

Abstract
Candida glabrata, the second most common cause of Candida infections, is associated with high rates of mortality and often exhibits resistance to the azole class of antifungal agents. Upc2 and Ecm22 in Saccharomyces cerevisiae and Upc2 in Candida albicans are the transcriptional regulators of ERG11, the gene encoding the target of azoles in the ergosterol biosynthesis pathway. Recently two homologs for these transcription factors, UPC2A and UPC2B, were identified in C. glabrata. One of these, UPC2A, was shown to influence azole susceptibility. We hypothesized that due to the global role for Upc2 in sterol biosynthesis in S. cerevisiae and C. albicans, disruption of UPC2A would enhance the activity of fluconazole in both azole-susceptible dose-dependent (SDD) and -resistant C. glabrata clinical isolates. To test this hypothesis, we constructed mutants with disruptions in UPC2A and UPC2B alone and in combination in a matched pair of clinical azole-SDD and -resistant isolates. Disruption of UPC2A in both the SDD and resistant isolates resulted in increased susceptibility to sterol biosynthesis inhibitors, including a reduction in fluconazole MIC and minimum fungicidal concentration, enhanced azole activity by time-kill analysis, a decrease in ergosterol content, and downregulation of baseline and inducible expression of several sterol biosynthesis genes. Our results indicate that Upc2A is a key regulator of ergosterol biosynthesis and is essential for resistance to sterol biosynthesis inhibitors in C. glabrata. Therefore, the UPC2A pathway may represent a potential cotherapeutic target for enhancing azole activity against this organism.
INTRODUCTION
Fungal infections caused by opportunistic organisms have continued to increase in recent decades and have become an important medical concern (1–3). Candida glabrata in particular has emerged over the past 2 decades as the predominant cause of yeast infections in diabetics (54%) and the elderly (51%) and is second to Candida albicans in most other patient populations (4–7). Candidemia mortality rates continue to rise, with rates for C. glabrata reported as high as 50% (8–10). The emergence of C. glabrata as a common pathogen is complicated by, and likely related to, its intrinsically low susceptibility to azole antifungals and its ability to rapidly develop high-level azole resistance during treatment (11–14).
Ergosterol is an essential component of the fungal cell membrane and is an important signaling molecule in the cell. It helps maintain membrane integrity and fluidity, which facilitates several membrane-bound enzymatic reactions. Ergosterol is not synthesized by the host, so the ergosterol biosynthesis pathway has long been a target for antifungal agents (Fig. 1). Compounds that inhibit this pathway are broadly categorized as sterol biosynthesis inhibitors (SBIs). The azole class of antifungals specifically targets lanosterol 14-alpha-demethylase (Erg11), which catalyzes the C-14 demethylation of lanosterol (15–17). Statins, such as lovastatin, inhibit the gene products of HMG1 and HMG2, which convert 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) to mevalonate, resulting in a reduction in the synthesis of cholesterol in mammals and the synthesis of ergosterol in yeasts (Fig. 1).
FIG 1.
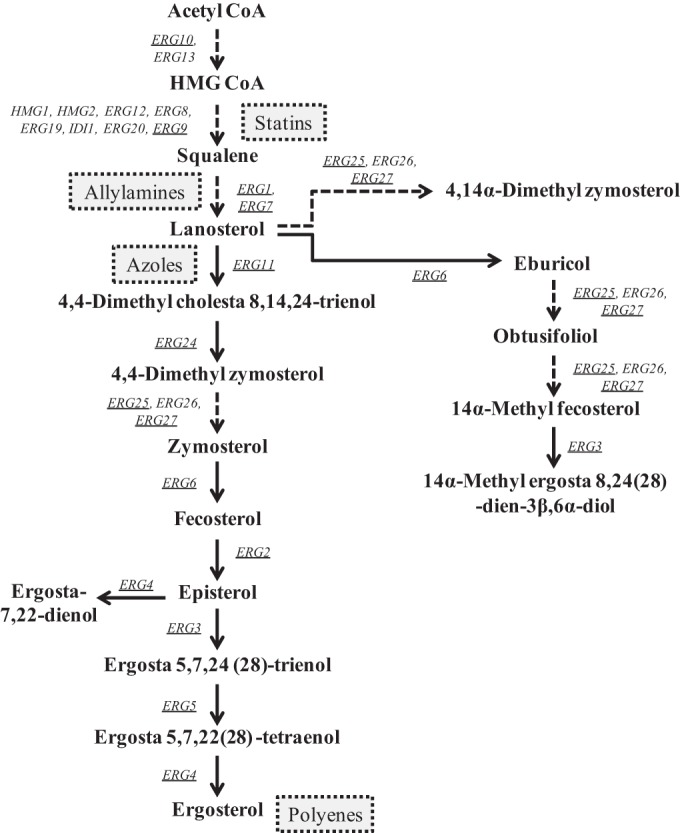
Representation of the ergosterol biosynthesis pathway in Candida glabrata. Genes (in italics) whose expression was monitored in this study are underlined. Sites of action for specific drug classes are indicated by dashed box. CoA, coenzyme A.
As has been observed in Saccharomyces cerevisiae and C. albicans, exposure to azoles also results in upregulation of genes from the ergosterol biosynthesis pathway (including ERG11) in C. glabrata (14–17). The predominant mechanism of azole resistance in C. glabrata is the constitutive overexpression of ATP-binding cassette (ABC) transporter genes, CDR1, PDH1, and SNQ2, which are under the transcriptional regulation of a hyperactive form of the transcription factor Pdr1. ERG gene expression and regulation could also be important in acquired azole resistance, as is the case in C. albicans, or may potentially contribute to the intrinsic reduced azole susceptibility in C. glabrata (14, 18–25).
Resistant isolates of S. cerevisiae often have increased expression of ERG11, as well as other ergosterol biosynthesis genes, which are under the control of the transcriptional regulators Upc2 and Ecm22 (26, 27). In C. albicans, a single homolog, Upc2, has been identified and found to be involved in ERG gene regulation (17, 25, 28–30). These transcriptional regulators are members of the well-characterized fungus-specific Zn2-Cys6 family of transcriptional activators (18). ERG11 is not an essential gene in C. glabrata. Disruption of this gene or pharmacologic inhibition of its product prevents ergosterol production, which causes accumulation of alternate sterols, including lanosterol, obtusifoliol, and 4,14α-dimethyl zymosterol, leading to decreased susceptibility to fluconazole, itraconazole, and amphotericin B (31, 32). Biosynthesis of these alternate sterols does not require ERG11 but does require other genes in the ergosterol biosynthesis pathway.
Nagi et al. recently described two Upc2 homologs in C. glabrata, Upc2A and Upc2B. Upc2A was shown to play a major role in the regulation of ERG2 and ERG3 in the ergosterol biosynthesis pathway (24). A strain with a disruption of UPC2A also exhibited 4-fold and 16-fold increases in fluconazole and lovastatin (a member of the statin class of cholesterol-lowering agents) susceptibilities, respectively (24). Given the global role of Upc2 in regulation of sterol biosynthesis, we reasoned that disruption of UPC2A might enhance the activity of fluconazole in azole-resistant as well as azole-susceptible dose-dependent (SDD) clinical isolates.
MATERIALS AND METHODS
Strains and growth media.
The matched fluconazole-SDD and -resistant clinical isolate set used in this study has been described previously (12, 19). All strains (Table 1) were maintained in YPD (1% yeast extract, 2% peptone, and 2% dextrose) broth at 30°C and stored as 40% glycerol stocks at −80°C. One Shot Escherichia coli TOP10 chemically competent cells (Invitrogen, Carlsbad, CA) were used as the host for plasmid construction and propagation. These strains were grown at 37°C in Luria-Bertani (LB) broth or on LB plates supplemented with 100 μg/ml of ampicillin (Sigma, St. Louis, MO) or 50 μg/ml of kanamycin (Fisher BioReagents, Fair Lawn, NJ).
TABLE 1.
