

Abstract
Determination of progesterone receptor (PR) status in hormone-dependent diseases is essential in ascertaining disease prognosis and monitoring treatment response. The development of a noninvasive means of monitoring these processes would have significant impact on early detection, cost, repeated measurements, and personalized treatment options. Magnetic resonance imaging (MRI) is widely recognized as a technique that can produce longitudinal studies, and PR-targeted MR probes may address a clinical problem by providing contrast enhancement that reports on PR status without biopsy. Commercially available MR contrast agents are typically delivered via intravenous injection, whereas steroids are administered subcutaneously. Whether the route of delivery is important for tissue accumulation of steroid-modified MRI contrast agents to PR-rich tissues is not known. To address this question, modification of the chemistry linking progesterone with the gadolinium chelate led to MR probes with increased water solubility and lower cellular toxicity and enabled administration through the blood. This attribute came at a cost through lower affinity for PR and decreased ability to cross the cell membrane, and ultimately it did not improve delivery of the PR-targeted MR probe to PR-rich tissues or tumors in vivo. Overall, these studies are important, as they demonstrate that targeted contrast agents require optimization of delivery and receptor binding of the steroid and the gadolinium chelate for optimal translation in vivo.
Introduction
Small molecule imaging probes have been extensively studied to monitor and quantify physiological processes.1,2 Steroid receptors are an example of a target for this type of probe because these proteins regulate a number of cell processes through transcriptional regulation of genes.3 The ability to noninvasively interrogate the function of these important receptors will guide the development of new therapeutic targets for hormone-dependent diseases such as endometriosis and breast, ovarian, uterine, and prostate cancers.4−6 Some commonly prescribed chemotherapeutics, such as the estrogen receptor (ER)-targeting tamoxifen, are designed to block the steroid receptor activity that facilitates tumor growth, and receptor status is frequently determined for these diseases prior to treatment.7,8 The presence of both receptors, PR and ER, in breast cancer correlates positively with patient survival rate, whereas the loss of steroid receptor expression coincides with the disease becoming more aggressive and drug resistant.9−11 Furthermore, triple-negative breast cancer, tumors that do not express PR, ER, or a third important marker, Her2, stands as the most aggressive subtype. Treatment of receptor-negative disease requires a wholly different approach when compared to that for receptor-expressing cells. Noninvasively determining receptor status may be critical in detecting new lesions, categorizing tumors, and determining when disease is becoming refractory with current treatment, thus improving disease prognosis. Due to this significant role of steroid receptors in tumor progression, these proteins represent excellent imaging targets for noninvasive molecular characterization and monitoring of cancers and benign disease states such as endometriosis.12,13
In vivo imaging of steroid receptors has been demonstrated using positron emission tomography (PET).12,14−18 However, rapid metabolism of probes by 20-hydroxysteroid dehydrogenase limited the use of these probes in human subjects.19−21 In addition, PET can be restricted by limited spatial and temporal resolution.2,19,22 In contrast, MRI has high temporal and spatial resolution without exposing patients to radiation.2 Typically, MRI uses contrast agents to increase local signal intensity by distinguishing tissues or organs that are magnetically similar but histologically distinct. These probes make use of a paramagnetic ion, Gd(III), that decreases the proton spin-lattice relaxation time (T1) of protons.23,24 This decrease in T1 is manifested as bright regions in an acquired T1-weighted MR image. Structural modification of the chelate provides the opportunity to target specific pathological processes.23,25−31
Imaging probes targeted for ER and PR have many similarities in their structure and function. The specificity for a given receptor lies in the targeting ligand that is used for synthesis. A number of steroid receptor-targeted MRI contrast agents have been developed.32−36 For example, ProGlo is a progesterone receptor (PR)-targeted agent that has shown promising cellular retention and selectivity in tissues and tumors with elevated PR expression.32 The use of steroid receptor-targeted MRI contrast agents has been limited to preclinical research. Challenges with previous generations of PR-targeted probes demonstrated the potential need for modification due to the inability to test them using intravenous (i.v.) injection, the most commonly employed method of administration for clinically used MR contrast probes.34
To determine the relationship among complex linker length and solubility, toxicity, tissue distribution, and MR contrast, we synthesized and biologically evaluated a series of PR-targeted MR imaging probes with variable linker length (Figure 1). By systematic alteration of the aliphatic linker between the Gd(III) chelate and the steroid, an increase in linker length could be correlated to an increase in hydrophobicity. The most hydrophilic complex exhibited the lowest toxicity in PR(+) human breast cancer cells. In vivo, complex 1 preferentially accumulated in tissues that expressed PR when injected either i.v. or intraperitoneally (i.p.). The highest concentration of 1 was observed in the uterus after i.p. injection, indicating that i.v. administration does not enhance PR-targeted MR probe delivery. Complex 1 was additionally found to enhance PR-rich tissues compared with muscle in T1-weighted images taken at 9.4 T. Overall, further optimization through rational probe design yielded PR-targeted contrast probes with increased water solubility and allowed for multiple routes of in vivo administration but ultimately did not appreciably alter delivery to target tissues or provide a significant advancement in contrast efficiency. These studies illustrate that optimization of a targeted MR agent should include consideration of solubility to change the route of delivery as well as steroid biology, which dictates tissue accumulation.
Figure 1.

Structures of PR-targeted contrast probes, 1–4. The alkane linker between the PR-targeting moiety and Gd(III) chelate was modified to elucidate the effect of linker length on toxicity, receptor binding, and tissue distribution in vivo.
Results
Synthesis of a Series of PR-Targeted MRI Contrast Agents with Varying Linker Lengths
Previous experiments have demonstrated that ProGlo successfully targets PR receptors in vivo while enhancing contrast in MR images of tumor xenografts.32,34 The hydrophobicity of ProGlo and solubility in aqueous media limited the method of in vivo administration.34,35 Here, we have focused on preserving the structural foundation of earlier generations of these probes while increasing solubility to determine if delivery through the blood enhances bioactivity (Figure 1).
Synthesis of complexes 1 and 4 were performed with minor modification to published procedures.32 The synthesis of 2 and 3 was initiated with the coupling of either 1,4-dibromobutane or 1,5-dibromopentane to 21-hydroxyprogesterone. In order not to inhibit the binding affinity of the steroid, attachment of the contrast probe was carried out on the D-ring (Figure S1).37−39
Relaxivity and Octanol–Water Partition Coefficients
The relaxivity of a MR contrast agent is defined by its ability to increase the relaxation rates of the surrounding water proton spins. The relaxivity values measured by the PR probes described were similar to clinically used Gd(III) contrast agents (Table 1). Octanol–water partition coefficients (logP values) were measured and reflect the hydrophobicity of the agents (Table 1). Compound 1 exhibited the most negative logP value (−1.06 ± 0.02).33,40 Not surprisingly, complex 4, which has a six carbon atom linker arm, was the most hydrophobic (logP value of 1.40 ± 0.08). Complexes 2 (four carbon linker) and 3 (five carbon linker) have intermediate logP values around 0, −0.08 ± 0.06 and 0.11 ± 0.01, respectively.
Table 1. Characterization of PR-Targeted Contrast Agents.
| agent | r1a | r2a | logP | toxicity LC50b | competitive PR binding IC50c |
|---|---|---|---|---|---|
| 1 | 5.2 ± 0.3 | 5.7 ± 0.3 | –1.06 ± 0.02 | 2.65 | 122 ± 26 |
| 2 | 6.9 ± 0.1 | 7.9 ± 0.1 | –0.08 ± 0.06 | 1.23 | 0.80 ± 0.06 |
| 3 | 6.7 ± 0.3 | 7.6 ± 0.3 | 0.11 ± 0.01 | 1.00 | 1.80 ± 0.44 |
| 4 | 6.4 ± 0.1 | 7.4 ± 0.1 | 1.40 ± 0.08 | 0.88 | 0.95 ± 0.34 |
| P4 | N.D. | N.D. | 2.88d | 0.002241 | 0.001632 |
r1 and r2 measured in mM–1 s–1 at 1.41 T, 37°.
LC50 measured in mM and determined by sinusoidal curve fitting of a dose response curve in GraphPad Prism in T47D PR(+) cells.
IC50 measure in μM and determined by the equation mP100% + (mP0% – mP100%)/(1 + 10 log (IC50 – X)), where Y = mP, X = Log [compound], mP100% = 100% inhibition, and mP0% = 0% inhibition.
