts such as E. coli or Pseudomonas putida strains was also not possible, a finding similar to our experience with RoxA. Only in species of the same genus, i.e., Streptomyces lividans and Streptomyces erythraea, was the expression of Lcp successful (12). However, the amount of produced Lcp was low and apparently not suited for purifying Lcp in sufficient quantities. Since our Xanthomonas ΔroxA(pNH1::roxA) strains produced 2 to 5 mg of RoxA per liter of culture fluid, as well as for RoxA orthologs from myxobacteria, we speculated that Xanthomonas sp. 35Y could be a suitable host for the heterologous expression of Lcp. To this end, we cloned a synthetic DNA sequence that coded for the entire amino acid sequence of Lcp from Streptomyces sp. K30, including the signal sequence for TAT-dependent secretion (17), into our pNH1 plasmid. The resulting pNH1::lcp plasmid was chromosomally anchored in a Xanthomonas ΔroxA background via PhiC31 integrase-mediated attB/attP recombination at the site of the former roxA locus (for further details, see Materials and Methods and also reference 4). When the recombinant Xanthomonas ΔroxA(pNH1::lcp) strain was grown on opaque latex overlay agar that had been supplemented with 0.1% l-rhamnose, the arising colonies produced small clearing zones and could be positively stained with fuchsin solution (Fig. 2A). The negative control (i.e., the Xanthomonas ΔroxA strain without pNH1::lcp) produced no clearing zone and could not be stained with fuchsin. We concluded that the Xanthomonas ΔroxA(pNH1::lcp) strain expressed and secreted functional Lcp protein. In agreement with this, a new protein band appeared after SDS-PAGE of cell-free supernatants at an apparent molecular mass of ∼40 kDa, and this value corresponded to the theoretical value for mature Lcp (41 kDa). Moreover, rubber-cleaving activity was detected in the cell-free culture fluid using the fuchsin assay or the HPLC method, whereas no activity was found in culture fluids of control cells (data not shown).
FIG 2.
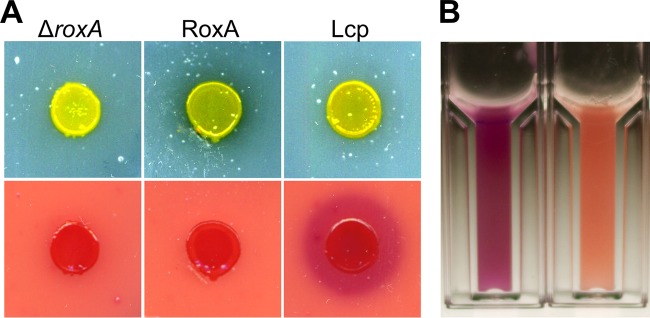
Growth of Xanthomonas sp. 35Y on latex agar and staining of degradation products by Schiff reagent. (A) ΔroxA Xanthomonas sp. (left), ΔroxA Xanthomonas harboring pNH1::roxA (middle), and ΔroxA Xanthomonas harboring pNH1::lcp (right) were grown on solid latex plates (top row) for 3 weeks at room temperature and subsequently stained with Schiff reagent (bottom row). There was no fuchsin staining for RoxA (bottom middle) because of uptake and the use of degradation product ODTD. Note the clearing zone for RoxA (top middle) and stained halo for Lcp (bottom right), presumably because of insufficient uptake of Lcp-derived degradation products that are considerably larger than ODTD. (B) Activity assay for purified Lcp after staining with Schiff reagent (cuvette on the left with Lcp, cuvette on the right without Lcp).
Development of a purification procedure for Lcp.
Preliminary experiments showed that Lcp is able to bind to ion exchange materials Q-Sepharose and hydroxyapatite and to elute from the column matrices by raising the concentrations of NaCl or potassium phosphate, respectively. Based on these results, a three-step purification protocol was developed as described in Materials and Methods and was applied to purify Lcp from combined cell-free culture fluids (the total culture volume was 12 liters) of a Xanthomonas ΔroxA(pNH1::lcp) culture on modified LB-rhamnose medium. Due to the low expression of Lcp at 30°C (probably due to the instability of Lcp at this temperature), the cells were grown at room temperature. The hydroxyapatite step turned out to be very efficient, and a sharp Lcp-containing peak was obtained at ∼75 mM potassium phosphate (buffer pH, 6) (Fig. 3A). In total, 1.5 mg of Lcp was obtained after the last purification step. SDS-PAGE of the final Lcp fraction indicated that Lcp was almost homogeneous, and only a few minor contaminating bands were detected after application of the sensitive silver-staining method (Fig. 3B). RoxA was purified from a Xanthomonas ΔroxA(pNH1::roxA) culture as described previously (4).
FIG 3.