Strains used in this study
| Strain | Parent | Genotype or description | Reference |
|---|---|---|---|
| SM1 | Azole-SDD clinical isolate | 12 | |
| SM1Δupc2A | SM1 | upc2AΔ::FRT | This study |
| SM1Δupc2B | SM1 | upc2BΔ::FRT | This study |
| SM1Δupc2AΔupc2B | SM1 | upc2AΔ::FRT/upc2BΔ::FRT | This study |
| SM1Δupc2A/UPC2A | SM1 | upc2AΔ::FRT-UPC2A | This study |
| SM1Δupc2AΔupc2B/UPC2A | SM1 | upc2AΔ::FRT-UPC2A/upc2BΔ::FRT | This study |
| SM3 | Azole-resistant clinical isolate | 12 | |
| SM3Δupc2A | SM3 | upc2AΔ::FRT | This study |
| SM3Δupc2B | SM3 | upc2BΔ::FRT | This study |
| SM3Δupc2AΔupc2B | SM3 | upc2AΔ::FRT/upc2BΔ::FRT | This study |
| SM3Δupc2A/UPC2A | SM3 | upc2AΔ::FRT-UPC2A | This study |
| SM3Δupc2AΔupc2B/UPC2A | SM3 | upc2AΔ::FRT-UPC2A/upc2BΔ::FRT | This study |
Gene disruption.
Mutant strains (Table 1) were generated by the SAT1-flipper gene disruption method (33). Briefly, approximately 900 bp of sequence immediately upstream of the target gene plus the first 50 bp of the open reading frame (ORF), referred to as the 5′ flanking region, was cloned upstream of the SAT1-flipper cassette in pSFS2A, and the final 50 bp of the target gene ORF plus approximately 900 bp of sequence immediately downstream of the target gene, referred to as the 3′ flanking region, was cloned downstream of the SAT1-flipper cassette. The disruption cassettes consisting of the SAT1-flipper and 5′ and 3′ flanking sequences of the either the UPC2A or UPC2B genes were excised from the final plasmid construct and gel purified. Primers used to construct the cassettes are listed in Table 2. C. glabrata cells were transformed by the lithium acetate method using approximately 1 μg of DNA. The transformed cells were allowed to recover for 6 h in YPD at 30°C before being plated on YPD agar plates containing 200 μg/ml of nourseothricin (Jena Biochemical, Germany) and incubated at 30°C. Positive transformants were selected within 24 h, and successful insertion of the disruption cassette at the target gene locus was confirmed by Southern hybridization. Subsequently, induction of the flipper recombinase gene in the disruption cassette was performed by overnight growth of the positive transformant clones in YPD at 30°C with shaking (under no selective pressure). Selection of cassette excision was then performed by plating a series of dilutions of this culture on YPD agar plates containing 25 μg/ml of nourseothricin and incubating them for up to 24 h at 30°C. Clones were selected due to size differences (slower-growing colonies usually indicated successful cassette excision) and confirmed by Southern hybridization (see Fig. S1 in the supplemental material).
TABLE 2.
Primers used in this study (grouped by application)
| Primer function and name | Primer sequencea |
|---|---|
| Cassettes for constructing mutants | |
| CgUPC2A-A | 5′-GTTAAAACGGGCCCATGATCGTCGTATCC-3′ |
| CgUPC2A-B | 5′-AATAGCTTTGGCTCGGTATCCTCTATGT-3′ |
| CgUPC2A-C | 5′-CTAGAGATGCGGCCGCTTATAGTGGTGGT-3′ |
| CgUPC2A-D | 5′-TATCGATTTAACCGCGGTGATATTCTTCCA-3′ |
| CgUPC2A-E | 5′-GTACCTAGTGGGCCCACCTTGATCTGT-3′ |
| CgUPC2A-F | 5′-AATAAAGGAAGTAAATTGCATATTTCAGAG-3′ |
| CgUPC2B-A | 5′-TGAGTTCAGTTTTATGGGCCCTTAGAAAG-3′ |
| CgUPC2B-B | 5′-AGTTGTACTAACCTTCTTACCTCGAGTATCTA-3′ |
| CgUPC2B-C | 5′-TAAGATATTGCCGCGGTTTACTAACGATGT-3′ |
| CgUPC2B-D | 5′-CAAAACTATAGGAGCTCGAAGGAATATGTG-3′ |
| qRT-PCR | |
| ACT1F | 5′-CGCTTTGGACTTCGAACAAGAA-3′ |
| ACT1R | 5′-GTTACCGATGGTGATGACTTGAC-3′ |
| ERG1F | 5′-GGTAAGAAGGTGCTGATTG-3′ |
| ERG1R | 5′-GATGTTGTTGATGGACTGG-3′ |
| ERG2F | 5′-AATCTGCTATTGCTGGTTGCC-3′ |
| ERG2R | 5′-CGCTGCCGTTGTGTTTGG-3′ |
| ERG3F | 5′-AAGCGTGTGAACAAGGAC-3′ |
| ERG3R | 5′-TACAATAGCAGACCGAAGAC-3′ |
| ERG4F | 5′-GCTGTACGCTAACGCTTGTG-3′ |
| ERG4R | 5′-CAGTGGCAGTATGTGTATGGAAC-3′ |
| ERG5F | 5′-CCCAGCCCTACACGACCCAGAAG-3′ |
| ERG5R | 5′-GGACCACAGCCGAAGACCAACC-3′ |
| ERG6F | 5′-TCCTCTTTCCACTTCTCCCGTTTC-3′ |
| ERG6R | 5′-GCCACCGACACCGCAACC-3′ |
| ERG7F | 5′-ATGCCTGATGGTGGTTGG-3′ |
| ERG7R | 5′-TCTTCATTTGGACACTTAGCC-3′ |
| ERG9F | 5′-CAGCACCAATGACACCAG-3′ |
| ERG9R | 5′-CCTCAGAATCAGATAGAAGACC-3′ |
| ERG10F | 5′-GTTATCGCTGGTGGTTGTG-3′ |
| ERG10R | 5′-CATTGGTTGGTGGTCGTAAG-3′ |
| ERG11F | 5′-TACCAAGCCATACGAGTTC-3′ |
| ERG11R | 5′-GGTCAAGTGGGAGTAAGC-3′ |
| ERG24F | 5′-ACTCGCTGGACTACTACTTC-3′ |
| ERG24R | 5′-CAACTTAGTGCCGTCTCTTAG-3′ |
| ERG25F | 5′-ATGTGTATGGATTACCTTGAGAC-3′ |
| ERG25R | 5′-GTAGTTACCGATGAAGTAGTGG-3′ |
| ERG27F | 5′-CCAGGAAAGTAGCCGTTATCAC-3′ |
| ERG27R | 5′-TCAACTCAACAACCTCTCTTACAC-3′ |
| 18SF | 5′-TCGGCACCTTACGAGAAATCA-3′ |
| 18SR | 5′-CGACCATACTCCCCCCAGA-3′ |
| PDR1F | 5′-TTTGACTCTGTTATGAGCGATTACG-3′ |
| PDR1R | 5′-TTCGGATTTTTCTGTGACAATGG-3′ |
| CDR1F | 5′-CATACAAGAAACACCAAAGTCGGT-3′ |
| CDR1R | 5′-GAGACACGCTTACGTTCACCAC-3′ |
| SNQ2F | 5′-CGTCCTATGTCTTCCTTACACCATT-3′ |
| SNQ2R | 5′-TTTGAACCGCTTTTGTCTCTGA-3′ |
| PDH1F | 5′-ACGAGGAGGAAGACGACTACGA-3′ |
| PDH1R | 5′-CTTTACTGGAGAACTCATCGCTGGT-3′ |
Underlined bases indicate introduction of restriction enzyme cloning sites to allow directional cloning into the SAT1-flipper cassette.
Likewise, reintroduction of the UPC2A gene into the UPC2A locus was accomplished as described above with a SAT1-flipper cassette in which the 5′ flanking sequence cloned upstream of the flipper cassette was replaced with a DNA fragment consisting of the 5′ flanking sequence and full-length ORF of UPC2A. Isolate-specific constructs were prepared to reintroduce the native UPC2A alleles back into their original locus. This cassette was excised, gel purified, and used for cell transformation as described above. Screening for positive clones was accomplished as described above (see Fig. S1).
Isolation of genomic DNA and Southern hybridization.
Genomic DNA from C. glabrata was isolated as described previously (34). For confirmation by Southern hybridization, approximately 10 μg of genomic DNA was digested with the appropriate restriction enzymes, separated on a 1% agarose gel containing ethidium bromide, transferred by vacuum blotting onto a nylon membrane, and fixed by UV cross-linking. Hybridization was performed with the Amersham ECL direct nucleic acid labeling and detection system (GE Healthcare, Pittsburgh, PA) as per the manufacturer's instructions.
Susceptibility testing.