Measured by shake flask method/mass.42
This trend in hydrophobicity is additionally mirrored in the HPLC retention times of each complex. An evaporative light scattering detector, ELSD, was used to obtain a trace of each complex upon elution from a C18 column. The mobile phase was a gradient ramp from 100% water to 100% acetonitrile. Complex 1 was detected at 9.9 min and was the most hydrophilic of the complexes. The addition of the aliphatic linker caused a significant increase in retention time, as corroborated in the logP values. Complexes 2 and 3 have similar retention times at 16.1 and 16.7 min, respectively. The most hydrophobic complex, 4, was retained longest on the column, with an elution time of 17.3 min.
As previously observed, the Gd(III) chelate has a pronounced effect on the binding affinity of progesterone to the receptor as compared to unmodified progesterone.32,34 A competitive binding assay quantified the effect of linker length on the affinity of the contrast agents for PR (Figure 2), and IC50 values are reported in Table 1. Complex 4 binds to PR with a similar affinity as that previously observed.32 Complex 2 and 3 bound PR with a similar affinity to that of 4. Complex 1 demonstrated the lowest affinity for the receptor among the series (p < 0.01), with almost 100-fold lower affinity compared to that of 2–4.
Figure 2.
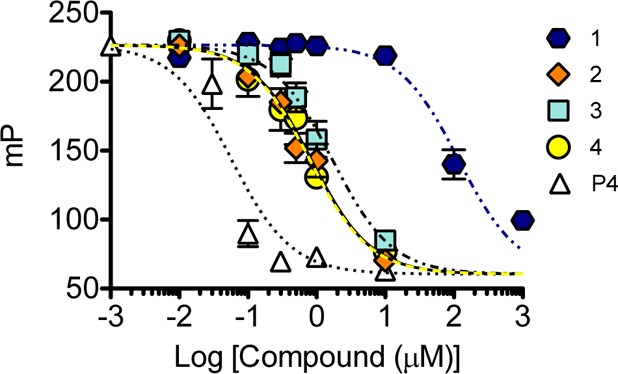
Relative binding affinity of complexes 1–4, compared with an unmodified progesterone control (P4), to progesterone receptor. As the linker length increases, the binding affinity of the probe is improved. Error bars indicate ±SEM.
Correlation Observed between LogP Values and Cytotoxicity
One aspect of enhanced water solubility for PR contrast agents may be that it reduces toxicity. MTS cell viability assays determine the cytotoxicity of the probes in vitro after incubation with varying concentrations of each complex in PR(+) T47D or PR(−) MDA-MB-231 cells (Figure 3). Cytotoxicity correlated to logP values in PR(+) T47D cells. Complex 1 exhibited the lowest toxicity at all of the concentrations tested and was the least hydrophobic. Complexes 2–4 exhibited similar toxicity in a trend following their logP values (reported in Table 1). In MDA-MB-231, all agents had similar toxicity profiles, implying that probe toxicity is associated with the presence of PR (Figure S2).
Figure 3.
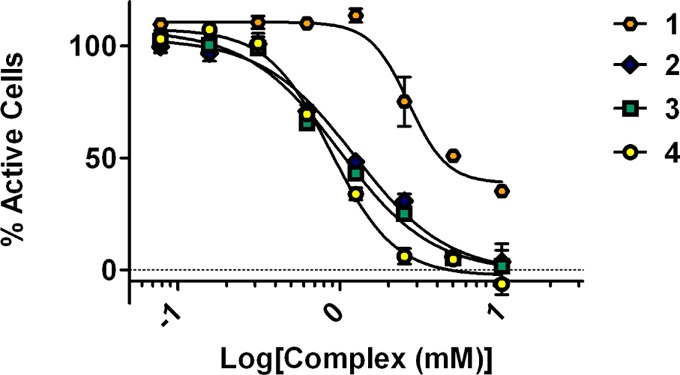
Cytotoxicity of complexes 1–4 as obtained through MTS assay on the PR(+) T47D cell line. Complex 1 is the least toxic, followed by 2, 3, and 4. Error bars indicate ±SEM.
Correlation Observed between LogP Values and Cellular Association
PR is an intracellular protein, and interaction between the contrast probes and PR requires cellular uptake utilizing a diffusion mechanism through the cellular membrane. Uptake experiments of the probes to PR(+) T47D and PR(−) MDA-MB-231 human breast cancer cells evaluated the cell permeability of the probes. Time-dependent uptake was not observed in the PR(−) cells for any of the reported complexes where Gd(III) concentrations remained consistent over all of the time points as measured by ICP-MS. Time-dependent accumulation was observed for 1 and 2 in PR(+) cells (Figure 4). Uptake of 3 and 4 in PR(+) cells increased dramatically after 1 h, with higher accumulation at 4 h before decreasing by 8 h. This trend in cellular accumulation correlated to hydrophobicity, where the most hydrophobic complex, 4, had the highest concentrations of probe in cells. In PR(+) cells, the concentrations of all of the steroid-conjugated probes are markedly higher than that found in PR(−) cells at all time points. This suggests that PR plays a role in the retention of these probes and that the ability to cross the cell membrane correlated with hydrophobicity.
Figure 4.
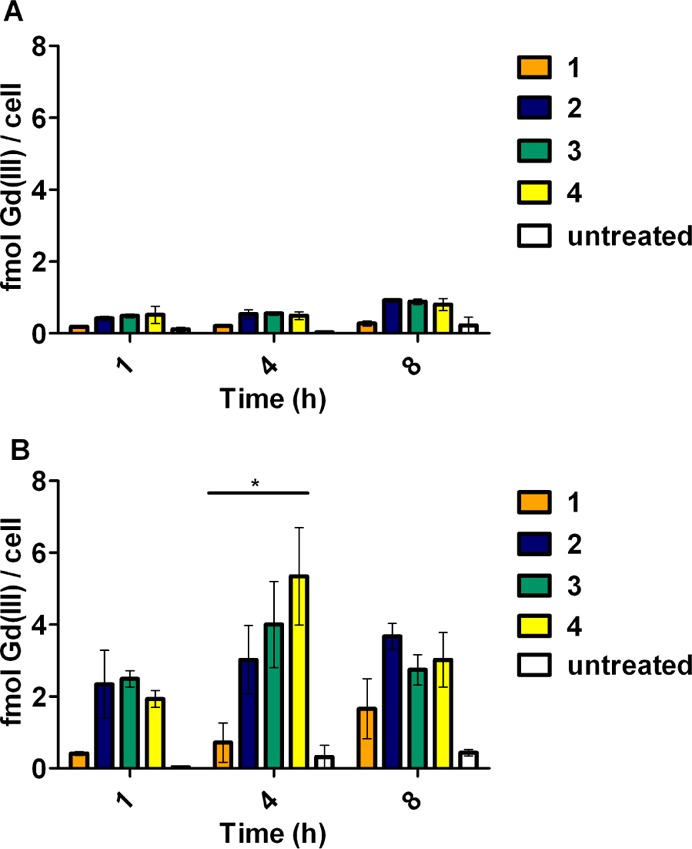
Complexes accumulate preferentially in cells that express PR. Time-dependent uptake experiments were performed in PR(−) MDA-MB-231 cells (A) and PR(+) T47D cells (B). Error bars indicate ±SEM. Statistical difference determined using two-way ANOVA test. * p < 0.05.
PR Activation Is Retained as Water Solubility Increases
To further demonstrate that the MR probes cross the cell membrane and bind PR, the ligand-activated transcription factor activity of PR was exploited using a luciferase reporter assay (Figure 5A). Upon activation, progesterone receptors dimerize and bind to a region of DNA referred to as the progesterone response element (PRE). Ligation of this DNA element to a segment of DNA encoding the luciferase gene can verify the activation of PR by these complexes through the formation of a functional transcription complex. It was expected that these agents have similar profiles of receptor activation as that of the previously developed PR-targeted MR complexes, as the chemical modification was performed in the same position.
Figure 5.