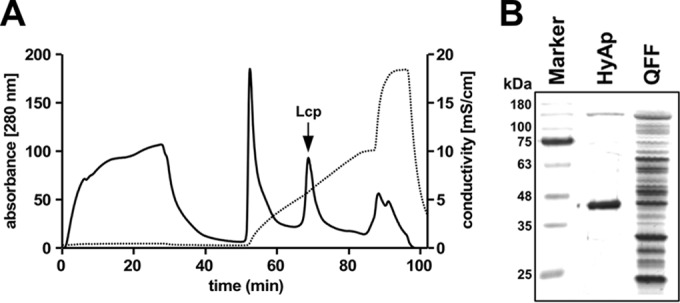
(A) Elution profile of Lcp-containing Q-Sepharose pool on hydroxyapatite. A 55-ml portion of a concentrated Lcp pool after chromatography on Q-Sepharose was applied to a 20-ml hydroxyapatite column that had been equilibrated with 1 mM potassium phosphate buffer (pH 6) and eluted with a 1 to 200 mM potassium phosphate gradient in equilibration buffer. Solid and dotted lines represent the absorbance at 280 nm and conductivity, respectively. Note that the second sharp peak of the gradient contained Lcp (arrow) and illustrates the separation power of hydroxyapatite. (B). SDS-PAGE of Lcp-containing fractions at different stages of purification. The gel was stained with silver. Molecular mass markers are indicated. HyAp, hydroxyapatite step; QFF, Q-Sepharose Fast Flow step.
RoxA and Lcp are indirectly responsible for clearing zones on opaque latex agar.
Xanthomonas sp. 35Y and Streptomyces sp. K30 both belong to the group of rubber-degrading bacteria that produce clearing zones during growth on opaque polyisoprene latex-containing media (1, 9, 20). Other rubber-degrading species, such as Gordonia westfalica and Gordonia polyisoprenivorans, grow adhesively on solid rubber and do not form clearing zones (10, 21), although they have lcp genes in their genomes. To investigate whether Lcp and/or RoxA are responsible for clearing zone formation, we applied purified Lcp and purified RoxA (5 μg of each) to an opaque polyisoprene agar plate and incubated the plate at room temperature. Remarkably, no clearing zones were formed by either Lcp or by RoxA, even if the plates were incubated for longer times. However, the enzymes had cleaved polyisoprene, as shown by positive staining with Schiff reagent. We conclude that the RoxA- and Lcp-derived degradation products contribute to the opaqueness of the plate because of the poor solubility of the cleavage products. Only if consuming cells were present nearby did a clearing zone become visible. In conclusion, clearing zones on opaque latex media are formed as a consequence of two events: the secretion of an active rubber-degrading enzyme and the uptake and consumption of the cleavage product by the cells.
RoxA and Lcp cleave polyisoprene to oligo-isoprenoids with different numbers of isoprene units.
Polyisoprene latex was incubated with purified RoxA or Lcp (each, 5 μg/ml) for 2 h at 37°C (RoxA) or room temperature (Lcp). In both cases, a purple color developed upon staining of the assay mixtures with Schiff reagent and indicated the presence of carbonyl groups in the products (Fig. 2B). No purple color was produced when heat-treated (10 min, 95°C) RoxA or Lcp was used. The RoxA- and Lcp-catalyzed cleavage products were solvent extracted with ethyl acetate, dissolved in methanol, and then separated by HPLC on a reversed-phase C8 column. A series of at least 11 different product peaks were detected after cleavage of the rubber by purified Lcp (18.8, 20.8, 22.0, 22.9, 23.6, 24.3, 25.2, 26.2, 27.6, 29.4, and 32.0 min), whereas only one major peak at 15.3 min was detected in RoxA-treated samples (Fig. 4A). The latter corresponded to ODTD, the well-known major cleavage product of RoxA (1). None of the peaks occurred without enzymes or with heat-treated enzymes. When oxygen was removed from the assay mixture (by flushing with nitrogen gas and subsequent incubation under nitrogen atmosphere), no cleavage products could be detected by HPLC analysis. These results confirmed that both RoxA and Lcp require molecular oxygen for the cleavage reaction.
FIG 4.