Susceptibility testing was performed by broth microdilution assay according to the CLSI guidelines outlined in document M27-A3, with a few modifications (35). Antifungal agents were obtained from the following sources: fluconazole was from LKT Laboratories (St. Paul, MN), miconazole was from MP Biomedicals, Inc. (Solon, OH), anidulafungin and voriconazole were from Pfizer (New York, NY), and ketoconazole, amphotericin B, lovastatin, and terbinafine were from Sigma (St. Louis, MO). All drugs were dissolved in dimethyl sulfoxide (DMSO) and diluted to yield a final DMSO concentration of ≤0.5% in all experiments. Growth of C. glabrata was not affected at this concentration of DMSO (data not shown). Cultures were diluted to 2.5 × 103 cells/ml in RPMI medium (Sigma) with 2% glucose and morpholinepropanesulfonic acid (MOPS), pH 7.0. Plates were incubated at 35°C for 24, 48, and 72 h. Absorbance at 620 nm was read with a microplate reader (Beckman Coulter, Inc., Fullerton, CA); background due to medium was subtracted from all readings. The MIC was defined as the lowest concentration inhibiting growth by at least 80% (or no visible growth for amphotericin B) relative to that of the drug-free control after incubation with drug for 24 h. While CLSI guidelines indicate an endpoint of 50% inhibition of growth, we chose to use a more stringent endpoint of 80% inhibition of growth in order to detect a more pronounced effect. Absorbance measured at 72 h was used to assess regrowth of the cultures (36). The minimum fungicidal concentration (MFC) was determined by spotting 5 μl from each well of the 48-h MIC plate onto YPD solid media and incubating the media at 30°C for an additional 24 h. The MFC was defined as the lowest concentration that exhibited complete inhibition of growth (37, 38).
Spot assays.
Colonies from cultures grown on Sabouraud dextrose (BD Biosciences, San Jose, CA) solid media for 24 h were diluted in sterile water to an optical density at 600 nm of 0.1. Cultures were serially diluted one to four in sterile water. Two microliters of each dilution was spotted on each of three solid media: YPD, YPD with 5 μg/ml of fluconazole, and YPD with 10 μg/ml of fluconazole. The serial dilutions were incubated at 30°C for 24 and 72 h. Plates were imaged at each time point, and degrees of growth inhibition were compared (20, 36, 39).
Time-kill analysis.
Time-kill experiments were adapted from the methods described by Klepser et al. (40) Briefly, isolates started from freezer stocks were subcultured twice on potato dextrose agar (BD Biosciences, San Jose, CA). Colonies were picked from the plates into sterile water and the densities adjusted to that of a 0.5 McFarland standard. The suspensions were then diluted 1:10 in RPMI medium to a final volume of 3 ml with or without 10 μg/ml of fluconazole. Cultures were incubated with agitation at 35°C. One-hundred-microliter samples were removed from each culture at 0, 6, 12, and 24 h and serially diluted in sterile water, and 50-μl aliquots were spread on potato dextrose agar. Plates were incubated at 35°C and colonies counted at 24 and 48 h. Each experiment was performed in triplicate.
Ergosterol quantification analysis.
Ergosterol was extracted and quantified as described by Arthington-Skaggs et al. (41). Briefly, a single colony of C. glabrata from a fresh YPD plate was used to inoculate 50 ml of RPMI medium (Sigma, St. Louis, MO) and incubated for 16 h with shaking at 35°C. Stationary-phase cells were collected by centrifugation for 5 min at 2,700 rpm and washed twice with sterile distilled water. The net weight of the pellet was determined. To each pellet, 3 ml of 25% alcoholic potassium hydroxide solution (25 g of KOH and 35 ml of sterile distilled water brought up to 100 ml with 100% ethanol) was added, followed by vortexing for 1 min. Cell suspensions were transferred to a sterile borosilicate glass screw-cap tube and incubated at 85°C for 1 h. Sterols were then extracted from cool tubes by the addition of a mixture of 3 ml of n-heptane and 1 ml of sterile distilled water, followed by vortexing for 3 min. The heptane layer was transferred to a clean sterile borosilicate glass tube and stored at −20°C for up to 24 h. One hundred microliters of sterol-heptane mixture was scanned spectrophotometrically between 240 nm and 300 nm with a DU530 Life Sciences UV spectrophotometer (Beckman Coulter, Brea, CA). The presence of ergosterol in the extracted sample resulted in a characteristic four-peak curve with peaks located at approximately 262, 270, 281, and 290 nm. The absence of detectable ergosterol was indicated by a flat line. n-Heptane was used to blank the spectrophotometer. A decrease in the height of the absorbance peaks correlated to a decrease in ergosterol content. Statistical significance was determined by using Student's t test (P < 0.05).
RNA isolation.
For RNA analysis, log-phase cultures grown in YPD medium at 30°C were adjusted to an optical density of 0.2 measured at 600 nm. Various concentrations of drug or the DMSO control were added, and the cultures were incubated for an additional 3 h to mid-log phase. RNA was extracted by the hot-phenol method (42), as previously described (43). RNA was treated with RQ1 DNase (Promega, Madison, WI). Quantity and purity were determined with a spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE).
Quantitative RT-PCR analysis.
Quantitative real-time PCR (RT-PCR) was conducted as described previously (43). Single-strand cDNA was synthesized from 2 μg of total RNA using the SuperScript First Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Relative quantitative real-time PCRs were performed in triplicate using the ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA). Independent PCRs were performed using the primers listed in Table 2 for both the genes of interest and either 18s rRNA or ACT1 using SYBR green PCR master mix (Applied Biosystems). Relative gene expression was calculated by the comparative threshold cycle (ΔΔCT) method. For expression of PDR1, CDR1, SNQ2, and PDH1, samples were normalized first to 18S rRNA expression and then to the untreated SM1 sample. For expression of genes in the ergosterol biosynthesis pathway, samples were normalized first to ACT1 and then to the untreated parent sample, SM1 or SM3.
RESULTS
Disruption of CgUPC2A results in increased susceptibility to azole antifungals and other sterol biosynthesis inhibitors in C. glabrata.
Using the C. glabrata genome database maintained by Génolevures (www.genolevures.org/cagl.html), BLASTP analysis was performed to search for homologs of either ScUpc2 or ScEcm22. Two potential homologs were identified, CAGL0C01199g and CAGL0F07865g, which were recently reported by Nagi et al. as UPC2A and UPC2B, respectively (24). UPC2A (CAGL0C01199g) alone, UPC2B (CAGL0F07865g) alone, and both genes together were disrupted in fluconazole-SDD clinical isolate, SM1, and tested against fluconazole (Table 3). As observed by Nagi et al., the disruption of UPC2A, alone or in combination with disruption of UPC2B, showed a marked increase in susceptibility to fluconazole, whereas the disruption of UPC2B alone did not. Complementation of UPC2A back into its native locus restored wild-type susceptibility. In addition to fluconazole, strains were tested against a panel of azoles, nonazole SBIs (terbinafine and lovastatin), and antifungal agents not involved in sterol biosynthesis (amphotericin B and anidulafungin) (Table 3). The additional azoles tested exhibited susceptibility patterns similar to that observed with fluconazole. Terbinafine inhibits squalene epoxidase and lovastatin inhibits HMG-CoA reductase, both early enzymes in the ergosterol biosynthesis pathway (Fig. 1). As was observed with the azole antifungals, strains with disruptions of UPC2A exhibited increased susceptibility to both of these agents (Table 3). As reported previously for C. albicans (29), disruption of either UPC2A or UPC2B had no effect on susceptibility to amphotericin B, which binds ergosterol in the fungal cell membrane. Likewise, disruption of UPC2 had no effect on susceptibility to the echinocandin anidulafungin, which affects cell wall stability by targeting β-1,3-glucan synthase (Table 3). Finally, reintroduction of UPC2A was able to restore wild-type susceptibilities. These results demonstrate that UPC2A influences the susceptibility of C. glabrata to a range of clinically used azoles as well as other SBIs.
TABLE 3.
Broth microdilution susceptibility testing of the susceptible dose-dependent clinical parent isolate SM1, mutant strains with disruptions of UPC2, and a revertant
| Strain | MIC (μg/ml)a |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| FLC | ITC | VRC | KTC | MC | AMB | LOVA | TRB | ANID | |
| SM1 | 8 | 2 | 2 | 2 | 0.5 | 2 | 200 | 512 | 0.016 |
| SM1Δupc2A | 0.5 | 0.0078 | 0.125 | 0.0625 | 0.01563 | 2 | 25 | 32 | 0.016 |
| SM1Δupc2B | 8 | 1 | 1 | 0.5 | 0.25 | 2 | 256 | 512 | 0.016 |
| SM1Δupc2AΔupc2B | 0.5 | 0.0078 | 0.125 | 0.01563 | 0.01563 | 2 | 16 | 8 | 0.016 |
| SM1Δupc2A/UPC2A | 8 | 2 | 2 | 2 | 0.5 | 2 | 200 | 512 | 0.016 |
FLC, fluconazole; ITC, itraconazole; VRC, voriconazole; KTC, ketoconazole; MC, miconazole; AMB, amphotericin B; LOVA, lovastatin; TRB, terbinafine; ANID, anidulafungin.