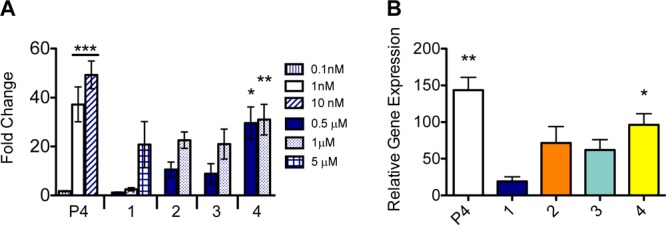
Complexes retain the ability to activate PR after chemical modification: the addition of the Gd(III) chelate to the PR-targeting moiety. Incubation with complexes resulted in transcriptional activation of the luciferase reporter gene (A). Transcription of an endogenous PR-inducible gene was monitored (B). Complex 4 was the most potent in both assays. Error bars indicate ±SEM. Statistical differences as indicated by asterisks measured by one-way ANOVA test. ** p < 0.05, * p < 0.05 (when compared to samples treated with only DMSO).
From the series of contrast agents, 4 activated transcription of the luciferase reporter gene most effectively (29.6-fold greater than solvent at 0.5 μM), whereas at the same concentration, 3 and 2 activated the reporter gene to a lesser extent (8.9- and 10.5-fold, respectively). Contrast agent 1 demonstrated the least transcriptional activation (1.2-fold). The most hydrophobic of compounds, 4 (ProGlo), was the most effective transcriptional activator, which is expected, as this complex displayed the highest binding affinity to PR. Due to the hydrophilicity and low toxicity of 1, higher concentrations of agent can be administered without harmful outcomes. The luciferase assay demonstrated that at this higher concentration of complex 1 a measurable higher activation of receptor (comparable to complexes 2 and 3) was observed. Activation of luciferase activity by progesterone and contrast agents is effectively blocked with the progesterone antagonist RU486, demonstrating that the activation of PR by these complexes is specific (Figure S3).
Transcriptional activation of an endogenous PR-regulated gene, ZBTB16, confirmed probe activation efficiency (Figure 5B). Similar to the luciferase reporter assay, 4 induced transcription of ZBTB16 at the highest rate, followed by 3, 2, and finally 1 being the least efficient while still maintaining the ability to activate transcription. The highly lipophilic 4 appears have the greatest ability to traverse the cell membrane and activate transcription.
Accumulation of Complex 1 in Uterus after in Vivo i.p. Injection
By evaluating the in vivo distribution of PR-targeted complexes, we were able to distinguish if i.v. or i.p. injections would be advantageous in terms of accumulation in PR-expressing tissues. Despite lower cellular retention and PR binding affinity, complex 1 was the only agent that was completely soluble in saline. Therefore, 1 was the only agent suitable for i.v. administration and was used to directly compare if i.v. or i.p. administration could influence cellular accumulation in PR-expressing organs. Given that most traditional MR contrast agents are administered i.v., yet most steroids are delivered i.p. or subcutaneously, a direct comparison of how the steroid modified agent would accumulate in PR-expressing tissues following standard i.p. and i.v. administration was performed. Following i.v. or i.p. injection, the organs from female CD-1 mice were harvested at 6 and 24 h. The total nanograms of Gd(III) per gram of tissue, as measured by ICP-MS, is shown in Figure 6.
Figure 6.
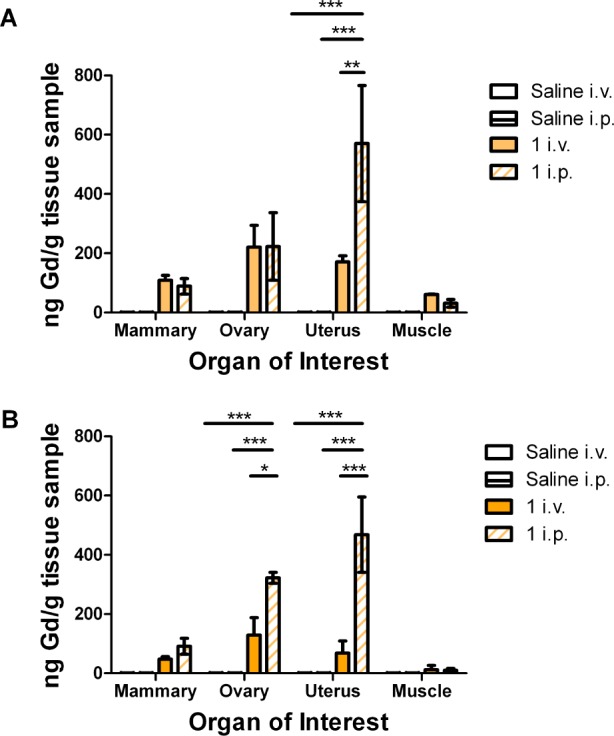
Complex 1 accumulates in tissues that have high concentrations of PR such as the uterus and ovary. 1 was injected either i.v. or i.p. into CD-1 female mice, and organs were harvested after 6 (A) or 24 (B) h. Data is presented normalized to saline ICP values. Error bars indicate ±SEM. Statistical differences determined through two-way ANOVA test. *** p < 0.0001, ** p < 0.001, * p < 0.05.
On the basis of the expression of PR, the organs of interest include the mammary gland, the ovaries, and the uterus. A section of muscle was used as a negative control, as the probe complexes should not accumulate in the muscle. The data is presented normalized to the average saline ICP values. The highest concentrations of Gd(III) from both routes of administration were found in the uterus and ovaries, similar to previous studies using MR and PET agents.12,34 The highest concentration of complex 1 was found in the uterus from the i.p. injection, significantly greater than corresponding accumulation due to i.v. administration (Figure 6 and Figure S4). In the ovary and mammary gland, 1 accumulated equivalently at 6 h but was greater after i.p. injection at 24 h. Taken together, these results demonstrate that i.v. injection does not increase delivery to PR-expressing tissues, suggesting i.v. administration is not necessary for PR-targeted MR probes.
Complex 1 Enhanced PR(+) Tissues in Vivo
To determine whether complex 1, dissolved in saline, would increase the contrast-to-noise ratio of PR(+) relative to PR(−) tumors in vivo, a preliminary xenograft tumor model in athymic nude mice was utilized. T47D (PR(+)) cells on the left and MDA-MB-231 (PR(−)) cells on the right were injected subcutaneously. Mice (n = 2) were injected i.p. with 1 and imaged at 6 h postinjection.
Contrast enhancement is observed above background muscle in the PR(+) tumor, Figure 7A. To ensure that the change in image contrast was due to the accumulation of Gd(III), the tumors were harvested, and Gd(III) concentration was quantified by ICP-MS, Figure 7B. The action of 1 did not surpass that of previously synthesized complex 4 which more effectively increased contrast resulting in a higher fold change between MR signal preinjection and at each time point.34
Figure 7.

In vivo imaging at 9.4 T after injection of complex 1 i.p. (A) MR image before injection, on the left, and 6 h after injection, on the right. PR(+) and PR(−) tumors are indicated by arrows. Positive enhancement is observed in the tumors. (B) Gd(III) concentration in the tissues harvested as quantified by ICP-MS. Gd(III) concentration was not significantly different between the tumor types.
Discussion and Conclusions
The presence and localization of steroid receptors (such as PR) are important diagnostic and prognostic markers in hormone-dependent diseases.40 The development of a noninvasive method to probe PR status would allow for repeat measurements, decreased patient discomfort, personalized treatment decisions, and improved characterization of the tumor mass. The previously developed PR-targeted MR probe, ProGlo, exhibited selective accumulation and MR enhancement of PR-rich tissues and tumors in vivo when delivered either i.p. or s.c.34 The insolubility of ProGlo and other PR-targeted contrast agents in aqueous solutions precluded the ability to investigate i.v. administration. Synthesis of a linker length series of PR-targeted MR probes was conducted to increase water solubility and lower toxicity and to determine if administration route is important for delivery. Of the three new MR probes, only complex 1 was completely water soluble, enabling the direct comparison between i.p. or i.v. injection for accumulation in PR-expressing tissues.
With increasing water solubility of PR-targeted probes, a mixture of positive and negative biological properties arose. As hydrophilicity increased, the cytotoxicity of the complexes decreased. Complex 4, ProGlo, was shown to be the most hydrophobic and exhibited the highest toxicity, whereas the water-soluble complex 1 demonstrated the lowest toxicity. The increased toxicity of ProGlo can be partially attributed to additional nonspecific interactions due to hydrophobicity; conversely, it can also be attributed to progesterone signaling, as progesterone itself can be toxic.35,41 However, with increasing water solubility, other biological properties such as receptor binding and the ability to cross the cell membrane were blunted. The more hydrophilic the compound, the more the likelihood that the probe will effectively be taken up into cells decreases, as observed with the cell accumulation assay. In addition, the binding of 1 to PR was decreased 100-fold compared to that of 2–4, potentially due to increased steric hindrance of the Gd(III) chelate. The decreased ability to cross the cell membrane and bind PR was reflected in the lower transcriptional activation. Taken together, an ideal amount of each characteristic, hydrophobicity to allow for cell uptake and hydrophilicity to increase probe safety, is essential for the optimized PR-targeted contrast probe.