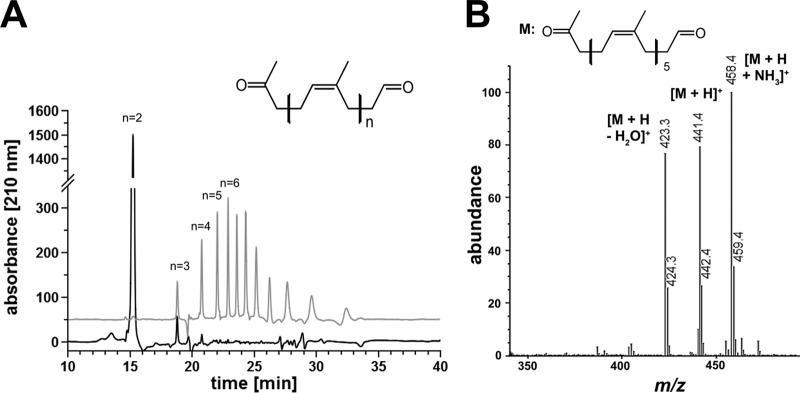
Separation of RoxA and Lcp-derived polyisoprene cleavage products. Polyisoprene latex was cleaved by RoxA and Lcp (each 5 μg) for 2 h at room temperature. Cleavage products were extracted with ethyl acetate, dissolved in methanol, and separated on a reversed-phase C8 HPLC column. (A) A UV (210-nm) absorbance spectrum of RoxA products (black line) and of Lcp products (gray line) elevated artificially by 50 absorption units for better visibility is shown. Note that of at least 11 species of degradation products, the first 4 were identified by MS analysis. The peaks at 23.6 min or later did not give signals in ESI-MS analysis, probably because of poor ionization and/or poor acceleration. The figure generally shows difference spectra (spectrum in the presence of enzyme minus the spectrum of the same experiment without the added enzyme). (B) HPLC-MS of Lcp-derived polyisoprene cleavage products. A part of the mass spectrum of the 22.0 min peak is shown. The m/z values of prominent signals are assigned. The minor side peaks at m/z 424.3, 442.4, and 459.4 represent 13C isotope peaks.
To determine the nature of the compounds behind the observed peaks of the Lcp-catalyzed reaction, the HPLC column was coupled with a mass spectrometry unit using the electrospray ionization (ESI)-MS technique. The peaks at 18.8, 20.8, 22.0, and 22.9 min corresponded to m/z values of 305, 373, 441, and 509, respectively. These values have an m/z increment of 68, and this mass difference matches the mass of one isoprene unit in polyisoprene. The m/z values of 305, 373, 441, and 509 correspond to protonated [M+H]+ oligo-isoprenoids with four (C20), five (C25), six (C30), or seven (C35) isoprene units, with the general structure CH3—CO—CH2—[CH2—CH=C(—CH3)—CH2]n—CH2—CHO, and all of them are larger than ODTD (C15; [M+H]+, m/z = 237) (Fig. 4B). When the Lcp-derived HPLC-ESI-MS spectrum was analyzed for other peaks that differed in m/z values from the above-mentioned values by 68, a minor peak at m/z of 237 (M+H+) corresponding to ODTD was also detected in the mass spectrum but was not visible in the UV spectrum. Apparently, ODTD is a minor trace product of Lcp-catalyzed polyisoprene cleavage that is only detectable by the sensitive ESI-MS method. The structure of the polyisoprene degradation product ODTD had been previously confirmed by nuclear magnetic resonance spectroscopy (1). At higher m/z values (>500) and at elution times where additional Lcp-derived UV product peaks were detected (23.6 min, 24.3 min, and later [see Fig. 4A]), no mass signals or only minor mass signals could be detected, and a direct proof of C40 and higher oligo-isoprenoids failed. We assume that the ionization of large oligo-isoprenoids is less efficient and/or that the mobilities of the respective higher oligo-isoprenoid ions were too poor to be detected. When purified RoxA was used to cleave polyisoprene, only one major peak at 15.3 min was detected by HPLC analysis, and this peak corresponded to protonated ODTD with an m/z value of 237, as described earlier (Fig. 4A). The minor peaks at 18.8 and 20.8 min correspond to ODTD analogs with one or two more isoprene units and represent minor products as described previously (1).
The Lcp-derived peaks at m/z values of 305 (18.8 min), 373 (20.8 min), 441 (22.0 min), and 509 (22.9 min) had side peaks with m/z values that were higher by 17 or lower by 18 (as shown, for example, for the m/z 441 peak in Fig. 4B). These side peaks corresponded to the respective ammonium [M+NH3]+ adduct molecules or to molecules in which a water molecule was removed [M+H-H2O]+. Analog side peaks were also observed for RoxA-derived ODTD earlier (22). Extension of the assay time or use of larger amounts of Lcp in the assay mixture did not result in a detectable difference of the product pattern. To identify the smallest degradation products of Lcp-catalyzed rubber cleavage, we isolated the cleavage products after incubation of polyisoprene latex with purified Lcp by solvent extraction. The products were emulsified in assay buffer (sample 1) and incubated with fresh Lcp at room temperature. After 2 h, the products were solvent extracted (sample 2), and both samples were analyzed by HPLC. The same 11 peaks displayed in Fig. 4A were detected in sample 1 and corresponded to the C20 tetra-isoprenoid and higher oligo-isoprenoids. Sample 2 revealed the same peaks, but the peak areas of the C20 tetra- and the C25 penta-isoprenoids were significantly larger (i.e., about double the peak areas) than in sample 1. We identified no significant increase in the trace amounts of ODTD (C15) or other smaller degradation products in the UV spectrum. We conclude that Lcp produces a large series of degradation products of different chain lengths and that the tetra-isoprenoid analog (C20 oligo-isoprenoid) is the smallest main end product.