Disruption of UPC2A results in enhanced activity of fluconazole against C. glabrata.
Fluconazole remains the most widely used antifungal agent prescribed for the treatment of Candida infections (44). We therefore wished to further investigate the extent to which UPC2A influences fluconazole susceptibility in C. glabrata. In isolate SM1, disruption of UPC2A alone or in combination with disruption of UPC2B resulted in a reduction not only in the fluconazole MIC (Table 3) but also in the fluconazole MFC, whereas disruption of UPC2B had no effect on either of these parameters (Fig. 2A). This phenotype reverted to that of the parent, SM1, when UPC2A was reintroduced. We used a 72-h endpoint for a broth microdilution assay in RPMI medium as a way to assess the ability of the organism to resume growth in the presence of fluconazole. The UPC2A mutant was unable to resume growth at this extended time point, whereas isolate SM1 was able to resume growth in concentrations up to 8 μg/ml (Fig. 2B). Serial dilutions of the strains with disruptions of UPC2A spotted on YPD solid medium containing 5 μg/ml or 10 μg/ml of fluconazole exhibited reduced growth, while SM1Δupc2B and the UPC2A revertants were no different than the parent, SM1. Moreover, the cultures were unable to recover after incubation for 72 h (Fig. 2C). Likewise, by time-kill analysis, disruption of UPC2A in isolate SM1 resulted in greater inhibition by 10 μg/ml of fluconazole than for the wild-type isolate (Fig. 2D). These results demonstrate that disruption of UPC2A greatly influences the ability of C. glabrata to grow in the presence of fluconazole.
FIG 2.

Enhanced activity of fluconazole in an SDD clinical isolate, SM1, with a disruption of UPC2A. (A) Minimum fungicidal concentrations were determined by spotting cultures on solid media after 48 h of growth in liquid RPMI medium. The MFC is the lowest concentration of fluconazole that inhibited growth completely when the strain was transferred to solid medium. (B) Broth microdilution assays were conducted according to CLSI guidelines. As a measure of the ability of the strains to regrow, the plates were allowed to incubate for 72 h prior to measurement of the optical density at 600 nm. (C) Serial dilutions of the indicated strains were incubated for 24 or 72 h on YPD solid media containing 0, 5, or 10 μg/ml of fluconazole. (D) Time-kill curves for UPC2A disruptants and revertants of a susceptible clinical isolate, SM1, treated with 10 μg/ml of fluconazole (open symbols) or a DMSO control (closed symbols). Cultures were grown in RPMI medium, with aliquots removed at the indicated time points and plated on potato dextrose agar for colony counts.
UPC2A is essential for resistance to sterol biosynthesis inhibitors in an azole-resistant clinical isolate of C. glabrata.
In order to determine if UPC2A or UPC2B influences resistance to SBIs, we disrupted these genes in a clinical isolate that exhibits high-level azole resistance due to overexpression of the CDR1, PDH1, and SNQ2 transporter genes as a result of an activating mutation in the transcription factor gene PDR1. The resulting isolate, SM3, was collected from the same patient as isolate SM1 after azole treatment failure and is described in detail elsewhere (12, 19). SM3 exhibited elevated MICs compared to those of SM1 for fluconazole, itraconazole, voriconazole, ketoconazole, and miconazole, as well as lovastatin and terbinafine. As was observed with isolate SM1, disruption of UPC2A alone or in combination with UPC2B resulted in increased susceptibility to all azoles and sterol biosynthesis inhibitors tested (Table 4). Indeed, disruption of UPC2A reduced the susceptibility of fluconazole to that observed in the wild-type isolate SM1, 8 μg/ml. Fluconazole MFCs of the strains lacking UPC2A decreased to 16 μg/ml, compared to >256 μg/ml for isolate SM3 and the revertants (Fig. 3A). After 72 h in RPMI medium, growth of this strain was unable to resume at a fluconazole concentration of ≥16 μg/ml (Fig. 3B). When serial dilutions of UPC2A disruptant strains were spotted on solid YPD medium containing 10 μg/ml of fluconazole, growth was inhibited (Fig. 3C). The growth inhibition was maintained after incubation for 72 h. By time-kill analysis, fluconazole concentrations of 10 μg/ml had no effect on isolate SM3 after 24 h, whereas disruption of UPC2A resulted in approximately a log fold reduction in growth after 24 h of fluconazole treatment (Fig. 3D). This was roughly equivalent to the effect of fluconazole on isolate SM1. These results demonstrate that UPC2A is essential for high-level fluconazole resistance in C. glabrata.
TABLE 4.
Broth microdilution susceptibility testing of the resistant clinical isolate SM3, mutant strains with disruptions of UPC2, and a revertant
| Strain | MIC (μg/ml)a |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| FLC | ITC | VRC | KTC | MC | AMB | LOVA | TRB | ANID | |
| SM3 | 256 | 8 | 8 | 8 | 2 | 2 | 200 | 512 | 0.016 |
| SM3Δupc2A | 8 | 0.125 | 0.25 | 0.25 | 0.062 | 2 | 25 | 32 | 0.016 |
| SM3Δupc2B | 256 | 16 | 4 | 8 | 2 | 2 | 256 | 512 | 0.016 |
| SM3Δupc2AΔupc2B | 8 | 0.25 | 0.25 | 0.25 | 0.062 | 2 | 64 | 16 | 0.016 |
| SM3Δupc2A/UPC2A | 256 | 8 | 8 | 8 | 2 | 2 | 200 | 512 | 0.016 |
FLC, fluconazole; ITC, itraconazole; VRC, voriconazole; KTC, ketoconazole; MC, miconazole; AMB, amphotericin B; LOVA, lovastatin; TRB, terbinafine; ANID, anidulafungin.
FIG 3.

Enhanced activity of fluconazole in a resistant clinical isolate, SM3, with a disruption of UPC2A. (A) Minimum fungicidal concentrations were determined by spotting cultures on solid media after 48 h of growth in liquid RPMI medium. The MFC is the lowest concentration of fluconazole that inhibited growth completely when the strain was transferred to solid medium. (B) Broth microdilution assays conducted according to CLSI guidelines. As a measure of the ability of the strains to regrow, the plates were allowed to incubate for 72 h prior to measurement of the optical density at 600 nm. (C) Serial dilutions of the indicated strains were incubated for 24 or 72 h on YPD solid media containing 0, 5, or 10 μg/ml of fluconazole. (D) Time-kill curves for UPC2A disruptants and revertants of a resistant clinical isolate, SM3, treated with 10 μg/ml of fluconazole (open symbols) or a DMSO control (closed symbols). Cultures were grown in RPMI media, with aliquots removed at the indicated time points and plated on potato dextrose agar for colony counts.
Disruption of UPC2A does not affect expression of PDR1, CDR1, SNQ2, or PDH1 in an azole-resistant clinical isolate of C. glabrata.
We observed that disruption of UPC2A increased susceptibility to fluconazole not only in clinical isolate SM1 but also in matched azole-resistant isolate SM3, which carries an activating mutation in PDR1 and overexpresses CDR1 and PDH1, as well as PDR1 itself. Recently, CaUpc2 was shown to bind in vivo to the promoter of CDR1, suggesting that C. glabrata Cdr1 may also be a direct target of CgUpc2A (45, 46). Those authors found that CaUpc2 is involved in maintaining baseline expression of CDR1 in C. albicans and observed a slight decrease in CDR1 expression in the absence of UPC2.
To determine what role, if any, the deletion of UPC2A in C. glabrata has on the expression of the efflux pump genes CDR1, PDH1, SNQ2, and PDR1, we examined the expression of these genes by qRT-PCR in strains SM1 and SM3, as well as their respective mutants with disruptions of UPC2A and the revertants grown for 3 h with or without 32 μg/ml of fluconazole. Disruption of UPC2A had no effect on the expression of these genes in the absence of fluconazole (Fig. 4). However, in fluconazole-treated cells, we did observe a reduction in the upregulation of all of the genes tested in the SM1Δupc2A mutant strain compared to the wild type (Fig. 4). Moreover, disruption of UPC2A in isolate SM3 had no effect on the constitutive overexpression of CDR1, PDH1, or SNQ2, suggesting that the reduced azole resistance observed in SM3 upon disruption of UPC2A is independent of these transporters.