The optimal delivery route for a PR-targeted MR probe is not straightforward. Typically, steroids are delivered s.c. or i.p. Alternatively, currently available MR contrast probes are commonly injected i.v. or administered orally. Given that a PR-targeted MR probe is mixture of the two aforementioned components explicitly testing the route of administration for effectively targeting PR-expressing tissues is a necessary foundation of this study.
Complex 1 was the only probe tested that could be completely dissolved into a saline solution and therefore was the only appropriate contrast agent to investigate the importance of delivery route. Although the binding and cellular association of this complex was significantly lower than the others under investigation, it was the least toxic. If i.v. delivery were essential to targeting tissues far from the peritoneal cavity such as mammary tissue, then this would allow for the use of a higher concentration of agent when performing in vivo studies with this probe, and, as mentioned, a higher concentration of this probe allows it to bind and activate PR comparably to the others. The mammary tissue, not located in the peritoneal cavity, showed no preference for delivery route. Intraperitoneal injections led to the highest concentration of Gd(III) to be found in the uterus compared with the ovary and mammary gland. This concentration of Gd(III) was highly retained even after 24 h, with no adverse effects observed in the animals. Intravenous administration allowed the probe to access and accumulate selectively in PR-rich tissues, albeit to a lesser extent than i.p. administration in tissues located within the peritoneal cavity. Intravenously delivered compounds must circulate systemically and likely become bound to serum hormone binding globulin. The observed increased accumulation in the uterus (at both 6 and 24 h) and ovary (at 24 h) following i.p. injection could be due to direct contact with the tissue, since both tissues are located in the peritoneal cavity, leading to an effectively higher concentration of 1 in the tissue.
In vivo imaging with complex 1 was not able to distinguish PR(+) from PR(–) tumors more effectively than previously tested complex 4. Analysis of Gd(III) concentration by ICP-MS reveals higher average retention of 1 in the PR(+) tumor compared with that in the PR(−) tumor, but not significantly. The increase in field strength (from 7 to 9.4 T) prevents an exact comparison between complex 4 and current work; however, at the same concentration and using the same xenograft model, complex 4 provided significant enhancement in contrast-to-noise of PR(+) versus PR(−) tumors in vivo. Mice recovered quickly after injection with 1, and no toxicity in the animal was observed.
In conclusion, a series of PR-targeted MR contrast agents with varying linker lengths was developed to impact solubility. These agents associated with cells and activated PR to varying extents. Toxicity and lipophilicity was shown to increase with increasing linker length. Despite the findings that complex 4 was more effective in binding and activating PR, the hydrophilic properties of complex 1 permit it to be used safely at higher concentrations and to investigate the route of delivery for targeting PR-expressing tissues. 1 accumulates in tissues that express a high concentration of PR, but i.v. delivery, made possible by changes to water solubility, did not improve targeting to PR-expressing tissues. MR contrast was enhanced at 9.4 T, but 1 did not display a marked improvement over the previously investigated 4. This demonstrates that focusing solely on the water solubility of the probe will not easily yield an optimized contrast agent for the imaging of hormone-dependent disease in vivo. Further optimization is required to develop the most effective probe to fulfill this urgent clinical need for noninvasive imaging of hormone-dependent diseases.
Experimental Procedures
General Materials and Methods
Unless otherwise noted, materials and solvents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO) and used without further purification. Gd(III)Cl3·6H2O and 1,4,7,10-tetraazacyclododecane (cyclen) were purchased from Strem Chemicals (Newburyport, MA) and used without further purification. All reactions were performed under an inert nitrogen atmosphere. THF, acetonitrile, and dichloromethane were purified using a Glass Contour Solvent system. Deionized water was obtained from a Millipore Q-Guard System equipped with a quantum Ex cartridge (Billerica, MA). Thin-layer chromatography (TLC) was performed on EMD 60F 254 silica gel plates. Visualization of the developed chromatogram was performed by CAM stain, potassium permanganate stain, and platinum stain. Standard grade 60 Å 230–400 mesh silica gel (Sorbent Technologies) was used for flash column chromatography. 1H and 13C NMR spectra were obtained on a Bruker 500 MHz Avance III NMR Spectrometer with deuterated solvent as noted. Electrospray ionization mass spectrometry (ESI-MS) spectra were acquired on a Varian 1200 L single-quadrupole mass spectrometer. High-resolution mass spectrometry data were acquired on an Agilent 6210 LC-TOF (ESI, APCI, APPI). Analytical reverse-phase HPLC-MS was performed on a Varian Prostar 500 system with a Waters 4.6 × 250 mm 5 μM Atlantis C18 column. This system is equipped with a Varian 380 LC ELSD system, a Varian 363 fluorescence detector, and a Varian 335 UV–vis detector. Preparative runs were performed on a Waters 19 × 250 mm Atlantis C18 column. Mobile phase consisted of water (solvent A) and HPLC-grade acetonitrile (solvent B) or 0.05% TFA in water (solvent C) and HPLC-grade acetonitrile (solvent B).
Synthesis
{1,4,7-Tris(carboxymethyl)-10-[10-(6-(2-((10R,13S,17S)-10,13-dimethyl-3-oxo-2,3,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethoxy)hexyl]-1,4,7,10-tetraazacyclododecanato}gadolinium (4, ProGlo)
The synthesis and purification of ProGlo was performed as previously described.32
{1,4,7-Tris(carboxymethyl)-10-[10-(2-((8S,9S,10R,13S,14S,17S)-10,13-dimethyl-3-oxo-2,3,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethyl]-1,4,7,10-tetraazacyclododecanato}gadolinium (1)
The synthesis and purification of 1 was performed as previously described.32
1,4,7-Tris(tert-butoxycarbonylmethyl)-1,4,7,10-tetraazacyclododecane (DO3A-tris-tert butyl ester, 9)
Cyclen (5 g, 29.0 mmol) and NaHCO3 (5.50 g, 65.5 mmol) were dissolved in anhydrous acetonitrile (150 mL). tert-Butylbromoacetate (9.60 mL, 65.0 mmol) was added dropwise under nitrogen to the solution, which was stirred at room temperature for 48 h. After filtration of the NaHCO3 and removal of the acetonitrile by rotary evaporation, the remaining crude product was dissolved in dichloromethane and washed with water. Recrystallization from toluene yielded an off-white solid (6.05 g, 40%). 1H NMR (500 MHz, chloroform-d) δ 3.38–2.88 (m, 21H), 1.47 (s, 27H). 13C NMR (125 MHz, chloroform-d) δ 170.76, 169.87, 82.07, 81.93, 58.49, 51.59, 51.17, 49.41, 47.79, 28.47, 28.43. ESI-MS m/z [M + H]+: 515.1.
{1,4,7-Tris(tert-butoxycarbonylmethyl)-1,4,7,10-tetraazacyclododecanato}gadolinium (Gd(III)-DO3A)
A solution of 9 (0.750 g, 1.46 mmol) in trifluoroacetic acid (2 mL) was stirred at room temperature for several hours. After removal of the trifluoroacetic acid, the crude free ligand was resuspended in water, and the pH was adjusted to 6. A solution of Gd(III)Cl3 in water was added slowly while maintaining the pH between 5.5 and 6.5 with 0.5 M NaOH. The solution was heated at 60 °C and stirred under nitrogen, and the pH was monitored and readjusted to maintain the pH between 5.5 and 6.5 with additional 0.5 M NaOH. The reaction mixture was lyophilized and purified by HPLC using a ramp from 0 to 100% B over 20 min to afford a white solid (0.470 g, 94%). HRMS (ESI) m/z: found, 502.09519 [M + H]+ (calcd, 502.09314 for C14H24N4O6Gd(III)).