When we added purified RoxA to the extracted product mixture of Lcp-catalyzed polyisoprene cleavage products and incubated the mixture for 2 h at room temperature in buffer, we determined that the C20 and higher oligo-isoprenoids had disappeared and that ODTD was the major end product. Neither RoxA nor Lcp was able to cleave ODTD to smaller products (not shown). In summary, the main end products of Lcp-catalyzed polyisoprene cleavage are ODTD analogs with four or more isoprene units and thus are considerably larger than RoxA-derived ODTD (tri-isoprenoid).
Properties of purified Lcp.
In contrast to RoxA, concentrated solutions of purified Lcp showed no red color, and no pseudoperoxidase activity was detected by the application of heme staining to SDS-PAGE-separated Lcp. In experiments with less-purified Lcp preparations, concentrated solutions revealed a minor absorption of ∼411 nm by UVvis spectroscopy. Reduction of such preparations by dithionite gave absorption maxima at approximately 429, 528, and 561 nm that are typical for reduced c-type cytochromes, and in SDS-PAGE analyses a minor contaminating band with pseudoperoxidase activity appeared below the 20-kDa standard band. This band corresponded to a Xanthomonas cytochrome and was also present in less-purified RoxA solutions (unpublished observations). Our data, along with the absence of a heme-binding motif in the amino acid sequence of Lcp, showed the absence of covalently attached heme groups in Lcp. Together with the differences in the amino acid sequences and in the determined cleavage products, our results confirmed that RoxA and Lcp fundamentally differ from each other.
Routinely, we performed activity assays by quantitative determination of cleavage products via HPLC. The HPLC assay has the advantage that many samples can be assayed simultaneously. We selected the area of the 22.9-min peak (C35 hepta-isoprenoid) or combined the areas of the first four product peaks (at 18.8 to 22.9 min, corresponding to C20 to C35 terpenoids) for product quantification in the case of Lcp assay and the area of the C15-ODTD peak in the case of RoxA. A linear dependence of the product area peak was determined for purified Lcp (≤5 μg of Lcp/ml) and from the incubation time up to 2 h at room temperature (data not shown). RoxA is a very stable enzyme, and no significant reduction in activity was determined after incubation of RoxA at 37°C for 22 h. In contrast, Lcp activity decreased by incubation at 37°C by 80% within the 22 h of incubation time (Fig. 5). Lcp was less inactivated at room temperature (∼70%) but was almost stable during storage on ice (≤5% inactivation).
FIG 5.
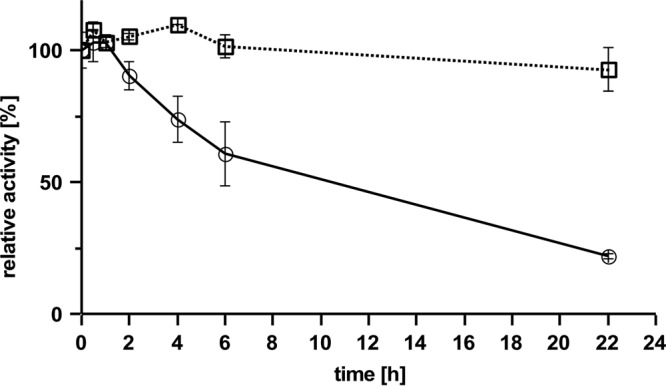
Stability of RoxA and Lcp. RoxA (□) and Lcp (○) were incubated at 37°C for various times, as indicated, before the residual activity was determined. 100% activity corresponds to an area of 1,280 (Lcp, 22.9-min peak) or 17,500 (RoxA, ODTD peak).
Similarly to RoxA, no addition of cofactors was necessary for the Lcp-catalyzed cleavage reaction. Besides the two substrates, polyisoprene and dioxygen, only appropriate physical conditions (aqueous buffer with a pH of around 7; temperature, ca. 25°C) were necessary. Maximum rubber-cleaving activities were determined between pH 6 and 8 in potassium phosphate buffer or in HEPES buffer (Fig. 6). Other buffers (Tris, carbonate-ammonium acetate, and glycylglycine) and pH values below pH 6 or above 8.5 resulted in only poor activities. Alternatively, we determined the Lcp activity by monitoring the decrease in dissolved dioxygen using an oxygen electrode. A time- and Lcp-dependent decrease in dissolved oxygen was determined, and a specific activity of 0.70 μmol of consumed dioxygen min−1 (mg of purified Lcp)−1 was calculated at room temperature (22°C). Purified RoxA had a lower activity of ∼0.26 μmol min−1 (mg of purified RoxA)−1 at room temperature. At 30°C, the specific activity of RoxA was almost double as high (0.48 μmol of consumed dioxygen min−1 mg−1).
FIG 6.