FIG 4.

Effect of UPC2A disruption on constitutive and fluconazole-induced gene expression. RNA was isolated from mid-log-phase cultures of parent clinical isolates, Δupc2A mutants, and revertants, and qRT-PCR analysis of relative gene expression was performed. Light bars represent untreated controls and dark bars represent treatment with 32 μg/ml of fluconazole for 3 h. (A) PDR1; (B) CDR1; (C) SNQ2; (D) PDH1. Data are shown as means ± SEs (n = 3).
Disruption of UPC2A results in a reduction in ergosterol levels in azole-SDD and -resistant C. glabrata isolates.
Upc2 has previously been shown to be involved in the regulation of sterol biosynthesis and uptake in both S. cerevisiae and C. albicans (17, 27). To determine whether the observed decrease in susceptibility to SBIs corresponds to a decrease in ergosterol biosynthesis, we determined the effect of disruption of UPC2A and UPC2B on total ergosterol content in clinical isolates SM1 and SM3. Sterols were extracted from the mutant and parent strains and measured at an absorption spectrum between 240 and 300 nm as described previously by Arthington-Skaggs et al., who demonstrated a direct correlation between ergosterol content and azole susceptibility in S. cerevisiae (41). As shown in Fig. 5, disruption of UPC2A alone or in combination with UPC2B resulted in a reduction in ergosterol levels in both azole-SDD isolate SM1 and azole-resistant isolate SM3. These results suggest that ergosterol biosynthesis is impaired in both SDD and resistant isolates of C. glabrata in the absence of UPC2A.
FIG 5.

Ergosterol quantification for UPC2A disruptants in an SDD clinical isolate, SM1 (A, C, and E), and matched resistant isolate SM3 (B, D, and F). Genotypes studied were as follows: wild-type UPC2A allele (solid circles), Δupc2a (open squares) (A and B), Δupc2b (open triangles) (C and D), and Δupc2a Δupc2b (open diamonds) (E and F). The results for the UPC2A disruptants shown in panels A, B, E, and F showed statistically significantly lower ergosterol content than was found in the wild type (P < 0.05). Knockout strains with the wild-type allele complemented back in showed no difference in ergosterol content compared to those of the wild-type isolates (data not shown). Strains were grown in RPMI medium for 16 h, followed by a heptane extraction and spectrophotometric scan between 240 and 300 nm (41). Each isolate was extracted and analyzed in three independent experiments.
UPC2 is essential for both baseline and sterol biosynthesis inhibitor-induced expression of ergosterol biosynthesis genes.
Disruption of UPC2A resulted in increased susceptibility to SBIs and a significant decrease in total ergosterol content. In order to determine if disruption of UPC2A influences baseline expression of genes involved in sterol biosynthesis, 13 ERG genes were examined by qRT-PCR. Disruption of UPC2A in both SM1 and SM3 resulted in ≥2-fold downregulation of all ERG genes examined. Reintroduction of UPC2A into its native locus partially or completely restored expression of most ERG genes (Fig. 6). There was no significant difference between SM1 and SM3 in expression of any of the ERG genes tested.
FIG 6.
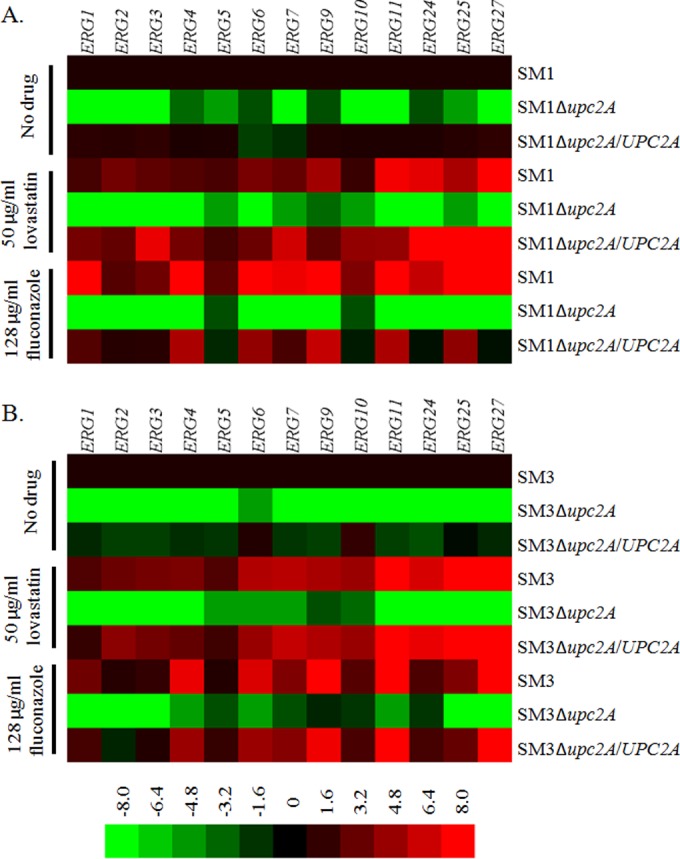
Effect of UPC2A disruption on both baseline and sterol biosynthesis inhibitor-induced expression of ergosterol biosynthesis genes. RNA was isolated from mid-log-phase cultures in parent clinical isolates, Δupc2A mutants, and revertants. qRT-PCR analysis of relative gene expression was performed. (A) SM1, Δupc2A mutant, and revertant gene expression. Gene expression was normalized to that in the untreated SM1 sample for each ergosterol biosynthesis gene. (B) SM3, Δupc2A mutant, and revertant gene expression. Gene expression was normalized to that in the untreated SM3 sample for each ergosterol biosynthesis gene.
Multiple fungal pathogens, including C. glabrata, upregulate ergosterol biosynthesis genes in response to treatment with SBIs, which likely contributes to the decreased susceptibility observed with these agents (24, 47). We used qRT-PCR to determine the effects of SBI treatment on strains with disruptions of UPC2A. Expression of ERG genes was measured for SM1 and SM3, as well as their respective UPC2A mutants and revertants, grown for 2 h in the presence or absence of 50 μg/ml of lovastatin or 128 μg/ml of fluconazole.
Similar to the case with S. cerevisiae (48), treatment of isolates SM1 and SM3 with lovastatin resulted in ≥2-fold upregulation of 12 of the 13 ERG genes examined in SM1 and all of the ERG genes examined in SM3. Treatment with fluconazole resulted in upregulation of all of these genes in SM1 and 10 of the 13 ERG genes examined in SM3. In the absence of UPC2A, lovastatin and fluconazole were unable to induce the expression of any of these ERG genes. Moreover, the majority of the 13 ERG genes were downregulated ≥2-fold even in the presence of drug treatment. In most cases, reintroduction of UPC2A into its native locus in both SM1 and SM3 restored the ability of SBI treatment to induce ERG gene expression (Fig. 6). These data demonstrate that UPC2A is required for both baseline and SBI induction of ERG gene expression in C. glabrata.
DISCUSSION
Despite the more recent development of the lipid-based formulations of amphotericin B and the echinocandins, fluconazole remains the most prescribed antifungal agent for the treatment of Candida infections. It is inexpensive, available as both intravenous and oral formulations, and associated with minimal adverse effects. Although its use is complicated by many drug-drug interactions, these are well understood and somewhat predictable. Indeed, fluconazole maintains excellent activity against many common and clinically important Candida species, such as C. albicans, C. parapsilosis, and C. tropicalis (49). Of particular concern, however, is C. glabrata, which has emerged as the second leading cause of candidiasis in the United States and which exhibits intrinsically low susceptibility to the azole antifungals (3, 50–55).
Furthermore, the development of high-level resistance to the azoles is common in C. glabrata and has been well documented for isolates from head and neck radiation patients, stem cell transplant patients, and human immunodeficiency virus (HIV) patients and for vulvovaginal candidiasis (11, 13, 39, 56, 57). The development of azole resistance has also been implicated in the fluconazole treatment failure and death of a patient suffering from C. glabrata candidemia (12). Such resistance appears to be almost exclusively due to activating mutations in the gene encoding the transcription factor Pdr1, leading to overexpression of multidrug resistance transporters Cdr1, Pdh1, and Snq2 (58–60). Strategies to preserve this antifungal class for use against C. glabrata by both enhancing activity and overcoming resistance are needed.