(10R,13S,17S)-17-(2-(4-Bromobutoxy)acetyl)-10,13-dimethyl-7,8,9,10,11,12,13,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3(2H)-one (5)
To a mixture of 21-hydroxyprogesterone (0.500 g, 0.151 mmol) and 40% KOH (750 μL) was added 1,4-dibromobutane (3.6 mL, 30.3 mmol) and NBu4OH (150 μL). The reaction mixture was stirred at room temperature for 16 h, diluted in dichloromethane, and washed with water three times. The organic layer was dried over sodium sulfate and concentrated by rotary evaporation. Flash chromatography in mobile phase 3:1 hexanes/ethyl acetate yielded the final product as a colorless oil (0.301 g, 43%). 1H NMR (500 MHz, CDCl3) δ 5.61 (s, 1H), 3.93 (dd, J = 28, 17.1 Hz, 2H), 3.40 (t, J = 6 Hz, 2H), 3.36 (t, J = 6.5 Hz, 2H), 2.50 (t, J = 9 Hz, 1H), 2.31–0.86 (complex, 23H), 1.08 (s, 3H), 0.57 (1, 3H). 13C NMR (125 MHz, CDCl3) δ 208.23, 199.27, 170.94, 123.72, 76.90, 70.36, 58.35, 55.96, 53.40, 44.37, 38.43, 38.39, 35.53, 35.38, 33.79, 33.70, 32.64, 31.76, 29.33, 28.00, 24.39, 22.69, 20.85, 17.23, 13.43. ESI-MS m/z [M + H]+: 465.45
(10R,13S,17S)-17-(2-(4-Bromopentoxy)acetyl)-10,13-dimethyl-6,7,8,9,10,11,12,13,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3(2H)-one (6)
A mixture of 21-hydroxyprogesterone (200 mg, 0.605 mmol), 1,5-dibromopentane (1.65 mL, 12.1 mmol), 40% KOH (300 μL), and tetrabutylammonium hydroxide (60 μL) was stirred for 16 h at room temperature. The reaction mixture was diluted in dichloromethane and washed with water three times. The organic layer was dried over sodium sulfate and concentrated by rotary evaporation. The crude residue was purified by flash chromatography with hexanes/ethyl acetate (2:1) as the mobile phase to afford 6 as a colorless oil (125 mg, 43%). 1H NMR (500 MHz, CDCl3) δ 5.67 (s, 1H), 3.96 (q, J = 17.2 Hz, 2H), 3.41 (t, J = 6.4 Hz, 2H), 3.38–3.31 (m, 2H), 2.56 (t, J = 9.1 Hz, 1H), 2.40–2.09 (m, 6H), 1.96 (dd, J = 9.6, 3.8 Hz, 1H), 1.90–1.74 (m, 5H), 1.72–1.16 (m, 14H), 1.12 (s, 3H), 1.05–0.86 (m, 2H), 0.65–0.58 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 207.55, 198.50, 169.94, 122.92, 75.79, 70.34, 61.56, 57.43, 55.12, 52.51, 43.53, 37.54, 34.61, 32.85, 31.74, 31.47, 30.82, 28.69, 27.74, 23.77, 23.47, 21.81, 19.97, 16.34, 12.55. ESI-MS m/z [M + H]+: 481.1.
Tri-tert-butyl 2,2′,2″-(10-(4-(2-((10R,13S,17S)-10,13-dimethyl-3-oxo-2,3,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethoxy)butyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate (7)
To a solution of 5 (0.301 g, 0.0648 mmol) in anhydrous acetonitrile was added 9 (0.401 g, 0.0778 mmol), K2CO3 (0.269 g, 1.95 mmol), and NBu4OH (15 μL). The mixture was heated at 60 °C for 48 h under N2. Following filtration of the K2CO3, the solvent was removed by rotary evaporation. The crude residue was purified by flash chromatography (20:1 dichloromethane/methanol) to afford the final product as a solid (0.142 g, 25%). 1H NMR (500 MHz, CDCl3) δ 5.56 (s, 1H), 3.90 (m, 2H), 3.30–0.79 (complex, 50H), 1.30 (m, 27H), 1.02 (s, 3H), 0.52 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 207.85, 199.27, 173.31, 172.29, 170.91, 170.38, 169.81, 123.65, 82.62, 82.39, 81.54, 81.36, 76.80, 70.97, 58.70, 58.40, 56.23, 55.82, 53.30, 50.96, 49.90, 48.83, 44.23, 38.34, 35.44, 35.27, 33.71, 32.54, 31.63, 29.43, 27.96, 27.74, 27.59, 27.34, 24.26, 23.91, 22.65, 22.52, 22.23, 20.74, 19.53, 17.14, 13.55, 13.33. ESI-MS m/z [M + H]+: 899.5, [M + Na]+: 921.5.
Tri-tert-butyl 2,2′,2″-(10-(5-(2-((10R,13S,17S)-10,13-dimethyl-3-oxo-2,3,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethoxy)pentyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate (8)
To a solution of 6 (0.368 g, 0.0770 mmol) in anhydrous acetonitrile were added 9 (0.475 g, 0.0923 mmol), K2CO3 (0.319 g, 2.31 mmol), and NBu4OH (15 μL). The mixture was heated at 60 °C for 48 h. Following filtration of the K2CO3, the solvent was removed by rotary evaporation. The crude residue was purified by flash chromatography (20:1 dichloromethane/methanol) to afford the final product as a solid (0.388 g, 55%). 1H NMR (500 MHz, CDCl3) δ 5.60 (s, 1H), 3.94 (m, 2H), 3.38–0.85 (complex, 52H), 1.34 (m, 27H), 1.07 (s, 3H), 0.56 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 208.11, 199.32, 172.42, 170.92, 169.84, 123.73, 82.60, 82.31, 81.59, 71.31, 58.46, 55.92, 55.57, 53.52, 53.40, 44.31, 38.42, 35.54, 35.37, 33.80, 32.64, 31.72, 29.41, 28.05, 28.00, 27.84, 27.68, 26.07, 24.36, 23.92, 22.73, 20.83, 17.23, 13.41. ESI-MS m/z [M + H]+: 914.5, [M + Na]+: 936.40
{2,2′,2″-(10-(4-(2-((10R,13S,17S)-10,13-Dimethyl-3-oxo-2,3,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethoxy)butyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate}gadolinium (2)
A solution of 7 (1 equiv) in formic acid was stirred at room temperature for 4 h. The reaction mixture was concentrated in vacuo and resuspended in water. Gd(III)Cl3·6H2O (1.1 equiv) was added, and the solution was heated at 60 °C and stirred under nitrogen. The pH was monitored and maintained between 5.5 and 6.5 (using 0.5 M NaOH). The reaction mixture was lyophilized and purified by HPLC using a ramp from 30 to 100% B over 20 min. Analytical HPLC-MS was used to confirm the purity and identity of the collected fractions. Pure fractions were freeze-dried and stored in a desiccator. ESI-MS m/z [M + H]+: 886.3.
2,2′,2″-(10-(5-(2-((10R,13S,17S)-10,13-Dimethyl-3-oxo-2,3,6,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethoxy)pentyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid (3)
A solution of 8 (1 equiv) in formic acid was stirred at room temperature for 3–4 h. The reaction mixture was concentrated in vacuo and resuspended in water. Gd(III)Cl3·6H2O (1.1 equiv) was added, and the solution was heated at 60 °C and stirred under nitrogen. The pH was monitored and maintained between 5.5 and 6.5 (using 0.5 M NaOH). The reaction mixture was lyophilized and purified by HPLC using a ramp from 30 to 100% B over 20 min. Analytical HPLC-MS was used to confirm the purity and identity of the collected fractions. Pure fractions were freeze-dried and stored in a desiccator. ESI-MS m/z [M + H]+: 899.5.