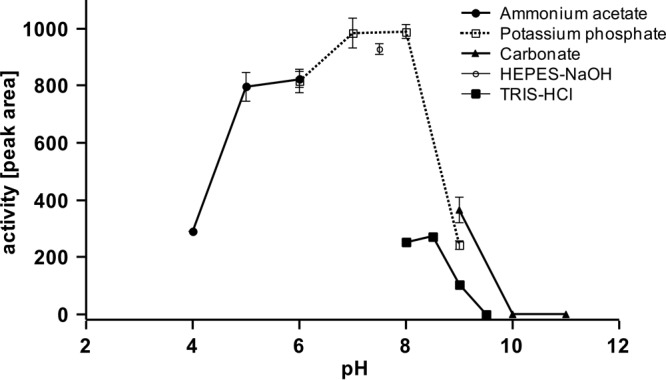
pH profile of Lcp-catalyzed cleavage of polyisoprene. Activity assays were performed as described in Materials and Methods using different buffer systems: 50 mM acetate buffer (●), 0.1 M potassium phosphate buffer (□), 50 mM HEPES buffer (○), 0.1 M Tris-HCl (◼), or 0.1 M carbonate buffer (▲). The area of the major product peak after HPLC analysis at 22.9 min is shown.
Low-molecular-weight substrate analogs with isoprenoid structure such as squalene (1,4-trans-isoprenoid), carotenoids, or α-tocopherol were not cleaved by Lcp. When polyisoprene latex was cleaved by Lcp in the presence of carotenoids or α-tocopherol (at a 1 mM end concentration), an inhibition of the cleavage reaction by ∼40% was determined (Fig. 7). Squalene inhibited the cleavage of polyisoprene only a little (∼90% residual activity). Reducing agents, such as dithiothreitol (DTT), strongly inhibited Lcp. The inhibition by DTT is remarkable because Lcp has no cysteine residues and therefore cannot have any essential disulfide bridges. Imidazole inhibited Lcp by 35 and 85% at 1 and 10 mM, respectively. Chelators (at 1 mM) such as sodium citrate, EDTA, ethyl xanthogenate, 4,5-dihydroxybenzene-1,3-disulfonate (tiron), 1,10-phenanthroline, or potassium ethyl 2,2′-bipyridine also had only minor effects on Lcp activity, and this indicated that Lcp activity is unlikely to depend on essential (metal) cations. Only sodium diethyl dithiocarbamate resulted in substantial inhibition at 1 mM (50% residual activity). Determination of the metal content of purified Lcp by ICP-MS did not result in the detection of iron, copper, or another transition metal (vanadium to zinc tested). However, because of a low protein concentration, the detection limits were relatively high, and the presence of up to 0.8 mol of Fe per mol of Lcp (0.2 mol of Cu per mol of Lcp) could not be excluded. Strong inhibitory effects were determined for ionic and nonionic detergents such as SDS, Triton X-100, or Tween 20. For the effects of other potential inhibitors, see Fig. 7.
FIG 7.
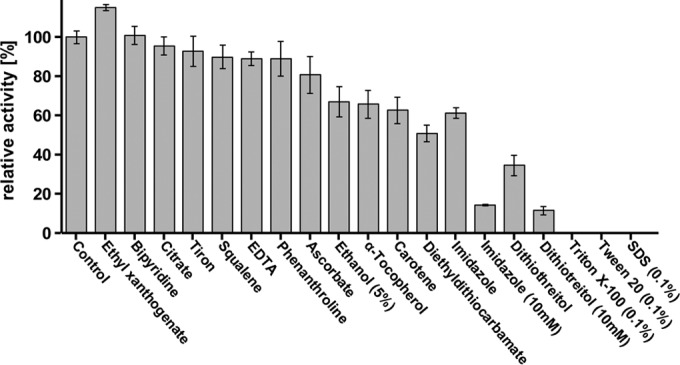
Effect of potential inhibitors on Lcp activity after a 1 mM concentration (unless stated otherwise) of the test compound was added to the assay mixture before the reaction was started by addition of purified Lcp protein. 100% value corresponds to 1,260 peak area in HPLC.
DISCUSSION
In this study, we succeeded to functionally express and purify Lcp of a rubber-degrading strain, Streptomyces sp. K30, in a ΔroxA background of Xanthomonas sp. From the combined culture fluids of a 12-liter culture, ∼1.5 mg of purified Lcp was obtained. A specific activity of 0.70 U/mg (consumed oxygen determined) was determined for purified Lcp at room temperature that was slightly higher than that obtained for RoxA at 30°C (0.48 U/mg). These low specific activities are in agreement with the relatively slow growth of rubber-degrading bacteria on polyisoprene (23, 24). The growth of many rubber-degrading bacteria takes weeks to months, especially if solid rubber pieces with small surface areas are used.