The azoles exert their antifungal action by binding to and inhibiting the activity of the cytochrome P450 enzyme, lanosterol demethylase, which is a key enzyme in the ergosterol biosynthesis pathway that is encoded by ERG11. In both S. cerevisiae and C. albicans, the zinc cluster transcription factor Upc2 has been shown to be a central regulator of not only ERG11 but also additional genes in the ergosterol biosynthesis pathway. When lanosterol demethylase is inhibited genetically or pharmacologically in C. glabrata, alternate sterols and toxic sterol precursors such as lanosterol, 4,14α-dimethyl zymosterol, and 14α-methyl ergosta 8,24(28)-dien-3β,6α-diol are produced, leading to reduced growth (31, 32) (Fig. 1). In S. cerevisiae, C. albicans, and C. glabrata, Upc2 has been shown to be a central regulator of baseline and inducible expression of the genes encoding enzymes of the ergosterol biosynthesis pathway (15, 24, 27, 29, 61). Moreover, disruption of UPC2 has been shown to increase the susceptibility of these organisms to the azole antifungals (24, 61, 62).
Given its global influence on sterol biosynthesis, we hypothesized that disruption of Upc2 might enhance the activity of the azole antifungals against azole-resistant as well as azole-SDD clinical isolates of C. glabrata. We have recently characterized a matched pair of azole-SDD and -resistant C. glabrata clinical isolates in which high-level azole resistance developed during fluconazole treatment (19). The resistant isolate in the pair carries an activating mutation in PDR1 leading to the overexpression of CDR1, PDH1, and SNQ2 and an increase in fluconazole MIC from 8 μg/ml to 256 μg/ml. Interestingly, disruption of UPC2A led to not only a reduction in the fluconazole MIC from 8 μg/ml to 0.5 μg/ml but also a reduction in the fluconazole MFC from >64 μg/ml to 1 μg/ml in the SDD isolate. Similar reductions in MIC were observed for other azoles and the SBIs terbinafine and lovastatin, but not for amphotericin B or the echinocandin anidulafungin, which act directly on ergosterol in the cell membrane or on the cell wall, respectively. Despite the reduction in ergosterol content, we expected no change in amphotericin B susceptibility based on previous findings for C. albicans and C. glabrata (24, 29, 63). We agree with the hypothesis of Silver et al. that this could potentially be due to the amount of remaining ergosterol being above the threshold necessary for amphotericin B to cause cell death (29).
As fluconazole is the most commonly prescribed azole antifungal, we examined its activity against these strains more closely. We used a 72-h endpoint in a broth microdilution assay to assess the ability of the organism to resume growth in the presence of fluconazole. Disruption of UPC2A greatly impeded the ability of isolate SM1 to resume growth in even relatively low concentrations of the drug. Likewise, when spotted on YPD plates containing either 5 or 10 μg/ml of fluconazole, isolate SM1 was unable to grow when UPC2A was disrupted. Similar results were observed by time-kill analysis in RPMI medium, in which isolate SM1 exhibited modest growth at 24 h in the presence of 10 μg/ml of fluconazole, whereas growth was inhibited in the absence of UPC2A. These results further demonstrate that UPC2A is required for optimal azole activity against C. glabrata.
We then examined the azole-resistant clinical isolate SM3 under the same conditions and observed remarkably similar results. Disruption of UPC2A led to increased susceptibility to fluconazole, resulting in an MIC equal to that of SDD isolate SM1. Moreover, the MFC for fluconazole was reduced from >256 μg/ml to 16 μg/ml. Disruption of UPC2A resulted in similar effects on susceptibility to other azoles and sterol biosynthesis inhibitors. Growth in the presence of fluconazole was likewise found to be greatly reduced when measured at 72 h in liquid RPMI medium, on YPD plates containing 10 μg/ml of fluconazole and by time-kill analysis at 24 h in 10 μg/ml of fluconazole. These results demonstrate that UPC2A is required for high-level azole resistance in C. glabrata.
One possible explanation for the increased fluconazole susceptibility observed in these isolates is a change in expression of the genes encoding the multidrug resistance transporters Cdr1, Pdh1, and Snq2. Indeed, it has been shown that in C. albicans, Upc2 binds the promoter of CDR1, suggesting that it may contribute to its regulation (30). Interestingly, the sterol response element recognized by Upc2, as defined by Znaidi et al., is present in the promoter region of CDR1 in C. glabrata as well (30). It has also been shown that fluconazole treatment activates the transcription factor Pdr1 and induces the expression of its targets, multidrug transporter genes (64). We therefore examined the influence of UPC2A on both baseline and inducible expression of PDR1, CDR1, PDH1, and SNQ2 in isolates SM1 and SM3. Disruption of UPC2A had no effect on baseline expression of these genes in isolate SM1 or on their constitutive overexpression in isolate SM3. Interestingly, disruption of UPC2A reduced the inducible expression of all four genes in response to fluconazole. This suggests that Upc2A is required for optimal activation of the Pdr1 transcriptional pathway. The diminished capacity of isolate SM1 to respond to fluconazole stress by upregulating these transporter genes may contribute to its increased susceptibility to the azoles and other sterol biosynthesis inhibitors.
Another possible explanation for the increased fluconazole susceptibility observed in these isolates is a change in the expression of genes involved in ergosterol biosynthesis that are regulated by UPC2A. Both SM1 and SM3 exhibited reduced cellular ergosterol when UPC2A was disrupted. Likewise, baseline expression as well as inducible expression by fluconazole or lovastatin of all ergosterol biosynthesis genes analyzed was reduced in the absence of UPC2A. Importantly, the azole target, ERG11, is included in this group of ergosterol biosynthesis genes, which would result in a reduced amount of target that would need to be inhibited in order to effectively inhibit ergosterol biosynthesis. These data suggest that the global impact on sterol biosynthesis in the absence of UPC2A greatly increases the vulnerability of C. glabrata to fluconazole, even in the setting of Pdr1 activation and overexpression of CDR1, PDH1, and SNQ2.
Additionally, the incorporation of alternate sterols into the cell membrane affects fluidity of the membrane and may compromise the ability of the efflux pumps to function properly. Krishnamurthy and Prasad demonstrated altered accumulation of various compounds when tested against a panel of S. cerevisiae ERG mutants expressing C. albicans Cdr1 (65). Altered membrane fluidity and asymmetry have also been associated with increased resistance to fluconazole in C. albicans independent of efflux pump function (66). Taken together, our findings suggest that Upc2A and the transcriptional activation pathway it regulates represent potential targets for overcoming azole antifungal resistance in C. glabrata.
Supplementary Material
ACKNOWLEDGMENTS
We thank Shelley Magill for providing the clinical isolates used in this study. We thank Qing Zhang for her valuable technical support in the laboratory.
This work was supported by NIH NIAID grant R01AI058145 (to P.D.R.) and a grant from the Children's Foundation Research Institute (to K.E.C.).
Footnotes
Published ahead of print 27 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02217-13.