Relaxivity
Solutions of 1–4 were prepared in 400 μL of Millipore water containing 1% DMSO for T1 and T2 acquisition to a concentration of 0.500 mM. DMSO was required for solubility of 2–4. DMSO can affect relaxivity in that protons are shifted downfield, but, as the concentration of DMSO used for these studies is so low, little effect from the solvent mixture is observed. Serial dilutions were performed to yield the experimental concentrations of 0.500, 0.250, 0.125, 0.063, and 0.031 mM. T1 and T2 relaxation times were measured on a Bruker mq60 NMR analyzer equipped with Minispec V2.51 Rev.00/NT software (Billerica, MA) operating at 1.41 T (60 MHz) and 37 °C. T1 relaxation times were measured using an inversion recovery pulse sequence (t1_ir_mb) using the following parameters: 4 scans per point, 10 data points for fitting, monoexponential curve fitting, phase cycling, 10 ms first pulse separation, and a recycle delay and final pulse separation ≥ 5T1. Measurements were performed in triplicate. The Gd(III) concentration of each solution was determined using ICP-MS on a computer-controlled (Plasmalab software) Thermo X series II ICP-MS (Thermo Fisher Scientific, Waltham, MA, USA) operating in standard mode equipped with an ESI SC-2 autosampler (Omaha, NE). The inverse of the relaxation time (1/T1, s–1) (taken as the average of the three replicates) was plotted against Gd(III) concentration (mM) and fitted to a straight line with R2 > 0.99. The slope of the fitted line was recorded as the relaxivity, r1.
Octanol–Water Partition Coefficients
Each complex (0.5 mg) was dissolved in 1 mL of a 1:1 mixture of water/1-octanol. After shaking the sample tube vigorously for 30 s, the tube was placed on a rotator for gentle mixing for 4 h. The tube was removed from the rotator, and complete separation of the aqueous and organic phases was allowed over 10 h. Fifty microliters was removed from each layer and subjected to ICP-MS to determine the Gd(III) concentration in each layer. The partition coefficient was calculated from the following equation
where logP is the logarithm of the partition coefficient, Co is the concentration of Gd(III) in the 1-octanol layer, and Cw is the concentration of Gd(III) in the water layer.
ICP-MS Sample Preparation and Instrument Parameters
For logP measurements, ACS reagent grade nitric acid (70%) was added to solutions of the agent in water or 1-octanol (for a 1.0:1.0 v/v sample/nitric acid) in 15 mL conical tubes and placed at 65 °C for 4 h to allow for complete sample digestion. For samples in 1-octanol, caps were removed from tubes and replaced to vent tubes every 30 min due to the buildup of pressure. Filtered, deionized H2O (18.2 MΩ·cm) and multielement internal standard containing Bi, Ho, In, Li, Sc, Tb, and Y (Inorganic Ventures, Christiansburg, VA) were added to produce a final solution of 3.0% nitric acid (v/v) and 5.0 ng/mL internal standard. Instrument calibration was accomplished by preparing individual-element Gd(III)(III) standards (Inorganic Ventures, Christiansburg, VA, USA) using concentrations of 0.7813, 1.563, 3.125, 6.250, 12.50, 25.00, 50.00, 100.0, and 200.0 ng/mL containing 3.0% nitric acid (v/v) and 5.0 ng/mL of the multielement internal standard.
General Cell Culture and Animal Studies
Dulbecco’s modified phosphate buffered saline (DPBS), media, sera, and dissociation reagents were purchased from Invitrogen (Carlsbad, CA). Cell culture consumables (flasks, plates, etc.) were purchased from VWR (Radnor, PA). Charcoal dextran stripped FBS was purchased from Atlanta Biologicals (Lawrenceville, GA). MDA-MB-231 cells were cultured using phenol red free α-MEM (modified to contain 20 ng/mL insulin) supplemented with 10% FBS (characterized) or with 10% charcoal dextran stripped FBS. T47D cells were cultured using phenol red free RPMI 1640 (modified to contain 1.0 mM sodium pyruvate, 1.0 mM HEPES, and 4.5 g/L glucose) supplemented with 10% FBS or 10% charcoal dextran stripped FBS. Prior to all experiments, cells were plated in the appropriate media containing FBS. After plating, this media was replaced with media containing charcoal dextran stripped FBS and incubated at 37 °C in a 5.0% CO2 incubator for 24 h at which point the media was replaced with fresh charcoal dextran stripped FBS containing media, and the cells were incubated an additional 24 h prior to beginning the experiment. MDA-MB-231 and T47D cells were harvested by incubation with 0.25% TrypLE for 10 min at 37 °C in a 5.0% CO2 incubator. All incubations were carried out at 37 °C in a 5.0% CO2 incubator unless otherwise specified.
Female CD-1 mice, acquired from Harlan (Indianapolis, IN), and female Balb/C athymic nude mice, acquired from Charles River (Wilmington, MA), were housed under pathogen-free conditions. All animal studies were conducted at University of Illinois at Chicago in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and established institutional animal use and care protocols.
Cell Counting and Percent Cell Viability Determination Using Guava EasyCyte Mini Personal Cell Analyzer (PCA) System
After cell harvesting, an aliquot (15 or 30 μL) of the cell suspensions was mixed with Guava ViaCount reagent (final sample volume of 150 μL) and incubated at room temperature for 5 min. After incubation, samples were vortexed for 10 s. Cells were counted, and percent cell viability was determined via manual analysis using a Guava EasyCyte Mini Personal Cell Analyzer (PCA) and ViaCount software module. For each sample, 1000 events were acquired with dilution factors that were determined on the basis of the optimum machine performance (∼25–70 cells/μL). Instrument reproducibility was assessed daily using GuavaCheck Beads and following the manufacturer’s suggested protocol using the Daily Check software module.
Cellular Uptake Studies
Cells (either T47D or MDA-MB-231) were plated at 12 000 cells per well in a 48-well plate. Contrast agents were dissolved in DMSO to obtain a stock solution of 100 mM. An incubation solution of 0.125 mM contrast agent was made by diluting the stock solution in the appropriate media (containing stripped FBS) for each cell line (T47D and MDA-MB-231). Cells were incubated with 150 μL of the 0.125 mM solution for 1, 4, and 8 h. After incubation, the media was removed, and the cells were rinsed twice with 0.500 mL of DPBS and harvested by incubation with 50 μL of 0.25% TrypLE for 10 min at 37 °C in a 5.0% CO2 incubator. Fifty microliters of media was added to each well, a 30 μL aliquot was removed for cell counting, and a 60 μL aliquot was analyzed for Gd(III) content by ICP-MS. Each condition was done in triplicate.
Cytotoxicity: MTS Assay
The CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) was used to measure cell viability. Cells, either T47D or MDA-MB-231, were plated at 500–1000 cells/well in 96–well plates and maintained in media containing charcoal stripped serum for 48 h before the experiment. 4-Hydroxytamoxifen, 1, 2, 3, 4, and Gd(III)-DO3A were dissolved in DMSO to make a stock solution of 100 mM. Solutions were diluted to the experimental concentrations (10.0, 5.00, 2.5, 1.25, 0.625, 0.313, 0.156 mM) in media. After 48 h of incubation with 50 μL of solution, the assay was run according to the manufacturer’s protocol. Absorbance at 490 nm was measured using a Synergy4 microplate reader (BioTek, Winooski, VT). Percent toxicity was calculated on the basis of the absorbance.
Luciferase Assay for PR Activation
T47D cells were grown in phenol red-free medium, and the cells were trypsinized and plated in 24-well plate (50 000 cells/well). Incubation of cells with pPRE-luciferase plasmid (100 ng/well, construct provided by Dr. Ken Korach, NIEHS, NIH), RSV-β-galactosidase (100 ng/well, provided by Dr. William T. Beck, University of Illinois at Chicago), and Lipofectamine 2000 (1 μL per well, Invitrogen, Carlsbad, CA) in Opti-MEM was performed overnight at 37 °C inside a humidified incubator. The cells were treated with complexes 1–4 or controls for an additional 24 h.