RoxA and Lcp have in common that both enzymes oxidatively attack the double bonds of rubber molecules to give cleavage products with aldehyde (—CH2—CHO) and keto (CH3—CO—CH2—) end groups and a certain number of isoprene units in between (1, 16, 25). RoxA produces only one major polyisoprene cleavage product (ODTD; C15 tri-isoprenoid); in contrast, Lcp cleaves rubber to multiple products ranging from C20 tetra-isoprenoid to at least C35 hepta-isoprenoid. Presumably, Lcp can also produce significantly larger degradation products that were not detectable by HPLC-MS analysis or were lost by the poor solubility during product extraction with ethyl-acetate and solubilization in methanol. Extension of the incubation time or addition of fresh Lcp enzyme during a polyisoprene cleavage reaction did not result in the appearance of degradation products smaller than the C20 tetra-isoprenoid and confirmed that the C20 tetra-isoprenoid is the smallest Lcp product (apart from the tiny traces of ODTD that were only seen in the highly sensitive mass-spec analysis). Previous determination of rubber intermediates from an Lcp-expressing wild-type culture of Nocardia farcinica E1 had revealed a large range of degradation products having molecular masses from approximately 900 to 4,500 Da and terminal aldehyde (—CH2—CHO, m/z 43) and keto (CH3—CO—CH2—, m/z 57) end groups (25). The same terminal aldehyde and keto end groups that had been previously determined for rubber cleavage products of RoxAs of Xanthomonas sp. 35Y and other Gram-negative bacteria (1, 6, 16) were also present in the degradation products of Lcp from Streptomyces sp. K30. We conclude that cleavage of polyisoprene by RoxAs and by Lcps generally occurs at the double bonds and that cleavage products only differ in the number of intact isoprene units between the terminal —CH2—CHO and CH3—CO—CH2— groups. An explanation for why Ibrahim et al. (25) did not identify the C20 to C35 oligo-terpenoid degradation products among the rubber degradation intermediates of whole-cell cultures might be the uptake and consumption of the smallest degradation products by the bacteria.
The different product spectra of Lcp compared to RoxA show that the active sites of both enzymes are different. The latter is deeply buried in the enzyme structure (5) and has no direct open access to the protein surface. An exo-type cleavage and a molecular ruler are postulated for RoxA to explain the uniform spacing between two adjacent cleavage sites of the polymeric substrate. Such a molecular ruler obviously does not exist in Lcp, and the variability of different cleavage products is in agreement with an endo-type cleavage mechanism. We assume that the active site of Lcp is more surface exposed and should be located close to the substrate binding site. The latter should comprise at least the length of the smallest degradation product (C20 isoprenoid). Depending on the binding of the polymer to the protein, the substrate can be cleaved at different relative positions with respect to the end of the polymer molecule, resulting in cleavage products that differ largely in the number of isoprene units.
The second major difference between RoxA and Lcp is the absence of essential cofactors in Lcp. We excluded the presence of covalently attached heme in purified Lcp, and we found no evidence for the dependence of Lcp activity from metals. We conclude that Lcp activity does not depend on known cofactors and thus differs from Fe-containing (heme) RoxA.
Lcp catalyzes the oxidative cleavage of polyisoprene with dioxygen as a cosubstrate. A direct reaction of molecular oxygen (triplet state) with a singlet organic substrate is not possible (“spin forbidden”) (26). Therefore, the Lcp-catalyzed oxygenase reaction should require either the presence of a transition metal resulting in orbital overlapping or the transfer of an electron to oxygen (e.g., by a reduced cofactor) to give an (enzyme-bound) reduced oxygen species. However, we found no evidence for the presence of a metal ion or a cofactor in Lcp, suggesting that Lcp might be a member of the growing group of so-called cofactor-independent oxygenases. (For reviews on cofactor-independent oxygenases, see references 14 and 15.) In this case, Lcp should be able to abstract a proton from the substrate to give a carbanion. The carbanion could then transfer an electron to dioxygen, and this would result in the formation of a substrate radical and a superoxide radical. Recombination of the substrate radical with the superoxide is a spin-allowed process and would give a peroxide intermediate. A prominent example for a cofactor-independent dioxygenase is 3-hydroxy-4-oxoquinaldine 2,4-dioxygenase (Hod) from Arthrobacter sp. Rü61a (27, 28). Analysis of the three-dimensional structure of Hod and the biochemical data of His251Ala Hod-muteins showed that His251 is the catalytically active base that accepts a proton from the substrate. The formed substrate anion then reacts with molecular oxygen without the need for a metal or a cofactor (29–31). Hod was partially inhibited by imidazole (as Lcp) in a concentration-dependent manner. However, the mechanism of inhibition remained unknown (29). Interestingly, Lcp proteins of different species have two conserved His residues (His198 and His203 in Lcp of Streptomyces sp. K30) (9) that are possible candidates to function as a base for proton abstraction from the substrate. The assumption of a histidine residue as the active site would be in agreement with the determined preferred pH value of Lcp between 6 and 8. Future research should target these residues to find experimental evidence for the involvement of these residues in catalysis.
ACKNOWLEDGMENTS
This study was supported by a grant of the Deutsche Forschungsgemeinschaft to D.J.
We thank Weber and Schaer company for providing polyisoprene and T. Gassner for assistance in some experiments. We also thank B. Nebel (Institute of Technical Biochemistry) for assistance with the HPLC-MS analysis and J. Altenbuchner (Institute of Industrial Genetics) for assistance with the experimental setup in Lcp expression.