REFERENCES
- 1.Odds FC. 1988. Candida and candidosis: a review and bibliography, 2nd ed. Bailliere Tindall, London, United Kingdom [Google Scholar]
- 2.Ostrosky-Zeichner L, Rex JH, Pappas PG, Hamill RJ, Larsen RA, Horowitz HW, Powderly WG, Hyslop N, Kauffman CA, Cleary J, Mangino JE, Lee J. 2003. Antifungal susceptibility survey of 2,000 bloodstream Candida isolates in the United States. Antimicrob. Agents Chemother. 47:3149–3154. 10.1128/AAC.47.10.3149-3154.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfaller MA, Messer SA, Hollis RJ, Jones RN, Doern GV, Brandt ME, Hajjeh RA. 1999. Trends in species distribution and susceptibility to fluconazole among blood stream isolates of Candida species in the United States. Diagn. Microbiol. Infect. Dis. 33:217–222. 10.1016/S0732-8893(98)00160-6 [DOI] [PubMed] [Google Scholar]
- 4.Bader MS, Lai SM, Kumar V, Hinthorn D. 2004. Candidemia in patients with diabetes mellitus: epidemiology and predictors of mortality. Scand. J. Infect. Dis. 36:860–864. 10.1080/00365540410021126 [DOI] [PubMed] [Google Scholar]
- 5.Dan M, Segal R, Marder V, Leibovitz A. 2006. Candida colonization of the vagina in elderly residents of a long-term-care hospital. Eur. J. Clin. Microbiol. Infect. Dis. 25:394–396. 10.1007/s10096-006-0150-y [DOI] [PubMed] [Google Scholar]
- 6.Goswami D, Goswami R, Banerjee U, Dadhwal V, Miglani S, Lattif AA, Kochupillai N. 2006. Pattern of Candida species isolated from patients with diabetes mellitus and vulvovaginal candidiasis and their response to single dose oral fluconazole therapy. J. Infect. 52:111–117. 10.1016/j.jinf.2005.03.005 [DOI] [PubMed] [Google Scholar]
- 7.Vermitsky JP, Self MJ, Chadwick SG, Trama JP, Adelson ME, Mordechai E, Gygax SE. 2008. Survey of vaginal-flora Candida species isolates from women of different age groups by use of species-specific PCR detection. J. Clin. Microbiol. 46:1501–1503. 10.1128/JCM.02485-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andes DR, Safdar N, Baddley JW, Playford G, Reboli AC, Rex JH, Sobel JD, Pappas PG, Kullberg BJ, Mycoses Study Group 2012. Impact of treatment strategy on outcomes in patients with candidemia and other forms of invasive candidiasis: a patient-level quantitative review of randomized trials. Clin. Infect. Dis. 54:1110–1122. 10.1093/cid/cis021 [DOI] [PubMed] [Google Scholar]
- 9.Almirante B, Rodriguez D, Park BJ, Cuenca-Estrella M, Planes AM, Almela M, Mensa J, Sanchez F, Ayats J, Gimenez M, Saballs P, Fridkin SK, Morgan J, Rodriguez-Tudela JL, Warnock DW, Pahissa A, Barcelona Candidemia Project Study Group 2005. Epidemiology and predictors of mortality in cases of Candida bloodstream infection: results from population-based surveillance, Barcelona, Spain, from 2002 to 2003. J. Clin. Microbiol. 43:1829–1835. 10.1128/JCM.43.4.1829-1835.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gudlaugsson O, Gillespie S, Lee K, Vande Berg J, Hu J, Messer S, Herwaldt L, Pfaller M, Diekema D. 2003. Attributable mortality of nosocomial candidemia, revisited. Clin. Infect. Dis. 37:1172–1177. 10.1086/378745 [DOI] [PubMed] [Google Scholar]
- 11.Gygax SE, Vermitsky JP, Chadwick SG, Self MJ, Zimmerman JA, Mordechai E, Adelson ME, Trama JP. 2008. Antifungal resistance of Candida glabrata vaginal isolates and development of a quantitative reverse transcription-PCR-based azole susceptibility assay. Antimicrob. Agents Chemother. 52:3424–3426. 10.1128/AAC.00462-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Magill SS, Shields C, Sears CL, Choti M, Merz WG. 2006. Triazole cross-resistance among Candida spp.: case report, occurrence among bloodstream isolates, and implications for antifungal therapy. J. Clin. Microbiol. 44:529–535. 10.1128/JCM.44.2.529-535.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Redding SW, Kirkpatrick WR, Saville S, Coco BJ, White W, Fothergill A, Rinaldi M, Eng T, Patterson TF, Lopez-Ribot J. 2003. Multiple patterns of resistance to fluconazole in Candida glabrata isolates from a patient with oropharyngeal candidiasis receiving head and neck radiation. J. Clin. Microbiol. 41:619–622. 10.1128/JCM.41.2.619-622.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vermitsky JP, Edlind TD. 2004. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob. Agents Chemother. 48:3773–3781. 10.1128/AAC.48.10.3773-3781.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies BS, Wang HS, Rine J. 2005. Dual activators of the sterol biosynthetic pathway of Saccharomyces cerevisiae: similar activation/regulatory domains but different response mechanisms. Mol. Cell. Biol. 25:7375–7385. 10.1128/MCB.25.16.7375-7385.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song JL, Harry JB, Eastman RT, Oliver BG, White TC. 2004. The Candida albicans lanosterol 14-alpha-demethylase (ERG11) gene promoter is maximally induced after prolonged growth with antifungal drugs. Antimicrob. Agents Chemother. 48:1136–1144. 10.1128/AAC.48.4.1136-1144.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White TC, Silver PM. 2005. Regulation of sterol metabolism in Candida albicans by the UPC2 gene. Biochem. Soc. Trans. 33:1215–1218. 10.1042/BST20051215 [DOI] [PubMed] [Google Scholar]
- 18.Akache B, Wu K, Turcotte B. 2001. Phenotypic analysis of genes encoding yeast zinc cluster proteins. Nucleic Acids Res. 29:2181–2190. 10.1093/nar/29.10.2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caudle KE, Barker KS, Wiederhold NP, Xu L, Homayouni R, Rogers PD. 2011. Genomewide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot. Cell 10:373–383. 10.1128/EC.00073-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhauser J, Rogers PD. 2008. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot. Cell 7:1180–1190. 10.1128/EC.00103-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heilmann CJ, Schneider S, Barker KS, Rogers PD, Morschhauser J. 2010. An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans. Antimicrob. Agents Chemother. 54:353–359. 10.1128/AAC.01102-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoehamer CF, Cummings ED, Hilliard GM, Morschhauser J, Rogers PD. 2009. Upc2p-associated differential protein expression in Candida albicans. Proteomics 9:4726–4730. 10.1002/pmic.200900176 [DOI] [PubMed] [Google Scholar]
- 23.Hoot SJ, Smith AR, Brown RP, White TC. 2011. An A643V amino acid substitution in Upc2p contributes to azole resistance in well-characterized clinical isolates of Candida albicans. Antimicrob. Agents Chemother. 55:940–942. 10.1128/AAC.00995-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagi M, Nakayama H, Tanabe K, Bard M, Aoyama T, Okano M, Higashi S, Ueno K, Chibana H, Niimi M, Yamagoe S, Umeyama T, Kajiwara S, Ohno H, Miyazaki Y. 2011. Transcription factors CgUPC2A and CgUPC2B regulate ergosterol biosynthetic genes in Candida glabrata. Genes Cells 16:80–89. 10.1111/j.1365-2443.2010.01470.x [DOI] [PubMed] [Google Scholar]
- 25.White TC, Marr KA, Bowden RA. 1998. Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin. Microbiol. Rev. 11:382–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shianna KV, Dotson WD, Tove S, Parks LW. 2001. Identification of a UPC2 homolog in Saccharomyces cerevisiae and its involvement in aerobic sterol uptake. J. Bacteriol. 183:830–834. 10.1128/JB.183.3.830-834.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vik A, Rine J. 2001. Upc2p and Ecm22p, dual regulators of sterol biosynthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 21:6395–6405. 10.1128/MCB.21.19.6395-6405.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. 2005. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob. Agents Chemother. 49:1745–1752. 10.1128/AAC.49.5.1745-1752.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silver PM, Oliver BG, White TC. 2004. Role of Candida albicans transcription factor Upc2p in drug resistance and sterol metabolism. Eukaryot. Cell 3:1391–1397. 10.1128/EC.3.6.1391-1397.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Znaidi S, Weber S, Al-Abdin OZ, Bomme P, Saidane S, Drouin S, Lemieux S, De Deken X, Robert F, Raymond M. 2008. Genomewide location analysis of Candida albicans Upc2p, a regulator of sterol metabolism and azole drug resistance. Eukaryot. Cell 7:836–847. 10.1128/EC.00070-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geber A, Hitchcock CA, Swartz JE, Pullen FS, Marsden KE, Kwon-Chung KJ, Bennett JE. 1995. Deletion of the Candida glabrata ERG3 and ERG11 genes: effect on cell viability, cell growth, sterol composition, and antifungal susceptibility. Antimicrob. Agents Chemother. 39:2708–2717. 10.1128/AAC.39.12.2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hull CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AG, Kelly DE, Kelly SL. 2012. Facultative sterol uptake in an ergosterol-deficient clinical isolate of Candida glabrata harboring a missense mutation in ERG11 and exhibiting cross-resistance to azoles and amphotericin B. Antimicrob. Agents Chemother. 56:4223–4232. 10.1128/AAC.06253-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reuss O, Vik A, Kolter R, Morschhauser J. 2004. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341:119–127. 10.1016/j.gene.2004.06.021 [DOI] [PubMed] [Google Scholar]