To measure luciferase production, cells were lysed in 100 μL of GME buffer [25 mM glycylglycine (pH 7.8), 15 mM MgSO4, 4 mM EGTA, 1 mM dithiothreitol, and 1% Triton X-100], and lysates were added to assay buffer (GME buffer, 16.5 mM KPO4, 2.2 mM ATP, and 1.1 mM dithiothreitol). Luciferase substrate was injected followed by a 30 s read by a FLUOstar OPTIMA (BMG Lab Tech, Offenburg, Germany). LacZ activity (50 μL lysate) was measured from cleavage of ONPG. The sample results were normalized to β-galactosidase to account for transfection efficiency by dividing the sum of the luciferase activity by the sum of the β-galactosidase activity.
qPCR
T47D cells were plated at 300 000 cells/well in 6–well plates in phenol red-free RPMI1640 media supplemented with 4.5 g/L glucose and 10% charcoal dextran stripped FBS. Twenty four hours later, cells were washed once with 1× PBS and incubated with contrast agents in DMSO or DMSO only in fresh phenol red-free RPMI1640 media supplemented with 4.5 g/L glucose and 10% charcoal dextran stripped FBS for 24 h. After treatment, cells were harvested in 0.5 mL of Trizol, and RNA was extracted using the manufacturer’s protocol. RNA was treated with DNaseI (New England Biolabs, Ipswich, MA) for 10 min at 37 °C followed by inactivation at 75 °C for 10 min. RNA samples (1 μg) were primed with random primers (Promega, Madison, WI) and then reverse transcribed using Revertaid Reverse Transcriptase (Fermentas, Glen Burnie, MD) according to the manufacture’s protocol. For PCR amplification, the cDNA was diluted 1:10, and 1 μL was used in an 11 μL reaction using FastStart Universal SYBR Green Master Mix (ROX) (Roche) in a VIIA7 Real Time PCR System (Life Technologies) under the following conditions: hold at 94 °C for 10 min, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The primers for ZBTB16 and GAPDH were used at 0.5 μM and were the same as described.35 Induction of ZBTB16 was normalized to GAPDH, and fold change was calculated using the ΔΔCt method.
Biodistribution
Animals were injected with 0.15 mmol/kg body weight of 1. After an incubation of 6 or 24 h, organs were harvested, and quantification of Gd(III) was performed using ICP-MS of acid-digested samples. For organ digestion, Teflon tubes were boiled in a mixture of ∼1–5% Alconox (w/v) and 3.0% (v/v) ACS reagent grade nitric acid (70%) to ensure complete removal of lipid and residual Gd(III). The tubes were washed with filtered, deionized H2O (18.2 MΩ·cm) and air-dried. Organs were weighed in clean Teflon tubes followed by the addition of 1 mL of ACS reagent grade nitric acid (70%) per gram of tissue. Samples were digested in a Milestone EthosEZ microwave digestion system (Shelton, CT, USA) with a 120 °C temperature ramp for 30 min (600 W) and a 120 °C hold for 30 min (400 W), followed by a 45 min exhaust cycle. The resultant liquefied organ samples were weighed, with a portion of each sample being placed in a clean preweighed 15 mL conical tube followed by addition of multielement internal standard and filtered, deionized H2O (18.2 MΩ·cm) to produce a final solution of 3.0% nitric acid (w/w) and 5 ng/mL multielement internal standard containing Bi, Ho, In, Li, Sc, Tb, and Y (Inorganic Ventures, Christiansburg, VA) and filtered, deionized H2O (18.2 MΩ·cm) to a final volume of 5 mL. Instrument calibration was performed by preparing individual-element Gd(III)(III) standard (Inorganic Ventures, Christiansburg, VA) using concentrations of 1.000, 5.000, 10.00, 20.00, 50.00, 100.0, and 200.0 ng/mL containing 3.0% nitric acid (v/v) and 5.0 ng/mL of the multielement internal standard.
Tumor Xenograft Model
A 17β-estradiol pellet (Innovative Research of America, Sarasota, FL, 70 day release, 0.72 mg/pellet) was implanted in the nape of the neck of athymic nude mice due to their intrinsic low circulating estradiol levels. This pellet ensures the growth of the estrogen-dependent T47D cells. Two days later, T47D and MDA-MB-231 ((1–2) × 106) cells were suspended in Matrigel (1:1 ratio by volume) and injected into the rear flanks (MDA-MB-231 cells on the right and T47D on the left). Mice were monitored for tumor growth every other day after inoculation. Mice were imaged when tumors were palpable.
In Vivo MR Imaging
Xenografted nude mice (n = 2) were injected i.p. with complex 1 at a concentration of 0.15 mmol/kg mouse weight dissolved in buffered saline solution. During imaging, mice were held under anesthesia (1–3% inhaled isoflurane). Mice were allowed to regain consciousness and recover in between imaging time points. Tubing containing heated water was placed under the animals to maintain a constant body temperature. All imaging experiments were performed on a 30 cm bore BioSpec 9.4 T MR imager fitted with 12 cm gradient inserts (Bruker BioSpin, Billerica, MA) using a 40 mm quadrature volume coil.
Standard T1-weighted rapid acquisition rapid echo (RARE) scans were used for imaging the xenografts. The parameters were as follows: TR = 1500 ms, TE = 8.5 ms, flip angle = 180.0°, FOV = 3 cm, matrix size = 256 × 256, slice thickness = 0.75 mm, and interslice distance = 0.75 mm. Images were analyzed using the ImageJ software package. Contrast-to-noise ratios (CNR) were calculated using the equation CNR = (SItissue – SImuscle)/σnoise, where SItissue is the signal intensity in the tissue of interest or tumor mass, SImuscle is the signal intensity in the muscle, and σnoise is the standard deviation of the noise. CNRs were averaged over two to three axial slices in which the tumors were clearly visible.
Acknowledgments
We thank Dan Lantvit for his advice and expertise concerning the design and execution of animal studies. This work was supported by NIH grant R01EB014806. T.R.T. acknowledges that this material is based on work supported by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1324585. This work was supported by the Northwestern University High Throughput Analysis Laboratory. Metal analysis was performed at the Northwestern University Quantitative Bioelemental Imaging Center, generously supported by NASA Ames Research Center NNA06CB93G. We would like to acknowledge Alex Waters for her imaging expertise. Imaging work was performed at the Northwestern University Center for Advanced Molecular Imaging, generously supported by NCI CCSG P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center.
Supporting Information Available
Synthetic scheme for PR-targeted contrast probes, luciferase assay with PR antagonist RU-486 results, and HPLC traces of pure compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Czernin J.; Weber W. A.; Herschman H. R. (2006) Molecular imaging in the development of cancer therapeutics. Annu. Rev. Med. 57, 99–118. [DOI] [PubMed] [Google Scholar]
- Weissleder R. (2006) Molecular imaging in cancer. Science 312, 1168–1171. [DOI] [PubMed] [Google Scholar]
- Kim J. J.; Kurita T.; Bulun S. E. (2013) Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer. Endocr. Rev. 34, 130–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q.; Li Z. (2013) Molecular imaging probes for diagnosis and therapy evaluation of breast cancer. Int. J. Biomed. Imaging 2013, 230487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K.; Mori M.; Uchiyama M.; Iwai K.; Iwasaka T.; Sugimori H. (1998) Prognostic significance of progesterone receptor immunohistochemistry in endometrial carcinoma. Gynecol. Oncol. 69, 220–225. [DOI] [PubMed] [Google Scholar]
- Heinlein C. A.; Chang C. (2004) Androgen receptor in prostate cancer. Endocr. Rev. 25, 276–308. [DOI] [PubMed] [Google Scholar]
- Ahmad N.; Kumar R. (2011) Steroid hormone receptors in cancer development: a target for cancer therapeutics. Cancer Lett. 300, 1–9. [DOI] [PubMed] [Google Scholar]
- Orlando L.; Schiavone P.; Fedele P.; Calvani N.; Nacci A.; Rizzo P.; Marino A.; D’Amico M.; Sponziello F.; Mazzoni E.; Cinefra M.; Fazio N.; Maiello E.; Silvestris N.; Colucci G.; Cinieri S. (2010) Molecularly targeted endocrine therapies for breast cancer. Cancer Treat. Rev. 36, S67–S71. [DOI] [PubMed] [Google Scholar]
- Uharcek P. (2008) Prognostic factors in endometrial carcinoma. J. Obstet. Gynaecol. Res. 34, 776–783. [DOI] [PubMed] [Google Scholar]