Footnotes
Published ahead of print 6 June 2014
REFERENCES
- 1.Braaz R, Fischer P, Jendrossek D. 2004. Novel type of heme-dependent oxygenase catalyzes oxidative cleavage of rubber (poly-cis-1,4-isoprene). Appl. Environ. Microbiol. 70:7388–7395. 10.1128/AEM.70.12.7388-7395.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jendrossek D, Reinhardt S. 2003. Sequence analysis of a gene product synthesized by Xanthomonas sp. during growth on natural rubber latex. FEMS Microbiol. Lett. 224:61–65. 10.1016/S0378-1097(03)00424-5 [DOI] [PubMed] [Google Scholar]
- 3.Schmitt G, Seiffert G, Kroneck PMH, Braaz R, Jendrossek D. 2010. Spectroscopic properties of rubber oxygenase RoxA from Xanthomonas sp., a new type of dihaem dioxygenase. Microbiology 156:2537–2548. 10.1099/mic.0.038992-0 [DOI] [PubMed] [Google Scholar]
- 4.Birke J, Hambsch N, Schmitt G, Altenbuchner J, Jendrossek D. 2012. Phe317 is essential for rubber oxygenase RoxA activity. Appl. Environ. Microbiol. 78:7876–7883. 10.1128/AEM.02385-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seidel J, Schmitt G, Hoffmann M, Jendrossek D, Einsle O. 2013. Structure of the processive rubber oxygenase RoxA from Xanthomonas sp. Proc. Natl. Acad. Sci. U. S. A. 110:13833–13838. 10.1073/pnas.1305560110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birke J, Röther W, Schmitt G, Jendrossek D. 2013. Functional identification of rubber oxygenase (RoxA) in soil and marine myxobacteria. Appl. Environ. Microbiol. 79:6391–6399. 10.1128/AEM.02194-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imai S, Ichikawa K, Muramatsu Y, Kasai D, Masai E, Fukuda M. 2011. Isolation and characterization of Streptomyces, Actinoplanes, and Methylibium strains that are involved in degradation of natural rubber and synthetic poly(cis-1,4-isoprene). Enzyme Microb. Technol. 49:526–531. 10.1016/j.enzmictec.2011.05.014 [DOI] [PubMed] [Google Scholar]
- 8.Imai S, Yoshida R, Endo Y, Fukunaga Y, Yamazoe A, Kasai D, Masai E, Fukuda M. 2013. Rhizobacter gummiphilus sp. nov., a rubber-degrading bacterium isolated from the soil of a botanical garden in Japan. J. Gen. Appl. Microbiol. 59:199–205. 10.2323/jgam.59.199 [DOI] [PubMed] [Google Scholar]
- 9.Rose K, Tenberge KB, Steinbüchel A. 2005. Identification and characterization of genes from Streptomyces sp. strain K30 responsible for clear zone formation on natural rubber latex and poly(cis-1,4-isoprene) rubber degradation. Biomacromolecules 6:180–188. 10.1021/bm0496110 [DOI] [PubMed] [Google Scholar]
- 10.Bröker D, Dietz D, Arenskötter M, Steinbüchel A. 2008. The genomes of the non-clearing-zone-forming and natural-rubber-degrading species Gordonia polyisoprenivorans and Gordonia westfalica harbor genes expressing Lcp activity in Streptomyces strains. Appl. Environ. Microbiol. 74:2288–2297. 10.1128/AEM.02145-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rose K, Steinbüchel A. 2005. Biodegradation of natural rubber and related compounds: recent insights into a hardly understood catabolic capability of microorganisms. Appl. Environ. Microbiol. 71:2803–2812. 10.1128/AEM.71.6.2803-2812.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yikmis M, Steinbüchel A. 2012. Historical and recent achievements in the field of microbial degradation of natural and synthetic rubber. Appl. Environ. Microbiol. 78:4543–4551. 10.1128/AEM.00001-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fetzner S. 2012. Ring-cleaving dioxygenases with a cupin fold. Appl. Environ. Microbiol. 78:2505–2514. 10.1128/AEM.07651-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fetzner S. 2007. Cofactor-independent oxygenases go it alone. Nat. Chem. Biol. 3:374–375. 10.1038/nchembio0707-374 [DOI] [PubMed] [Google Scholar]
- 15.Fetzner S, Steiner RA. 2010. Cofactor-independent oxidases and oxygenases. Appl. Microbiol. Biotechnol. 86:791–804. 10.1007/s00253-010-2455-0 [DOI] [PubMed] [Google Scholar]
- 16.Tsuchii A, Takeda K. 1990. Rubber-degrading enzyme from a bacterial culture. Appl. Environ. Microbiol. 56:269–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yikmis M, Arenskotter M, Rose K, Lange N, Wernsmann H, Wiefel L, Steinbüchel A. 2008. Secretion and transcriptional regulation of the latex-clearing protein, Lcp, by the rubber-degrading bacterium Streptomyces sp. strain K30. Appl. Environ. Microbiol. 74:5373–5382. 10.1128/AEM.01001-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hambsch N, Schmitt G, Jendrossek D. 2010. Development of a homologous expression system for rubber oxygenase RoxA from Xanthomonas sp. J. Appl. Microbiol. 109:1067–1075. 10.1111/j.1365-2672.2010.04732.x [DOI] [PubMed] [Google Scholar]