- 34.Amberg DC, Burke DJ, Strathern JN. 2006. Isolation of yeast genomic DNA for Southern blot analysis. CSH Protoc. 10.1101/pdb.prot4149 [DOI] [PubMed] [Google Scholar]
- 35.CLSI. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard M27-A3, 3rd ed. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 36.Epp E, Vanier G, Harcus D, Lee AY, Jansen G, Hallett M, Sheppard DC, Thomas DY, Munro CA, Mullick A, Whiteway M. 2010. Reverse genetics in Candida albicans predicts ARF cycling is essential for drug resistance and virulence. PLoS Pathog. 6:e1000753. 10.1371/journal.ppat.1000753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Onyewu C, Blankenship JR, Del Poeta M, Heitman J. 2003. Ergosterol biosynthesis inhibitors become fungicidal when combined with calcineurin inhibitors against Candida albicans, Candida glabrata, and Candida krusei. Antimicrob. Agents Chemother. 47:956–964. 10.1128/AAC.47.3.956-964.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Espinel-Ingroff A. 1998. Comparison of in vitro activities of the new triazole SCH56592 and the echinocandins MK-0991 (L-743,872) and LY303366 against opportunistic filamentous and dimorphic fungi and yeasts. J. Clin. Microbiol. 36:2950–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. 1999. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 43:2753–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klepser ME, Ernst EJ, Lewis RE, Ernst ME, Pfaller MA. 1998. Influence of test conditions on antifungal time-kill curve results: proposal for standardized methods. Antimicrob. Agents Chemother. 42:1207–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arthington-Skaggs BA, Jradi H, Desai T, Morrison CJ. 1999. Quantitation of ergosterol content: novel method for determination of fluconazole susceptibility of Candida albicans. J. Clin. Microbiol. 37:3332–3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmitt ME, Brown TA, Trumpower BL. 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 18:3091–3092. 10.1093/nar/18.10.3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu TT, Lee RE, Barker KS, Lee RE, Wei L, Homayouni R, Rogers PD. 2005. Genome-wide expression profiling of the response to azole, polyene, echinocandin, and pyrimidine antifungal agents in Candida albicans. Antimicrob. Agents Chemother. 49:2226–2236. 10.1128/AAC.49.6.2226-2236.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azie N, Neofytos D, Pfaller M, Meier-Kriesche HU, Quan SP, Horn D. 2012. The PATH (Prospective Antifungal Therapy) Alliance(R) registry and invasive fungal infections: update 2012. Diagn. Microbiol. Infect. Dis. 73:293–300. 10.1016/j.diagmicrobio.2012.06.012 [DOI] [PubMed] [Google Scholar]
- 45.Hoot SJ, Brown RP, Oliver BG, White TC. 2010. The UPC2 promoter in Candida albicans contains two cis-acting elements that bind directly to Upc2p, resulting in transcriptional autoregulation. Eukaryot. Cell 9:1354–1362. 10.1128/EC.00130-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliver BG, Song JL, Choiniere JH, White TC. 2007. cis-Acting elements within the Candida albicans ERG11 promoter mediate the azole response through transcription factor Upc2p. Eukaryot. Cell 6:2231–2239. 10.1128/EC.00331-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henry KW, Nickels JT, Edlind TD. 2000. Upregulation of ERG genes in Candida species by azoles and other sterol biosynthesis inhibitors. Antimicrob. Agents Chemother. 44:2693–2700. 10.1128/AAC.44.10.2693-2700.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith SJ, Crowley JH, Parks LW. 1996. Transcriptional regulation by ergosterol in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 16:5427–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pfaller MA, Messer SA, Woosley LN, Jones RN, Castanheira M. 2013. Echinocandin and triazole antifungal susceptibility profiles for clinical opportunistic yeast and mold isolates collected from 2010 to 2011: application of new CLSI clinical breakpoints and epidemiological cutoff values for characterization of geographic and temporal trends of antifungal resistance. J. Clin. Microbiol. 51:2571–2581. 10.1128/JCM.00308-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bodey GP, Mardani M, Hanna HA, Boktour M, Abbas J, Girgawy E, Hachem RY, Kontoyiannis DP, Raad II. 2002. The epidemiology of Candida glabrata and Candida albicans fungemia in immunocompromised patients with cancer. Am. J. Med. 112:380–385. 10.1016/S0002-9343(01)01130-5 [DOI] [PubMed] [Google Scholar]
- 51.Ghannoum MA, Rice LB. 1999. Antifungal agents: mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin. Microbiol. Rev. 12:501–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pfaller MA, Jones RN, Doern GV, Sader HS, Hollis RJ, Messer SA. 1998. International surveillance of bloodstream infections due to Candida species: frequency of occurrence and antifungal susceptibilities of isolates collected in 1997 in the United States, Canada, and South America for the SENTRY Program. The SENTRY Participant Group. J. Clin. Microbiol. 36:1886–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuman P, Sobel JD, Ohmit SE, Mayer KH, Carpenter CC, Rompalo A, Duerr A, Smith DK, Warren D, Klein RS. 1998. Mucosal candidal colonization and candidiasis in women with or at risk for human immunodeficiency virus infection. HIV Epidemiology Research Study (HERS) Group. Clin. Infect. Dis. 27:1161–1167 [DOI] [PubMed] [Google Scholar]
- 54.Trick WE, Fridkin SK, Edwards JR, Hajjeh RA, Gaynes RP, National Nosocomial Infections Surveillance System Hospitals 2002. Secular trend of hospital-acquired candidemia among intensive care unit patients in the United States during 1989–1999. Clin. Infect. Dis. 35:627–630. 10.1086/342300 [DOI] [PubMed] [Google Scholar]
- 55.Vazquez JA, Sobel JD, Peng G, Steele-Moore L, Schuman P, Holloway W, Neaton JD. 1999. Evolution of vaginal Candida species recovered from human immunodeficiency virus-infected women receiving fluconazole prophylaxis: the emergence of Candida glabrata? Terry Beirn Community Programs for Clinical Research in AIDS (CPCRA). Clin. Infect. Dis. 28:1025–1031 [DOI] [PubMed] [Google Scholar]
- 56.Bennett JE, Izumikawa K, Marr KA. 2004. Mechanism of increased fluconazole resistance in Candida glabrata during prophylaxis. Antimicrob. Agents Chemother. 48:1773–1777. 10.1128/AAC.48.5.1773-1777.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richter SS, Galask RP, Messer SA, Hollis RJ, Diekema DJ, Pfaller MA. 2005. Antifungal susceptibilities of Candida species causing vulvovaginitis and epidemiology of recurrent cases. J. Clin. Microbiol. 43:2155–2162. 10.1128/JCM.43.5.2155-2162.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, Sanglard D. 2009. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 5:e1000268. 10.1371/journal.ppat.1000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai HF, Krol AA, Sarti KE, Bennett JE. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 50:1384–1392. 10.1128/AAC.50.4.1384-1392.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD, Rogers PD. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol. Microbiol. 61:704–722. 10.1111/j.1365-2958.2006.05235.x [DOI] [PubMed] [Google Scholar]
- 61.Hoot SJ, Oliver BG, White TC. 2008. Candida albicans UPC2 is transcriptionally induced in response to antifungal drugs and anaerobicity through Upc2p-dependent and -independent mechanisms. Microbiology 154:2748–2756. 10.1099/mic.0.2008/017475-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barker KS, Pearson MM, Rogers PD. 2003. Identification of genes differentially expressed in association with reduced azole susceptibility in Saccharomyces cerevisiae. J. Antimicrob. Chemother. 51:1131–1140. 10.1093/jac/dkg217 [DOI] [PubMed] [Google Scholar]
- 63.Flowers SA, Barker KS, Berkow EL, Toner G, Chadwick SG, Gygax SE, Morschhauser J, Rogers PD. 2012. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot. Cell 11:1289–1299. 10.1128/EC.00215-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thakur JK, Arthanari H, Yang F, Pan SJ, Fan X, Breger J, Frueh DP, Gulshan K, Li DK, Mylonakis E, Struhl K, Moye-Rowley WS, Cormack BP, Wagner G, Naar AM. 2008. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452:604–609. 10.1038/nature06836 [DOI] [PubMed] [Google Scholar]
- 65.Krishnamurthy SS, Prasad R. 1999. Membrane fluidity affects functions of Cdr1p, a multidrug ABC transporter of Candida albicans. FEMS Microbiol. Lett. 173:475–481. 10.1016/S0378-1097(99)00237-2 [DOI] [PubMed] [Google Scholar]
- 66.Kohli A, Smriti Mukhopadhyay K, Rattan A, Prasad R. 2002. In vitro low-level resistance to azoles in Candida albicans is associated with changes in membrane lipid fluidity and asymmetry. Antimicrob. Agents Chemother. 46:1046–1052. 10.1128/AAC.46.4.1046-1052.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.