- Boruban M. C.; Altundag K.; Kilic G. S.; Blankstein J. (2008) From endometrial hyperplasia to endometrial cancer: insight into the biology and possible medical preventive measures. Eur. J. Cancer Prev. 17, 133–138. [DOI] [PubMed] [Google Scholar]
- Cui X.; Schiff R.; Arpino G.; Osborne C. K.; Lee A. V. (2005) Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J. Clin. Oncol. 23, 7721–7735. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Zhou H. B.; Dence C. S.; Carlson K. E.; Welch M. J.; Katzenellenbogen J. A. (2010) Development of [F-18]fluorine-substituted Tanaproget as a progesterone receptor imaging agent for positron emission tomography. Bioconjugate Chem. 21, 1096–1104. [DOI] [PubMed] [Google Scholar]
- Singh M.; Zaino R. J.; Filiaci V. J.; Leslie K. K. (2007) Relationship of estrogen and progesterone receptors to clinical outcome in metastatic endometrial carcinoma: a Gynecologic Oncology Group Study. Gynecol. Oncol. 106, 325–333. [DOI] [PubMed] [Google Scholar]
- Hospers G. A.; Helmond F. A.; de Vries E. G.; Dierckx R. A.; de Vries E. F. (2008) PET imaging of steroid receptor expression in breast and prostate cancer. Curr. Pharm. Des. 14, 3020–3032. [DOI] [PubMed] [Google Scholar]
- Kiyono Y.; Mori T.; Okazawa H. (2012) Positron emission tomography radiopharmaceuticals for sex steroid hormone receptor imaging. Curr. Med. Chem. 19, 3266–3270. [DOI] [PubMed] [Google Scholar]
- Dehdashti F.; Laforest R.; Gao F.; Aft R. L.; Dence C. S.; Zhou D.; Shoghi K. I.; Siegel B. A.; Katzenellenbogen J. A.; Welch M. J. (2012) Assessment of progesterone receptors in breast carcinoma by PET with 21–18F-fluoro-16α,17α-[(R)-(1′-α-furylmethylidene)dioxy]-19-norpregn-4-ene-3,20-dione. J. Nucl. Med. 53, 363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler A. M.; Chan S. R.; Sharp T. L.; Fettig N. M.; Zhou D.; Dence C. S.; Carlson K. E.; Jeyakumar M.; Katzenellenbogen J. A.; Schreiber R. D.; Welch M. J. (2012) Small-animal PET of steroid hormone receptors predicts tumor response to endocrine therapy using a preclinical model of breast cancer. J. Nucl. Med. 53, 1119–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomper M. G.; Katzenellenbogen J. A.; Welch M. J.; Brodack J. W.; Mathias C. J. (1988) 21-[18F]Fluoro-16α-ethyl-19-norprogesterone: synthesis and target tissue selective uptake of a progestin receptor based radiotracer for positron emission tomography. J. Med. Chem. 31, 1360–1363. [DOI] [PubMed] [Google Scholar]
- Paquette M.; Ouellet R.; Archambault M.; Croteau É.; Lecomte R.; Bénard F. (2012) [18F]-Fluoroestradiol quantitative PET imaging to differentiate ER+ and ERα-knockdown breast tumors in mice. Nucl. Med. Biol. 39, 57–64. [DOI] [PubMed] [Google Scholar]
- Vijaykumar D.; Mao W.; Kirschbaum K. S.; Katzenellenbogen J. A. (2002) An efficient route for the preparation of a 21-fluoro progestin-16α,17α-dioxolane, a high-affinity ligand for PET imaging of the progesterone receptor. J. Org. Chem. 67, 4904–4910. [DOI] [PubMed] [Google Scholar]
- Couture J. F.; Legrand P.; Cantin L.; Luu-The V.; Labrie F.; Breton R. (2003) Human 20α-hydroxysteroid dehydrogenase: crystallographic and site-directed mutagenesis studies lead to the identification of an alternative binding site for C21-steroids. J. Mol. Biol. 331, 593–604. [DOI] [PubMed] [Google Scholar]
- Weissleder R. (2002) Scaling down imaging: molecular mapping of cancer in mice. Nat. Rev. Cancer 2, 11–18. [DOI] [PubMed] [Google Scholar]
- Caravan P. (2007) Physicochemical Principles of MR Contrast Agents. In Molecular and Cellular MR Imaging (Modo M. M. J., and Bulte J. W. M., Eds.) pp 13–36, Chapter 2, Taylor & Francis Group, LLC, Boca Raton, FL. [Google Scholar]
- Caravan P.; Farrar C. T.; Frullano L.; Uppal R. (2009) Influence of molecular parameters and increasing magnetic field strength on relaxivity of gadolinium- and manganese-based T1 contrast agents. Contrast Media Mol. Imaging 4, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravan P. (2006) Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem. Soc. Rev. 35, 512–523. [DOI] [PubMed] [Google Scholar]
- Frullano L.; Meade T. J. (2007) Multimodal MRI contrast agents. JBIC, J. Biol. Inorg. Chem. 12, 939–949. [DOI] [PubMed] [Google Scholar]
- Major J. L.; Meade T. J. (2009) Bioresponsive, cell-penetrating, and multimeric MR contrast agents. Acc. Chem. Res. 11, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastarone D. J.; Harrison V. S.; Eckermann A. L.; Parigi G.; Luchinat C.; Meade T. J. (2011) A modular system for the synthesis of multiplexed magnetic resonance probes. J. Am. Chem. Soc. 133, 5329–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade T. J.; Taylor A. K.; Bull S. R. (2003) New magnetic resonance contrast agents as biochemical reporters. Curr. Opin. Neurobiol. 13, 597–602. [DOI] [PubMed] [Google Scholar]
- Allen M. J.; Meade T. J. (2004) Magnetic resonance contrast agents for medical and molecular imaging. Met. Ions Biol. Syst. 42, 1–38. [PubMed] [Google Scholar]
- El Khouli R. H.; Macura K. J.; Kamel I. R.; Jacobs M. A.; Bluemke D. A. (2011) 3-T dynamic contrast-enhanced MRI of the breast: pharmacokinetic parameters versus conventional kinetic curve analysis. AJR, Am. J. Roentgenol. 197, 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Burdette J. E.; MacRenaris K. W.; Mustafi D.; Woodruff T. K.; Meade T. J. (2007) Rational design, synthesis, and biological evaluation of progesterone-modified MRI contrast agents. Chem. Biol. 14, 824–834. [DOI] [PubMed] [Google Scholar]
- Osborne C. K.; Schiff R.; Arpino G.; Lee A. S.; Hilsenbeck V. G. (2005) Endocrine responsiveness: understanding how progesterone receptor can be used to select endocrine therapy. Breast 14, 458–465. [DOI] [PubMed] [Google Scholar]
- Sukerkar P. A.; MacRenaris K. W.; Meade T. J.; Burdette J. E. (2011) A steroid-conjugated magnetic resonance probe enhances contrast in progesterone receptor expressing organs and tumors in vivo. Mol. Pharmaceutics 8, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukerkar P. A.; MacRenaris K. W.; Townsend T. R.; Ahmed R. A.; Burdette J. E.; Meade T. J. (2011) Synthesis and biological evaluation of water-soluble progesterone-conjugated probes for magnetic resonance imaging of hormone related cancers. Bioconjugate Chem. 22, 2304–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais A.; Biton I. E.; Margalit R.; Degani H. (2013) Characterization of estrogen-receptor-targeted contrast agents in solution, breast cancer cells, and tumors in vivo. Magn. Reson. Med. 70, 193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusher S. J.; Raaijmakers H. C. A.; Vu-Pham D.; Kazemier B.; Bosch R.; McGuire R.; Azevedo R.; Hamersma H.; Dechering K.; Oubrie A.; van Duin M.; de Vlieg J. (2012) X-ray structures of progesterone receptor ligand binding domain in its agonist state reveal differing mechanisms for mixed profiles of 11β-substituted steroids. J. Biol. Chem. 287, 20333–20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madauss K. P.; Deng S. J.; Austin R. J.; Lambert M. H.; McLay I.; Pritchard J.; Short S. A.; Stewart E. L.; Uings I. J.; Williams S. P. (2004) Progesterone receptor ligand binding pocket flexibility: crystal structures of the norethindrone and mometasone furoate complexes. J. Med. Chem. 47, 3381–3387. [DOI] [PubMed] [Google Scholar]
- Williams S. P.; Sigler P. B. (1998) Atomic structure of progesterone complexed with its receptor. Nature 393, 392–396. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. (2007) The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 6, 881–890. [DOI] [PubMed] [Google Scholar]
- Pedram B.; Oeveren A. v.; Mais D. E.; Marschke K. B.; Verbost P. M.; Groen M. B.; Zhi L. (2008) A tissue-selective nonsteroidal progesterone receptor modulator: 7,9-difluoro-5-(3-methylcyclohex-2-enyl)-2,2,4-trimethyl-1,2-dihydrochromeno[3,4-f]quinoline. J. Med. Chem. 51, 3696–3699. [DOI] [PubMed] [Google Scholar]
- Núñez F. A. A.; Yalkowsky S. H. (1997) Correlation between log P and ClogP for some steroids. J. Pharm. Sci. 86, 1187–1189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.