- 19.Yikmis M, Steinbüchel A. 2012. Importance of the latex-clearing protein (Lcp) for poly(cis-1,4-isoprene) rubber cleavage in Streptomyces sp. K30. Microbiologyopen 1:13–24. 10.1002/mbo3.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jendrossek D, Tomasi G, Kroppenstedt RM. 1997. Bacterial degradation of natural rubber: a privilege of actinomycetes? FEMS Microbiol. Lett. 150:179–188. 10.1016/S0378-1097(97)00072-4 [DOI] [PubMed] [Google Scholar]
- 21.Linos A, Berekaa MM, Reichelt R, Keller U, Schmitt J, Flemming HC, Kroppenstedt RM, Steinbüchel A. 2000. Biodegradation of cis-1,4-polyisoprene rubbers by distinct actinomycetes: microbial strategies and detailed surface analysis. Appl. Environ. Microbiol. 66:1639–1645. 10.1128/AEM.66.4.1639-1645.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braaz R, Armbruster W, Jendrossek D. 2005. Heme-dependent rubber oxygenase RoxA of Xanthomonas sp. cleaves the carbon backbone of poly(cis-1,4-isoprene) by a dioxygenase mechanism. Appl. Environ. Microbiol. 71:2473–2478. 10.1128/AEM.71.5.2473-2478.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bode HB, Zeeck A, Plückhahn K, Jendrossek D. 2000. Physiological and chemical investigations into microbial degradation of synthetic poly(cis-1,4-isoprene). Appl. Environ. Microbiol. 66:3680–3685. 10.1128/AEM.66.9.3680-3685.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berekaa MM, Linos A, Reichelt R, Keller U, Steinbüchel A. 2000. Effect of pretreatment of rubber material on its biodegradability by various rubber degrading bacteria. FEMS Microbiol. Lett. 184:199–206. 10.1111/j.1574-6968.2000.tb09014.x [DOI] [PubMed] [Google Scholar]
- 25.Ibrahim EMA, Arenskotter M, Luftmann H, Steinbüchel A. 2006. Identification of poly(cis-1,4-isoprene) degradation intermediates during growth of moderately thermophilic actinomycetes on rubber and cloning of a functional lcp homologue from Nocardia farcinica strain E1. Appl. Environ. Microbiol. 72:3375–3382. 10.1128/AEM.72.5.3375-3382.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bugg T. 2003. Dioxygenase enzymes: catalytic mechanisms and chemical models. Tetrahedron 59:7075–7101. 10.1016/S0040-4020(03)00944-X [DOI] [Google Scholar]
- 27.Bauer I, Max N, Fetzner S, Lingens F. 1996. 2,4-Dioxygenases catalyzing N-heterocyclic-ring cleavage and formation of carbon monoxide. Purification and some properties of 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase from Arthrobacter sp. Rü61a and comparison with 1H-3-hydroxy-4-oxoquinoline 2,4-dioxygenase from Pseudomonas putida 33/1. Eur. J. Biochem. 240:576–583 [DOI] [PubMed] [Google Scholar]
- 28.Frerichs-Deeken U, Fetzner S. 2005. Dioxygenases without requirement for cofactors: identification of amino acid residues involved in substrate binding and catalysis, and testing for rate-limiting steps in the reaction of 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase. Curr. Microbiol. 51:344–352. 10.1007/s00284-005-0065-3 [DOI] [PubMed] [Google Scholar]
- 29.Hernandez-Ortega A, Quesne MG, Bui S, Heuts DPHM, Steiner RA, Heyes DJ, de Visser SP, Scrutton NS. 2014. Origin of the proton-transfer step in the cofactor-free (1H)-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase: effect of the basicity of an active site His residue. J. Biol. Chem. 289:8620–8632. 10.1074/jbc.M113.543033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steiner RA, Janssen HJ, Roversi P, Oakley AJ, Fetzner S. 2010. Structural basis for cofactor-independent dioxygenation of N-heteroaromatic compounds at the alpha/beta-hydrolase fold. Proc. Natl. Acad. Sci. U. S. A. 107:657–662. 10.1073/pnas.0909033107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thierbach S, Bui N, Zapp J, Chhabra SR, Kappl R, Fetzner S. 2014. Substrate-assisted O2 activation in a cofactor-independent dioxygenase. Chem. Biol. 21:217–225. 10.1016/j.chembiol.2013.11.013 [DOI] [PubMed] [Google Scholar]